
Abstract
We undertook this phase I study to investigate the feasibility of the combination of temozolomide (TMZ) and lapatinib (LP) and to define the maximum tolerated dose (MTD) of LP in patients with relapsed high-grade gliomas. Eligible patients were enrolled in this dose escalation study of LP. TMZ was administered at a fixed dose of 200 mg/m2 d1–d5 every 28 days. Starting dose of LP was set at 1,000 mg daily continuously, escalated by 250 mg in cohorts of minimum three patients. Translational research investigations were also undertaken in available biopsy material. Between January 2009 and December 2010, 16 patients were entered into the study at three LP levels: 1,000 mg sid (11 patients), 1,250 mg sid (4 patients) and 1,500 mg sid (1 patient). A total of 55 cycles had been delivered. Fourteen patients had stopped treatment because of disease progression, and two because of toxicity. Three patients received 10, 11 and 17 cycles of treatment. Dose-limiting hematological toxicity was observed in 2 patients at the second LP dose level of 1,250 mg sid. MTD was defined at LP 1,000 mg sid. Median progression-free survival (PFS) and survival were 2.4 and 5.9 months, respectively. EGFR amplification and EGFRvIII expression were not related to PFS. Combination of TMZ and LP is feasible with manageable toxicity. The activity of this combination in patients with recurrent glioblastoma multiforme is further investigated in a recently initiated phase II trial.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gliomas are the most common primary central nervous system tumors, with an incidence of about 25,000–30,000 new cases annually. More than half of all gliomas, including glioblastoma multiforme (GBM) and anaplastic astrocytoma (AA), exhibit aggressive clinical behavior. GBM and AA are clinically managed in a similar fashion; however, despite all efforts, their prognosis remains dismal [25]. As recently published data showed, overall survival of patients with newly diagnosed GBM, the most common malignant glioma, was 42.4 % at 6 months, 17.7 % at 1 year, and 3.3 % at 2 years, despite access to state-of-the-art surgery, imaging, radiotherapy and chemotherapy [34].
Current therapeutic strategies include surgical resection followed by radiotherapy and adjuvant temozolomide (TMZ), which contributes to a small but statistically significant survival benefit [30]. However, recurrence inevitably occurs in the majority of patients, followed by death from the disease. Therefore, the need of novel active treatments remains a pressing issue.
Amplification, overexpression and mutations of the EGFR family members are quite common in gliomas, and the concept of targeted treatment has been applied in clinical research for some time now [10]. Gefitinib and erlotinib have already been tested in gliomas, and data show that in contrast to the lung cancer pattern of response, amplification and mutations of the EGFR gene are not predictive of response in glioma patients [22, 23, 32]. However, recent data demonstrated that coexpression of EGFRvIII and PTEN by glioblastoma cells is associated with responsiveness to EGFR kinase inhibitors [6].
We undertook a phase I trial of temozolomide and lapatinib, an orally active small molecule that reversibly inhibits EGFR and HER2 tyrosine kinases, to investigate the feasibility of the combination and characterize their toxicity profile in patients with recurrent high-grade gliomas following initial standard treatment.
Available tumor tissues were also profiled for standard genetic glioma markers, as well as for a panel of mRNA targets [24]. The latter were selected taking into account (a) the frequently observed EGFR amplification and the expression of its truncated variant, EGFRvIII in glioblastoma, (b) the EGFR signalling through STAT3/STAT5, which has been proposed as a for therapeutic targeting [21, 26, 37], and (c) the characterization of currently discussed molecular glioma subtypes [4, 33].
Materials and methods
Patient selection
Adult patients with confirmed histological diagnosis of grade IV glioblastoma multiforme or grade III anaplastic astrocytoma, who relapsed after optimal treatment and for whom there were no other standard treatments available, were included. All patients were required to have a WHO performance status of ≤2 and be >18 years old with measurable disease at screening, according to RANO criteria [36]. Adequate hematological function (absolute neutrophil count [ULN] ≥1.5 × 109/L, platelet count ≥100 × 109/L and hemoglobin ≥9 g/dL), liver function (total bilirubin <1.5 × upper limit of normal [ULN], AST, ALT <2.5 × ULN) and renal function (serum creatinine ≤1.25 × ULN or calculated creatinine clearance ≥50 mL/min) were mandatory for study entry. Patients were also required to have normal cardiac function (left ventricular ejection fraction within institutional normal range) and in the past 6 months have had no serious cardiac illness or medical condition, including but not confined to: history of documented congestive heart failure, high-risk uncontrolled arrhythmias, angina pectoris requiring antianginal medication, clinically significant valvular heart disease, evidence of transmural infarction on ECG and poorly controlled hypertension (e.g., systolic >180 mmHg or diastolic greater than 100 mmHg). If a patient required anticoagulant therapy, the patient was allowed to remain on study provided that he/she was carefully monitored.
