
Abstract
Nintedanib (BIBF 1120) is a small, orally available, triple angiokinase inhibitor in phase III development (various indications) that targets VEGFR 1–3, FGFR 1–3, and PDGFR-α/β. This open-label, uncontrolled, phase II study assessed the efficacy and safety of nintedanib in patients with recurrent glioblastoma multiforme (GBM) who had previously failed radiotherapy plus temozolomide as first-line therapy (STUPP), or the same regimen with subsequent bevacizumab-based therapy as second-line treatment (BEV). Patients with a performance status of 0–1, histologically proven GBM, and measurable disease (by RANO) were enrolled. Nintedanib was given orally at a dose of 200 mg twice daily (bid), with magnetic resonance imaging undertaken every 8 weeks. The primary endpoint was objective response rate. The study was stopped prematurely following a preplanned futility analysis after inclusion of 13 patients in the STUPP arm and 12 in the BEV arm. Best response was stable disease (SD) in three patients (12 %); all other patients progressed within the first four 28-day cycles. One patient in the BEV arm has had SD for 17+ months. Median progression-free survival was 1 month and median overall survival was 6 months. Nintedanib had an acceptable safety profile, with no CTCAE grade 3–4 adverse events. Common adverse events were CTCAE grade 1–2 fatigue, loss of appetite, diarrhea, and nausea. Single-agent nintedanib (200 mg bid) demonstrated limited, but clinically non-relevant antitumor activity in patients with recurrent GBM who had failed 1–2 prior lines of therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most common primary malignant neoplasm of the central nervous system (CNS) in adults [1]. Current primary treatment is maximal surgical resection followed by the ‘STUPP’ regimen [2]: brain irradiation (60 Gy in 30 fractions) concurrent with temozolomide (TMZ) over 6 weeks, followed by adjuvant TMZ monotherapy for up to 6 months [3, 4]. Median overall survival (OS) associated with this regimen was 14.6 months [2]. Patients who progress through initial treatment, however, have a dismal prognosis with a median OS of about 6 months [5]. For these patients there is no established treatment.
GBM is a highly vascularized tumor and vascular endothelial growth factor (VEGF) is highly expressed in GBM, where it induces angiogenesis primarily by stimulating VEGF receptor-2 (VEGFR-2) on tumor vessels [6]. Additionally, overexpression or amplification of the epidermal growth factor receptor (EGFR) and its truncated variant, EGFRvIII, is often seen in primary GBM [7], while overexpression of the platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), p53, and IDH1 is more common in secondary GBM [8–10]. Activation and overexpression of these signaling molecules is believed to facilitate angiogenesis and growth in this highly aggressive malignancy. PTEN mutations, contributing to abnormally high activity in the PI3 K/AKT/mTOR pathway, are also often seen in GBM tumors [11].
Although the angiogenic process is poorly understood, inhibition of the VEGF axis, in combination with other implicated growth factor receptors might have a synergistic inhibitory effect on tumor angiogenesis [12]. Targeting VEGF signaling alone with the monoclonal antibody, bevacizumab (which is approved for use in recurrent GBM in the USA), has resulted in modest clinical benefits (response rates of 28–35 % and median OS of 7.7–9.2 months when used in monotherapy) in patients with recurrent GBM in first/second relapse [13–15]. These limited effects may be a consequence of escape mechanisms for tumor angiogenesis, whereby PDGFRs and FGFRs compensate for the disruption of the VEGF pathway [16]. A multitargeted approach may therefore be more beneficial.