Drugs and several herbal constituents (e.g., bergamontin and glabridin), which are inducers or inhibitors of CYP3A4, were prohibited during and 10 days prior to the initiation of the treatment. Available archived tumor tissue for subsequent translational research studies was mandatory for study entry.
The clinical protocol and the related translational research studies were approved by the Hellenic Cooperative Oncology Group (HeCOG) Protocol Review Committee, the Institutional Review Boards of Papageorgiou and Ioannina Hospitals and the National Organization for Medicines. The trial was included in the Australian New Zealand Clinical Trials Registry (ANZCTR) and allocated the following Registration Number: ACTRN12611000418976. Upon participation in the trial, all patients provided a study specific written informed consent and a separate informed consent for providing biological material for future research studies. The study was conducted according to the Declaration of Helsinki and the guidelines for Good Clinical Practice.
Dosage and dose escalation schema
Patients were enrolled in the phase I study in cohorts of at minimum three patients. Patients were to receive TMZ 200 mg/m2 once daily for 5 consecutive days in cycles of 28 days. The TMZ dose was fixed at each dose level. The starting dose of lapatinib was 1,000 mg, administered once daily. Three dose levels of lapatinib were planned to be explored (1,000, 1,250 and 1,500 mg). Patients would receive LP/TMZ combination until disease progression or unacceptable toxicity. They were advised to take the LP and TMZ tablets on an empty stomach (either 1 h before or 1 h after meals). Antiemetic prophylaxis with a 5HT3 inhibitor was used for the 5 days of temozolomide administration. Cohorts of three patients received escalating doses of lapatinib until the maximum tolerated dose (MTD) was determined, as recorded in week 4 of the treatment (end of the 1st treatment cycle of the combination, day 28 of the treatment).
The MTD was defined as the dose preceding that at which two of a maximum six patients experienced dose-limiting toxicity (DLT). If one DLT was observed within a cohort, then the cohort was expanded to include three more patients. If no further DLTs were observed, dose escalation proceeded. If a second DLT was observed within the expanded cohort, then the MTD was reached. Patients who developed a DLT were withdrawn from the trial but were offered the option to continue treatment at a lower dose, off-protocol, if they had derived benefit from this therapy. The dose level below the MTD was then expanded. At this dose, a minimum of eight patients had to be treated before that dose was defined to be the recommended dose for a subsequent phase II trial (<30 % DLTs).
Patients continued on their initial dose until the development of unacceptable toxicity or disease progression. Dose escalation between cohorts was done according to the modified continual reassessment method, combined with review of available clinical and laboratory data. The evaluable patient population for the determination of the MTD consisted of patients who had met the minimum safety evaluation requirements of the study. Antitumor efficacy was assessed after two cycles of treatment.
Evaluation of toxicity
Toxicity was graded according to the National Cancer Institute Common Toxicity Criteria. Complete blood count, biochemical analyses and clinical toxicity assessments were performed weekly during all treatment courses. Dose escalation and determination of DLT and MTD were performed on the basis of toxicity occurring during the first cycle of treatment.
Dose limiting toxicity
DLT was defined as any grade 3, grade 4 or life threatening toxicity requiring hospitalization. Specifically a DLT was defined as: ANC <0.5 × 109/L (grade 4) for over seven consecutive days, febrile neutropenia [ANC < 1.0 × 109/L (grade 3) when accompanied by fever], hemoglobin <6.5 g/dL (grade 4) and grade 4 thrombocytopenia (platelet count <25 × 109/L). Any grade 3–4 non-hematological toxicity except alopecia, nausea, vomiting and fever, which could be rapidly controlled with appropriate measures, also consisted a DLT.
Evaluation of response
Patients completing at least two cycles of treatment with at least one follow-up tumor assessment were considered evaluable for response. An initial tumor assessment for all patients was performed within 4 weeks prior to treatment initiation, and thereafter every two cycles while on therapy and every 2 months thereafter until progression of disease. Response was documented using the RANO response criteria for high-grade gliomas [36]. Confirmation of objective responses was required in all cases at a minimum time interval of 4 weeks. Duration of response was calculated from the time the objective response was documented until the date of disease progression. Stable disease was measured from the start of the treatment until the criteria for progression were met.
Translational research
These investigations were undertaken at the Laboratory of Molecular Oncology of the Hellenic Foundation for Cancer Research, Aristotle University of Thessaloniki School of Medicine.
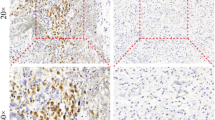
For the assessment of PTEN protein expression, a tissue microarray (TMA) block, containing two 1.5-mm tissue cores from the tumor area was created from the original blocks using the Beecher Instruments MTA-1 Tissue Arrayer (Beecher Instruments, Sun Prairie, WI). Serial 3-μm-thick TMA sections, mounted on adhesion microscope slides, were cut at the Laboratory of Molecular Oncology of the Hellenic Foundation for Cancer Research, Aristotle University of Thessaloniki School of Medicine. The IHC labeling was performed using the Bond Max™ (Leica Microsystems, Wezlar, Germany) and i6000 (Biogenex, San Ramon, CA) autostainers. Sections were stained with an antibody against PTEN, clone 6H2.1, Dako, Glostrup, DK, at dilution 1:300, for 30 min. DAB (3,3-diaminobenzidine) was used as a chromogen and Harris hematoxylin as a counterstain. The quality of IHC staining was evaluated using internal positive controls (endothelial cells). PTEN staining was scored according to Mellinghoff et al. [16], using a scale of 0–2. Vascular endothelial cells were used as control markers of staining intensity. Tumors with PTEN scores of 0 or 1 were considered to have PTEN protein loss.
Histologically adequate tumor material (routinely processed formalin-fixed paraffin-embedded [FFPE] tissue) was available from 16 patients.
DNA and RNA was extracted upon enrichment in tumor cells, while DNA was also extracted from peripheral blood samples. For DNA extraction, the QIAamp mini kit (Qiagen, Hilden, Germany) was used. Upon overnight tissue fragment lysis with proteinase K at 56 °C, RNA was extracted with TRIZOL-LS (Life Technologies, Paisley, UK) and reverse transcribed with random primers and the Superscript III system (Life Technologies). Commercial reagents were applied according to the instructions of the manufacturers. FFPE-DNA samples were checked for amplification capacity with the BIOMED2 multiplex DNA control PCR assay prior to undertaking molecular investigations. Molecular templates were of inadequate quality in one of the 16 cases; hence, molecular data are reported for 15 tumors only.
MGMT promoter methylation was assessed with the SALSA MS-MLPA KIT ME011 (MRC, Amsterdam, NL, Holland). Normal blood DNA and a reactive lymph node FFPE-DNA sample were used as controls. This method gave informative results, i.e., all peaks present in the non-digested sample with acceptable internal controls, in 10 of the 15 cases (66.6 %), in accordance with FFPE-DNA amplification capacity (≥300 bp for these 10 samples, <300 for the remaining 5 samples). The results were evaluated according to [13]: relative peak values ≤0.25 were characterized as unmethylated, relative peak values >0.25 as methylated, and relative peak values >0.75 as completely methylated. The procedure was repeated twice to assess concordance of results (retention in the same category of methylation status).
Chromosomal 1p/19q deletion was investigated with a previously published loss of heterozygocity (LOH) protocol [3]. Briefly, peripheral blood and tumor DNA samples from the same patient were compared for the presence of four microsatellite markers, D1S2845 (1pter), D1S216 (1p31), D1S2696 (1p13) and D19S589 (19q13), with capillary electrophoresis and analyzed with the GeneMapper software. Twelve tumors were informative for this parameter.
Mutation analysis for IDH1 exon 2 (including codon for p.R132) and IDH2 exon 4 (including p.R172) was accomplished with dd-sequencing (sense and antisense) in an ABI3130XL genetic analyzer. This approach was informative in 13 of the15 tumors (87 %).
Copy number variation (CNV) was assessed for the EGFR (7p12), MET (7q31), ERBB2 (17q21.1) and CCND1 (11q13) genes. For each gene, two genomic targets were amplified, to ensure informative results. The method involves a duplex real-time PCR reaction including (a) for the target gene, TaqMan® minor groove binding (MGB) probes, FAM™ labeled and (b) for the reference gene, Taqman VIC®-TAMRA™ labeled probes, both assays with unlabeled primers. Because these assays detect tiny parts of the whole gene, we selected two assays for each target gene, located at distant parts of the genomic sequence. The ready-made assays used and their ID numbers (Applied Biosystems/Life Technologies) were: for EGFR exon 2, Hs01426560_cn; for EGFR exon 10, Hs02822119_cn; for ERBB2 exon 13, Hs01839367_cn; for ERBB2 exon 21, Hs00709630_cn; for CCND1 intron 2 Hs03802861_cn, and exon 3 Hs01133305_cn; and for MET, exon 8 Hs02633538_cn, and intron 9 Hs05005398_cn. The TaqMan® Copy Number Reference Assay RNase P was used as endogenous reference. Reactions were run in duplicates in an ABI7900 system. Three normal blood DNA samples were included in each run as calibrator samples, along with a no-template control (NTC). Results (predicted copy numbers [CN]) were obtained automatically with the CopyCaller™ Software v1.0, in comparison to averaged calibrator values upon setting the evaluation threshold at Ct = 33. Z-scores for all accepted samples and CN range for duplicates were <1. No amplification was observed for NTCs. Out of the two assays that were used per target, the more stable one (lower CN range among replicate runs of the three normal samples) was used as a guide in ambiguous cases with more >2 CN difference. Amplification (CN gain) was considered for cases with >3 CN; cases with 2.5–3 CN were considered to be ambiguous (low amplification/polysomy). All 15 tumor samples were informative for CN evaluation with this approach.