Nintedanib (BIBF 1120; Boehringer Ingelheim, Germany) is a potent, oral triple angiokinase inhibitor in phase III development that targets VEGFRs, PDGFRs, FGFRs, Flt3, RET, and Src [16]. In preclinical models, the specific and simultaneous abrogation of these pathways resulted in effective growth inhibition of both endothelial and perivascular cells [16]. Nintedanib may therefore be a more effective inhibitor of angiogenesis than agents that solely target the VEGF pathway and thus may have a potential role in GBM. Phase I/II monotherapy studies in various tumour types (including advanced non-small cell lung cancer and ovarian cancer) and idiopathic pulmonary fibrosis show that nintedanib is generally well tolerated with mild-to-moderate adverse effects, such as gastrointestinal symptoms and reversible elevations of liver enzymes [17–20]. Initial signs of clinical activity, including encouraging tumor stabilization rates of 46–76 %, have also been observed during phase I/II development [17, 20, 21]. From these studies, the recommended dose for continuous daily monotherapy is 200–250 mg twice daily (bid) [17, 21] .
As nintedanib might present as a new treatment option for GBM, this phase II study was initiated to investigate the antitumor activity and safety of this agent in patients with primary GBM who had failed primary or secondary treatment for recurrent disease.
Patients and methods
Patient selection
Adult patients aged >18 years with an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1 and histologically proven GBM were enrolled. All patients had previously failed standard radiotherapy plus TMZ as first-line therapy (STUPP), or the same regimen with subsequent bevacizumab-based therapy as second-line treatment (BEV). Patients also had available archival tissue blocks, measurable disease (according to Revised Assessment in Neuro-Oncology [RANO] guidelines), progressive disease (documented by a magnetic resonance imaging [MRI] scan obtained within 14 days prior to enrolment), and had provided informed consent. Patients undergoing recent resection (>6 weeks) for recurrent tumors were eligible, but residual disease was required for inclusion. Fertile women had to use appropriate contraceptive methods.
Key exclusion criteria included: prior treatment with nintedanib or any other VEGFR inhibitor, except bevacizumab; inadequate hematologic function (absolute neutrophil count <1,500/ml, platelets <100,000/ml, or hemoglobin <9.0 g/dl); insufficient coagulation (international normalized ratio ≥2 or prothrombin time/partial thromboplastin time >50 % of the deviation of the institutional upper limit of normal [ULN]); inadequate hepatic function (alanine transaminase/aspartate transaminase >1.5× ULN or total bilirubin outside of normal limits); significant cardiovascular disease (uncontrolled hypertension, unstable angina, history of myocardial infarction in the 12 months prior to the start of treatment, congestive heart failure > New York Heart Association Class 2, pericardial effusion, or serious cardiac arrhythmia); hemorrhagic or thromboembolic events in the past 6 months; and major injuries in the 10 days prior to start of the study. Patients with active serious infection requiring systemic antimicrobial therapy, or patients with active or chronic hepatitis C or B infection were also ineligible.
This investigator-initiated study (clinicaltrial.gov identifier: NCT01251484) was supported by a research grant from Boehringer Ingelheim, who provided the study medication. The trial was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Guidelines for Good Clinical Practice. Protocol approval was obtained from the local Ethics Committee and Danish Medicines Agency.
Treatment plan
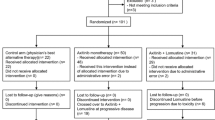
This was an investigator-initiated, single-center, non-randomized, open-label, two-stage phase II study of single-agent nintedanib in patients with recurrent primary GBM who had failed primary (first-line radiotherapy plus TMZ; STUPP arm) or secondary (STUPP regimen followed by second-line bevacizumab-based therapy; BEV arm) treatment for recurrent disease. The study was designed so that 32 patients would be enrolled into both the STUPP and BEV arms. The two-stage design included a stopping rule if three or fewer patients achieved an objective response once 16 patients had been enrolled into each arm.
All patients were treated continuously with nintedanib at a dose of 200 mg bid (two capsules twice daily). One cycle was 28 days, with treatment continued until disease progression (PD) or unacceptable toxicity.
Evaluation of response
Neuro-imaging with MRI, including pre-contrast T1, T2/FLAIR and post-contrast T1, with two orthogonal planes and T2-weighted flair, was performed at baseline, before the first treatment cycle, and every 8 weeks thereafter. Tumor size was measured in two dimensions, according to RANO criteria [22].