Relative mRNA expression was assessed with RTQ-PCR, using premade Taqman-MGB assays in an ABI7900HT system under default conditions. Samples were tested in 10 μl reactions and were run in duplicates. The assays used were: EGFR, Hs00193306_m1; DARPP32 (PPP1R1B), Hs00259967_m1; STAT1, Hs01014005_m1; STAT3, Hs01047580_m1; STAT5A, Hs00234181_m1; and STAT5B, Hs00560035_m1. EGFRvIII expression was assessed with a custom designed Taqman-MGB assay by using Primer Express® v1.3, spanning exons 1^8 (sequencing validated amplicon length: 102 bp; primers/probe available upon request). Exclusion criteria for RQ analysis were endogenous control (GUSB, assay 4333767F) CT values higher than 36 for each duplicate; and deltaCTs higher than 0.5 per duplicate pair. According to this evaluation, all 15 tumor samples (100 %) were found eligible for further analysis. Relative quantification (RQ) was assessed in a linear mode as (40 dCT) [9], whereby dCT = (avg. CT target) − (avg. CT GUSB).
Statistical analysis
The primary endpoint for the phase I study was to determine the MTD of LP when given in combination with TMZ. Patient demographics, tumor characteristics and lab data (IHC and mRNA) were presented in frequency tables using descriptive statistics. Associations between binary variables were examined using Fisher’s exact test, while the Mann–Whitney test was used to compare group of cases in terms of continuous variables. Progression-free survival (PFS) was calculated from date of study entry to the date of documented tumor progression or death from any cause. Survival was calculated from the date of study entry to the date of death from any cause. Alive patients or patients lost to follow-up were censored on the last date they were known to be alive. Survival and PFS were estimated by the Kaplan–Meier method. Toxicity is presented in a frequency table according to grade, along with the corresponding percentages (N %).
Results
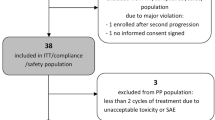
Between January 2009 and December 2010, 16 patients (9 males and 7 females) with a median age of 60.3 years entered the study at three LP levels: 1,000 mg sid (11 pts), 1,250 mg sid (4 pts) and 1,500 mg sid (1 pt). At the time of the analysis, a total of 68 cycles had been delivered with a median cycles per patient value of 2.5 (range: 1–17). All patients eventually discontinued treatment, two due to adverse events and the other 14 patients because of disease progression. Patient characteristics are shown in Table 1.
Dose escalation and maximum tolerated dose
Three patients were entered in the first dose level (LP 1,000 mg sid). No DLT toxicity was observed, and therefore the study moved forward to the second dose level (LP 1,250 mg sid), where three patients were initially entered. One patient had a DLT and subsequently one more patient entered the same dose level. A second DLT was reported. However, just before the end of the second dose level, one patient was dosed at the next dose level. This case was a protocol violation. Since the first dose level was the MTD, as per protocol design, 8 more patients were entered at this level to further characterize the toxicity profile of the drug combination.
Dose limiting toxicity
Hematological toxicity was the only DLT toxicity seen in two patients, at the second dose level, during the first cycle of their treatment (Table 2). Specifically, the first patient experienced grade 3 neutropenia and grade 4 leukopenia and the second patient had grade 4 thrombocytopenia. Treatment was discontinued in both patients. The first patient had a rapid disease progression and died a week after discontinuation of treatment, while the second patient died because of disease progression 8 months later, without receiving any further treatment. Neither was considered to be treatment-related death. No non-hematological DLT toxicity was reported. Febrile neutropenia requiring hospitalization was observed in one patient at the expanded first dose level. This episode occurred at the second cycle of treatment.
Most common toxicities
Hematological toxicity was the most common side effect observed. Eleven patients (68 %) developed hematological toxicity of any grade. Other common side effects, either clinical or laboratory, were minimal and are all depicted in the Appendix Table. Grade 3 CNS toxicity was observed in two patients, and although it was initially reported as a serious adverse event, after assessment it turned out to be disease related. Almost all side effects resolved to at least grade 1, either spontaneously or after temporary (less than 2 weeks) withdrawal of study medications.
Overall study treatment was well tolerated. Most patients discontinued treatment due to disease progression. In the expanded first cohort, one patient experienced 2 DLTs (febrile neutropenia grade 3 and thrombocytopenia grade 3) in the second cycle of treatment. Since DLTs were observed in less than 30 % of patients in the expanded first cohort, TMZ and LP 1,000 mg sid is the recommended dose for further phase II studies evaluating the combination.
Efficacy
Half of the patients received more than two cycles of the study regimens. One patient, treated at the second dose level, had early disease progression in the first cycle and therefore treatment had to be stopped. One patient who developed DLT at the second dose level, resulting in discontinuation of the treatment due to the observed toxicity, also had rapid disease progression and died 8 months later without receiving any further treatment. All eight patients who received more than two cycles of the study drugs were treated at the first dose level. There was one partial response documented, while five patients had stable disease. Three patients received treatment for 10, 11, and 17 cycles, with a PFS of 9, 10 and 17 months, respectively (Table 3). Median survival for all patients was calculated to be 5.0 months (range: 1.15–20.95), while median PFS was 2.4 months (range: 0.95–17.25) (Fig. 1).

Kaplan–Meier curves for PFS (left) and survival (right) in all patients
Translational analysis results
MGMT promoter methylation was observed in 2 of the 10 informative tumors and was considered to be incomplete; in both cases, only the 193 bp probe was found methylated with peak values of 0.43 and 0.64, respectively. No complete 1p/19q deletions were observed in the 12 informative tumors, although partial deletions were observed in two cases (Table 3). None of the tumors examined harbored IDH1 or IDH2 mutations or copy number gains for ERBB2, MET and CCND1.
EGFR gene copy number gains, corresponding to gene amplification, were observed in 9 of the 15 tumors (60 %) (Table 3). EGFRvIII expression was considered to be positive (RQ > 30) in 7 tumors and occurred in strong association with EGFR gene amplification, since all EGFRvIII positive tumors were EGFR amplified (Fisher’s exact test, p = 0.007). In some EGFRvIII positive tumors (cases GM007 and GM013 in Table 3), EGFR amplification could only be evaluated with the exon 10 assay; in these cases, CNV test results were negative with the exon 2 assay. In the same line, in 3 additional tumors, the predicted CN for exon 2 appeared distinctly lower than that for exon 10. Such discrepancies can be explained, considering that exon 2 is within the area deleted in the rearranged EGFR gene that expresses the truncated vIII variant. It thus appears that the application of EGFR CNV testing for distinct areas of the gene may indicate EGFRvIII expression, which might be interesting to validate in larger sample series for screening purposes.
EGFR mRNA expression was generally high in the GBM cases examined and was strongly positively associated with EGFR gene amplification (Mann–Whitney test, p = 0.009). STAT1 and STAT3 mRNA expression was also high, STAT5B intermediate, while DARPP32 and STAT5A mRNA expression was relatively low (Table 3). Neither EGFR gene amplification nor EGFRvIII expression were associated with relative transcript levels of the DARPP32/STAT genes.
Profiling of all mRNAs examined (Table 3) revealed distinct subgroups among the small number of tumors in this study. In three tumors, all transcripts were barely detectable (green cluster in Table 3), 5 tumors were EGFR amplified/EGFRvIII positive and expressed low DARPP32/all STAT genes (orange cluster), while 6 tumors were mostly EGFRvIII negative and expressed relatively high DARPP32/STAT5 and low STAT3 mRNA (blue cluster). Interestingly, patients with tumors in the blue cluster had longer PFS when compared to all other patients; the three patients who received the highest number of treatment cycles were also included in this group. However, numbers were too small to perform statistical analyses.
PTEN protein expression was assessed in 13 cases; in three cases biopsy material was inadequate. PTEN protein expression was observed in seven cases, whereas five cases had PTEN loss. EGFR amplification was observed in five cases with PTEN protein expression and three cases with PTEN loss. From the seven EGFRvIII positive tumors, four expressed PTEN (Table 3).