Safety assessment
All adverse events occurring during the course of the trial and for up to 2 months after the last dose of study medication were collected, documented, and reported. Toxicity was graded every 2 weeks for the first two cycles and every 4 weeks thereafter, according to National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 [23]). A toxicity and dose-reduction management plan was followed according to the investigators’ brochure.
Biomarker Analyses
Although biomarker analyses were pre-planned, utilizing archival tissue blocks and collected plasma samples to assess vascular and immune system parameters, none were undertaken due to early termination of the study.
Statistical analysis
The primary endpoint was the objective tumor response rate (complete remission [CR] + partial remission [PR]), assessed according to RANO criteria [22]. Secondary endpoints included progression-free survival (PFS), OS, safety, and evaluation of biomarkers.
It was estimated that a total of 32 patients per arm would be required to provide 76 % power to be able to detect a treatment effect on response rate at a one-sided significance level of α = 0.05. The proportion of responders under the null hypothesis (no drug effect) was assumed to be 10 %, while under the alternative hypothesis (drug effect), the proportion was assumed to be 25 %.
A preplanned safety analysis was undertaken when the first 10 patients had received a minimum of two cycles (8 weeks) of treatment. A futility analysis was also planned once 16 patients were evaluable for response in each arm (stage 1), or earlier if clinically indicated. An objective response in at >3/16 patients in either arm was required to proceed to stage 2, where a total of 32 patients would be recruited per arm. This would result in a probability of early termination of >75 % when the true response rate was <10 %.
Response rates were calculated in the intention-to-treat (ITT) population, with confidence intervals estimated using the Clopper-Pearson method. PFS and OS analyses were also performed in the ITT population using the Kaplan–Meier log-rank method. PFS was defined as the time between the start of treatment and first occurrence of disease progression or death; data were censored at last follow-up if the patient remained alive without PD. Safety data are summarized for all treated patients. The Kaplan–Meier method was used to analyze time-to event data and the log-rank test was used to compare PFS and OS (SPSS version 18.0) analyses.
Results
Patient characteristics
Twenty five patients were enrolled between January 2011 and July 2011. Baseline demographics and patient characteristics are shown in Table 1.
The study was stopped prematurely for futility after inclusion of 13 patients in the STUPP arm and 12 patients in the BEV arm, since no responses were observed in either arm. Median follow-up was 14 months (range 10–15 months) and median number of treatment cycles administered was 2 (range 1–17, Table 2).
Efficacy
The objective response rate (CR + PR) to nintedanib 200 mg bid among all patients in the ITT population was 0 % (Table 2). The best radiographic response was SD in three patients (12 %): two patients in the STUPP arm and one patient in the BEV arm. PD was observed in the remaining 22 patients (88 %). One patient in the BEV arm has had SD for 17 months (Fig. 1), while all other patients progressed within the first four cycles. Analyses of possible correlations with biomarkers were not performed due to the lack of response.

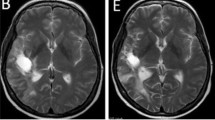
Baseline MRI (January 2011) from a patient with stable disease for 17 months showing contrast-enhanced lesion in the left temporo-parietal region (2.7 × 2.5 × 2.4 cm [AP × SS × CC] (a). T2 changes covered 8.8 cm anterior-posterior (b). After 12 cycles (February 2012) contrast-enhanced lesion in the left temporo-parietal region stable disease (2.7 × 2.5 × 2.7 cm [AP × SS × CC] (c). T2 changes covered 8.0 cm (d). At progression after 17 months the contrast-enhanced lesion increased in size (e), whereas the T2 changes remained unchanged (f). However, the patient also progressed clinically
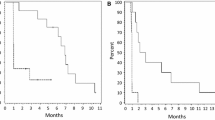
Six-month PFS was 4 % (n = 1; 95 % CI 0.1–20.4 %). Median PFS was estimated to be 1 month (95 % CI 0.7–1.3 months), and no significant differences were observed between the two arms. Estimated median OS was 6 months (95 % CI, 3.6–8.4 months). A significant difference in OS was observed between the STUPP and BEV arms (median OS: 8.1 [95 % CI 1.5–14.8] vs. 1.3 [95 % CI 0.0–4.9] months, respectively [p < 0.02]), (Fig. 2). Six patients were alive at the time of the analyses, five in STUPP and one in BEV arm.

A significant difference in overall survival was observed between the STUPP (solid line) and BEV (dotted line) arms (median OS: 8.1 [95 % CI 1.5–14.8] vs 1.3 [95 % CI 0.0–4.9] months, respectively [p < 0.02], log-rank test). Six patients were alive at the time of the analyses, five in STUPP and one in BEV arm
Only patients who maintained a good performance status (0–1) were retreated at progression after receiving nintedanib. Patients in the STUPP arm were subsequently treated with bevacizumab or TMZ induction therapy. Eligible patients in the BEV arm went on to receive re-irradiation in an ongoing trial. Among five patients in the STUPP arm who were subsequently treated with bevacizumab, four responded with a response duration between 4 and >10 months. Among two patients who received TMZ, one achieved PR for >6 months prior to progression. Only two patients in the BEV arm were retreated after nintedanib; both were re-irradiated, but neither responded to treatment.
Adverse events
Single-agent nintedanib treatment was well tolerated (Table 3). Grade 3–4 adverse events were not observed and there were no study-related deaths or episodes of intracranial hemorrhage. The most frequently reported adverse events were grade 1–2 fatigue (n = 24; 96 %), loss of appetite (n = 6; 24 %), diarrhoea (n = 9; 36 %), and nausea (n = 5; 20 %). There were no significant differences in adverse events between the STUPP and BEV arms, and no dose reductions were reported.
Discussion
Data from this phase II trial show that nintedanib 200 mg bid is well tolerated, but with limited, clinically non-relevant activity in patients with recurrent GBM who have failed primary or secondary treatment for recurrent disease. At the time of the futility analysis (which resulted in early closure of the trial), no responses had been reported and just 3/25 patients had achieved tumor stabilization. Median PFS was 1 month and median OS was 6 months. As expected, patients who had received nintedanib as third-line therapy had significantly shorter median OS than those who had received the drug as second-line therapy (2 vs 10 months, respectively; p < 0.02), reflecting the poorer prognosis of patients with more refractory disease. Despite this, one patient did achieve an extended period of disease stabilization (17 + months), which suggests that some patients may benefit from treatment. It also suggests that time to progression or SD may be better endpoints in studies of angiogenesis inhibitors.
This is the first study to evaluate objective responses to single-agent nintedanib in patients with recurrent GBM. In preclinical models, nintedanib predominately inhibits VEGFR 1–3 and PDGFR-α/β at nanomolar concentrations, with sustained VEGFR blockade lasting for >32 h [16, 24]. In addition, drug activity has been observed in cells that are predominately activated through FGFRs [16]; although the exact mechanism that underlies this effect is unknown [24]. Inhibition of pro-angiogenic receptor tyrosine kinases, such as PDGFR and FGFR, also appears to be relevant [25]. However, the lack of activity observed in the present study indicates that a potential blockade of the activity of VEGFR, PDGFR, and FGFR kinases by nintedanib does not inhibit angiogenesis in patients with recurrent GBM. One reason for this could be that glioma cells rely on other redundant pathways to support angiogenesis [26]. Consistently, a number of previous studies utilizing small molecule, dual-VEGFR/PDGFR inhibitors (sunitinib, sorafenib, vatalanib, cediranib, and pazopanib) also failed to demonstrate any clinically relevant activity in recurrent GBM [27–31]. Other authors have reported lower response rates and shorter disease control rates using bevacizumab after patients have relapsed on another prior anti-VEGF therapy [32]. This could not be confirmed in our study, since we observed several, durable responses to bevacizumab in patients previously treated with nintedanib.