Discussion
The survival outcomes for high-grade gliomas remain poor [34]. Failing initial treatment with surgery and chemoradiotherapy with TMZ, patients with recurrent tumors have limited therapeutic options and dismal prognosis [34]. Targeting the vascular endothelial growth factor and epidermal growth factor pathways has recently emerged as an increasingly interesting option in the treatment of recurrent disease [8, 28]. However, the employment of EGFR tyrosine kinase inhibitors in the treatment of recurrent gliomas has met limited utility. Despite the fact that EGFR is frequently overexpressed or mutated in glioblastoma tumor cells, response rates to single-agent EGFR tyrosine kinases, such as gefitinib and erlotinib, range between 0 and 15 % [8]. PTEN activity is necessary for EGFR tyrosine kinase inhibitors to be active, and PTEN mutations, which are common in these tumors, likely contribute to the limited activity observed with this class of agents to date [7].
Lapatinib is a dual-action tyrosine kinase inhibitor, with activity in both HER2/ERBB2 and EGFR receptors. LP in combination with capecitabine is an active regimen in metastatic breast cancer patients who express the HER2/new protein [20]. The clinical use of LP in recurrent gliomas has so far been limited. Thiessen and colleagues reported the results of a phase I/II trial of LP monotherapy in recurrent GBM; treatment with single-agent LP did not show significant activity in GBM patients and the trial was terminated early because of lack of efficacy [31].
Rechallenge with TMZ at the time of tumor progression has previously been reported in small case series. Alternative dosing schedules have been used in the form of metronomic administration, which showed remarkable efficacy in GBM patients due to an antiangiogenic effect [15, 29]. The rationale for TMZ rechallenge in previously TMZ-treated patients includes not only the potential antiangiogenic action of continuously delivered drug, but also the potential to overcome TMZ resistance via the MGMT repair pathway [17]. Nevertheless, in our study, MGMT promoter methylation was observed in only 2 out of the 10 tumors assessed and was considered to be incomplete.
This is the first reported trial of LP and TMZ in recurrent high-grade gliomas, as of date. In our study, sixteen patients were treated at two dose levels. The combination of TMZ at standard dose and LP at 1,000 mg sid seems to be feasible with a favorable toxicity profile in the pretreated population. Hematological toxicity was the only DLT observed, which was easily manageable. Approximately one-third of the patients had clinical benefit from the treatment. A phase II trial has recently started recruitment in order to assess the efficacy of this combination.
Several genes, including ERBB2, MET, CCND1 and EGFR, are altered in gliomas [2]. These alterations tend to occur in a defined order during the progression to a high-grade tumor. Loss or mutation of PTEN and amplification of EGFR are predominant characteristics of higher-grade tumors [16]. Combined allelic loss of 1p/19q is characteristic of oligodendrogliomas and of better prognosis, while partial losses may be observed in astrocytic tumors as well [18]. 1p/19q loss and mutations of the isocitrate dehydrogenase genes (IDH1 and 2) are early phenomena in glial tumor development and progression [14]. IDH mutations mark astrocytic and oligodendrocytic gliomas with a better prognosis, being more frequent in grade II/III and clinically secondary glioblastomas while seldom in primary glioblastomas [35, 38]. In GBM, co-expression of PTEN and EGFRvIII is associated with better response to EGFR TKIs [16], while MET and CCND1 overexpression is associated with shorter PFS and adverse prognosis.
In the present high-grade glioma series, no ERBB2, MET or CCND1 gene gains were observed. Further, consistent with the astrocytic morphology of the tumors, only parital 1p and 19q losses were observed, which are not related to patient prognosis, while all patients including those with histologically secondary glioblastomas had IDH-negative disease. Hence, we cannot comment on a possible effect of these markers on patient outcome.
In line with a high incidence of EGFR gene alterations in glioblastomas, the majority of the tumors examined exhibited EGFR copy gains. EGFR amplification is observed in half of primary glioblastomas and is associated with poor prognosis [11, 12, 27], while half of EGFR-amplified tumors harbor the truncated EGFRvIII mutant, the absence of which may be associated with a better prognosis in GBM [19]. Herein we show that EGFR mRNA expression was pronounced in cases with EGFR gene copy gains corresponding to EGFR amplification; in addition, 7 out of 9 EGFR amplified tumors also expressed EGFRvIII. However, these EGFR parameters were individually not associated with a benefit from LP treatment in our series.
Three patients received 10 or more cycles of treatment with a remarkable disease control for several months; two had EGFR gene amplification, and in the third case the EGFR gene was not amplified. From the two EGFR amplified cases, one was PTEN positive and EGFRvIII negative (11 months PFS) and the second one was found PTEN negative and EGFRvIII positive (17 months PFS). In the EGFR non-amplified case, EGFRvIII was negative with PTEN expression being not informative (9 months PFS). Nevertheless, due to the small number of studied cases, it is unclear whether these findings are associated with response to treatment and disease outcome. Similar to our results, in the study by Thiessen et al. [31], only three out of 16 cases had co-expression of EGFRvIII and PTEN, but it was also unclear whether this co-expression contributed to disease outcome.
Beyond the above markers that are usually examined in association with EGFR inhibition, herein we show that all three patients with a relatively more favorable PFS expressed DARPP32 and STAT5A/B mRNA; by contrast, those with the worst registered PFS did not express these genes. STAT5 proteins may be downstream targets of EGFRvIII [5], while DARPP32 (PPP1R1B) is a key molecule marking neuronal differentiation [1]. According to a proposed molecular glioma subtyping [33], gliomas with the most favorable outcome belong to the proneural subtype (oligodendrocytic profile, IDH-positive disease), followed by the neural, the classical, and the most aggressive mesenchymal subtypes. In this sense, DARPP32 mRNA may be a marker of the neural subtype, which is characterized by an expression profile resembling neurons and cultured astrocytes. It should be noted that all these data were determined in primary tumors, i.e., before radiation and temozolomide, with the status of these markers being unclear in glioblastoma tissues after progression upon standard treatment. Nevertheless, it seems worth further investigating whether DARPP32 and STAT5 expression interferes with lapatinib efficiency or whether it is an intrinsic characteristic of glioblastomas with a more favorable prognosis.
LP combined with capecitabine chemotherapy is efficaceous in HER2-expressing metastatic breast cancer patients. There is also evidence that this combination has remarkable efficacy in the hard-to-treat subpopulation with brain metastases. It has been hypothesized that LP can theoretically cross the blood–brain barrier and, as such, may exert its activity. Nevertheless, only modest activity has been documented with single agent LP in intracranial disease, however this was augmented by combining LP with capecitabine. In our study we did show a modest activity in a limited number of patients with recurrent gliomas treated with the LP and TMZ combination. In line with this, we could speculate that the amount of active drug that had crossed the blood–brain barrier was uncertain, and thus it is unclear whether the optimal drug concentration had reached the tumor. Applying pharmacokinetic studies of LP in CSF in future clinical trial designs, although difficult to carry out, may help to answer these questions.
In conclusion, combination of TMZ and LP seems to be feasible and well tolerated with manageable, predominantly hematological toxicity in patients with recurrent high-grade gliomas. The activity of this combination in patients with recurrent GBM is currently under investigation in a recently initiated phase II trial.
References
Aubry L, Bugi A, Lefort N, Rousseau F, Peschanski M, Perrier AL (2008) Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci USA 105:16707–16712
Bai RY, Staedtke V, Riggins GJ (2011) Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol Med 17:301–312
Boulay JL, Miserez AR, Zweifel C, Sivasankaran B, Kana V, Ghaffari A, Luyken C, Sabel M, Zerrouqi A, Wasner M, Van Meir E, Tolnay M, Reifenberger G, Merlo A (2007) Loss of NOTCH2 positively predicts survival in subgroups of human glial brain tumors. PLoS ONE 2:e576
Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A (2010) The transcriptional network for mesenchymal transformation of brain tumours. Nature 463:318–325
Chumbalkar V, Latha K, Hwang Y, Maywald R, Hawley L, Sawaya R, Diao L, Baggerly K, Cavenee WK, Furnari FB, Bogler O (2011) Analysis of phosphotyrosine signaling in glioblastoma identifies STAT5 as a novel downstream target of DeltaEGFR. J Proteome Res 10:1343–1352
Custodio A, Calles A, Perez-Segura P (2010) Response to erlotinib in recurrent glioblastoma multiforme showing coexpression of EGFRvIII and PTEN. Clin Transl Oncol (Official Publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico) 12:310–314
Friedman HS, Bigner DD (2005) Glioblastoma multiforme and the epidermal growth factor receptor. New Eng J Med 353:1997–1999
Gadji M, Crous AM, Fortin D, Krcek J, Torchia M, Mai S, Drouin R, Klonisch T (2009) EGF receptor inhibitors in the treatment of glioblastoma multiform: old clinical allies and newly emerging therapeutic concepts. Eur J Pharmacol 625:23–30
Hennig G, Gehrmann M, Stropp U, Brauch H, Fritz P, Eichelbaum M, Schwab M, Schroth W (2010) Automated extraction of DNA and RNA from a single formalin-fixed paraffin-embedded tissue section for analysis of both single-nucleotide polymorphisms and mRNA expression. Clin Chem 56:1845–1853
Huncharek M, Kupelnick B (2000) Epidermal growth factor receptor gene amplification as a prognostic marker in glioblastoma multiforme: results of a meta-analysis. Oncol Res 12:107–112
Hurtt MR, Moossy J, Donovan-Peluso M, Locker J (1992) Amplification of epidermal growth factor receptor gene in gliomas: histopathology and prognosis. J Neuropathol Exp Neurol 51:84–90
Jaros E, Perry RH, Adam L, Kelly PJ, Crawford PJ, Kalbag RM, Mendelow AD, Sengupta RP, Pearson AD (1992) Prognostic implications of p53 protein, epidermal growth factor receptor, and Ki-67 labelling in brain tumours. Br J Cancer 66:373–385
Jeuken JW, Cornelissen SJ, Vriezen M, Dekkers MM, Errami A, Sijben A, Boots-Sprenger SH, Wesseling P (2007) MS-MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest 87:1055–1065
Juratli TA, Kirsch M, Robel K, Soucek S, Geiger K, von Kummer R, Schackert G, Krex D (2012) IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol 108:403–410
Kong DS, Lee JI, Kim JH, Kim ST, Kim WS, Suh YL, Dong SM, Nam DH (2010) Phase II trial of low-dose continuous (metronomic) treatment of temozolomide for recurrent glioblastoma. Neuro-oncology 12:289–296
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. New Eng J Med 353:2012–2024
Mrugala MM, Chamberlain MC (2008) Mechanisms of disease: temozolomide and glioblastoma–look to the future. Nat Clin Pract Oncol 5:476–486
Ohgaki H, Kleihues P (2011) Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol 28:177–183
Omuro AM (2008) What is the relevance of determining EGFR-variant-III status in glioblastomas? Nat Clin Pract Oncol 5:188–189
Opdam FL, Guchelaar HJ, Beijnen JH, Schellens JH (2012) Lapatinib for advanced or metastatic breast cancer. Oncologist 17:536–542
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD (2010) A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neurooncology 12:95–103
Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon JE 2nd, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS (2004) Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol (Official Journal of the American Society of Clinical Oncology) 22:133–142
Riemenschneider MJ, Jeuken JW, Wesseling P, Reifenberger G (2010) Molecular diagnostics of gliomas: state of the art. Acta Neuropathol 120:567–584
Rock K, McArdle O, Forde P, Dunne M, Fitzpatrick D, O’Neill B, Faul C (2012) A clinical review of treatment outcomes in glioblastoma multiforme—the validation in a non-trial population of the results of a randomised Phase III clinical trial: has a more radical approach improved survival? Br J Radiol 1017:729–733
Scaltriti M, Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res (An official journal of the American Association for Cancer Research) 12:5268–5272
Schlegel J, Stumm G, Brandle K, Merdes A, Mechtersheimer G, Hynes NE, Kiessling M (1994) Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J Neurooncol 22:201–207
Specenier P (2012) Bevacizumab in glioblastoma multiforme. Expert Rev Anticancer Ther 12:9–18
Stockhammer F, Misch M, Koch A, Czabanka M, Plotkin M, Blechschmidt C, Tuettenberg J, Vajkoczy P (2010) Continuous low-dose temozolomide and celecoxib in recurrent glioblastoma. J Neurooncol 100:407–415
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Eng J Med 352:987–996
Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, McIntosh L, Eisenhauer E (2010) A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol 65:353–361
van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T (2009) Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol (Official Journal of the American Society of Clinical Oncology) 27:1268–1274
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Weller M, Stupp R, Hegi M, Wick W (2012) Individualized targeted therapy for glioblastoma: fact or fiction? Cancer J 18:40–44
Weller M, Wick W, von Deimling A (2011) Isocitrate dehydrogenase mutations: a challenge to traditional views on the genesis and malignant progression of gliomas. Glia 59:1200–1204
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, Tsien C, Mikkelsen T, Wong ET, Chamberlain MC, Stupp R, Lamborn KR, Vogelbaum MA, van den Bent MJ, Chang SM (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28:1963–1972
Worm K, Dabbagh P, Schwechheimer K (1999) Reverse transcriptase polymerase chain reaction as a reliable method to detect epidermal growth factor receptor exon 2–7 gene deletion in human glioblastomas. Hum Pathol 30:222–227
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Acknowledgments
Authors would like to thank Dr. Anna Kalogera-Fountzila, Consultant Radiologist, Ass. Professor of Radiology, Radiology Department, AHEPA University Hospital, Aristotle University of Thessaloniki School of Medicine, for the independent review of images and her extremely helpful assistance. Authors would also like to thank Anastasia Eleftheraki, Statistician, Hellenic Cooperative Oncology Group, Athens, Greece.
Conflicts of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Appendix
Rights and permissions
About this article
Cite this article
Karavasilis, V., Kotoula, V., Pentheroudakis, G. et al. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. J Neurol 260, 1469–1480 (2013). https://doi.org/10.1007/s00415-012-6812-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-012-6812-z