Restricted delivery of targeted agents across the blood–brain barrier may also reduce their efficacy against brain tumors. Several tyrosine kinase inhibitors, including cediranib, gefitinib, erlotinib, and lapatinib, have been shown to be substrates for two important efflux transporters at the blood–brain barrier, P-gp/Bcrp, which restricts their CNS penetration [33–36]. Consistently, preclinical whole-body autoradiography studies show that nintedanib (molecular weight 539 Da) does not cross the blood–brain barrier in normal rat brain (investigator’s brochure U03-1563), as is generally expected for molecules >300 Da. Nonetheless, molecules >300 Da can reach intracranial tumor lesions with disrupted blood–brain barriers in tumor vessels, as is the case for bevacizumab [37]. Studies are now required to investigate if adequate concentrations of nintedanib are achievable in the brain or if nintedanib’s lack of activity is due to compensatory pro-angiogenic or other mechanisms.
Several publications have shown that anti-VEGF therapy, either with bevacizumab alone or in combination with chemotherapy induces responses in 25–68 % of patients with recurrent GBM [14, 38–41]. Response duration can be months, but most patients eventually become resistant to treatment and their tumors progress [14, 38–41].The reasons for this are not entirely clear; although it has been postulated the recurrent phenotype might be more invasive and infiltrative than de novo GBM. In addition, resistance might develop due to up-regulation of PDGF and recruitment of pericytes, which stabilizes the neovasculature. It has therefore been suggested that a combination of VEGFR inhibition and PDGFR inhibition might overcome this resistance [42]. Furthermore, inhibition of FGF’s pro-angiogenic effect might facilitate inhibition of tumor neovasculization [34]. Unfortunately, the lack of response to nintedanib does not support these propositions. However, the lack of activity is consistent with that seen with other multikinase inhibitors [41, 43].
Phase I studies have shown that the maximum tolerated dose of nintedanib is 200–250 mg (administered once or twice daily) with other studies demonstrating moderate response rates at doses up to 250 mg bid in various metastatic tumors [17–19, 21]. In the present study, we used a dose of 200 mg bid, which is within the range of the optimal dose. At this dose, nintedanib was well tolerated. The reported incidence of adverse events was low compared to published reports in other malignancies [19]. Additionally, we did not observe many of the common toxicities usually associated with anti-VEGF therapy, which is consistent with previous data on nintedanib [17–19, 21].
Conclusion
Nintedanib is well tolerated in patients with recurrent GBM who have failed 1–2 prior lines of therapy, but does not demonstrate any clinically relevant antitumor activity.
References
Bruce J, Kennedy B. (2012) Glioblastoma Multiforme http://emedicine.medscape.com/article/283252-overview
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
National Comprehensive Cancer Network (NCCN) (2012) Clinical Practice Guidelines in Oncology: Central Nervous System Cancers, Version 2.2012
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK (1999) Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 17:2572–2578
Takano S, Yoshii Y, Kondo S, Suzuki H, Maruno T, Shirai S, Nose T (1996) Concentration of vascular endothelial growth factor in the serum and tumor tissue of brain tumor patients. Cancer Res 56:2185–2190
Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB (2001) PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 93:1246–1256
Hesselager G, Uhrbom L, Westermark B, Nister M (2003) Complementary effects of platelet-derived growth factor autocrine stimulation and p53 or Ink4a-Arf deletion in a mouse glioma model. Cancer Res 63:4305–4309
Iacob G, Dinca EB (2009) Current data and strategy in glioblastoma multiforme. J Med Life 2:386–393
Morrison RS, Yamaguchi F, Bruner JM, Tang M, McKeehan W, Berger MS (1994) Fibroblast growth factor receptor gene expression and immunoreactivity are elevated in human glioblastoma multiforme. Cancer Res 54:2794–2799
Kita D, Yonekawa Y, Weller M, Ohgaki H (2007) PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol 113:295–302
Cao H, Zhang H, Zheng X, Gao D (2007) 3D QSAR studies on a series of potent and high selective inhibitors for three kinases of RTK family. J Mol Graph Model 26:236–245
Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733–4740
Jakobsen JN, Hasselbalch B, Stockhausen MT, Lassen U, Poulsen HS (2011) Irinotecan and bevacizumab in recurrent glioblastoma multiforme. Expert Opin Pharmacother 12:825–833
Vredenburgh JJ, Desjardins A, Herndon JE, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Wagner M, Bailey L, Bigner DD, Friedman AH, Friedman HS (2007) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25:4722–4729
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ (2008) BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 68:4774–4782
Mross K, Stefanic M, Gmehling D, Frost A, Baas F, Unger C, Strecker R, Henning J, Gaschler-Markefski B, Stopfer P, de RL, Kaiser R (2010) Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res 16:311–319
Ledermann JA, Hackshaw A, Kaye S, Jayson G, Gabra H, McNeish I, Earl H, Perren T, Gore M, Persic M, Adams M, James L, Temple G, Merger M, Rustin G (2011) Randomized phase II placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor BIBF 1120 after chemotherapy for relapsed ovarian cancer. J Clin Oncol 29:3798–3804
Reck M, Kaiser R, Eschbach C, Stefanic M, Love J, Gatzemeier U, Stopfer P, von PJ (2011) A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer. Ann Oncol 22:1374–1381
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Kluglich M, du Bois RM (2011) Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 365:1079–1087
Okamoto I, Kaneda H, Satoh T, Okamoto W, Miyazaki M, Morinaga R, Ueda S, Terashima M, Tsuya A, Sarashina A, Konishi K, Arao T, Nishio K, Kaiser R, Nakagawa K (2010) Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol Cancer Ther 9:2825–2833
Quant EC, Wen PY (2011) Response assessment in neuro-oncology. Curr Oncol Rep 13:50–56
U.S.Department of Health and Human Services, National Institutes of Health, National Cancer Institute (2009) Common Terminology Criteria for Adverse Events (CTCAE) version 4.02
Santos ES, Gomez JE, Raez LE (2012) Targeting angiogenesis from multiple pathways simultaneously: BIBF 1120, an investigational novel triple angiokinase inhibitor. Invest New Drugs 30:1261–1269
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19:329–337
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318:287–290
Batchelor T, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, et al (2010) The efficacy of cediranib as monotherapy and in combination with lomustine compared to lomustine alone in patients with recurrent glioblastoma: a phase III randomized study. Neuro-Oncol 12:iv69–iv78
Conrad C, Friedman H, Reardon D, Provenzale J, Jackson E, Serajuddin H, Laurent D, Chen B, Yung WKA (2004) A phase I/II trial of single-agent PTK 787/ZK 222584 (PTK/ZK), a novel, oral angiogenesis inhibitor, in patients with recurrent glioblastoma multiforme (GBM). ASCO 22:1512
Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, Deangelis LM, Abrey LE, Zhang WT, Prados MD, Fine HA (2010) Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06–02). Neuro Oncol 12:855–861
Pan E, Yu D, Yue B, Potthast L, Chowdhary S, Smith P, Chamberlain M (2012) A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J Neurooncol 110(1):111–118
Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, Marcello J, Herndon JE, McLendon RE, Janney D, Friedman AH, Bigner DD, Friedman HS (2011) Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol 101:57–66
Scott BJ, Quant EC, McNamara MB, Ryg PA, Batchelor TT, Wen PY (2010) Bevacizumab salvage therapy following progression in high-grade glioma patients treated with VEGF receptor tyrosine kinase inhibitors. Neuro Oncol 12:603–607
Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF (2010) Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 334:147–155
Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796
Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37:439–442
Wang T, Agarwal S, Elmquist WF (2012) Brain distribution of cediranib is limited by active efflux at the blood-brain barrier. J Pharmacol Exp Ther 341:386–395
Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF (2011) Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med 13:e17
Chi AS, Sorensen AG, Jain RK, Batchelor TT (2009) Angiogenesis as a therapeutic target in malignant gliomas. Oncologist 14:621–636
Chowdhary S, Wong ET (2008) Bevacizumab combined with irinotecan for recurrent glioblastoma multiforme–improvement over available therapy? Nat Clin Pract Neurol 4:242–243
Reardon DA, Desjardins A, Peters KB, Gururangan S, Sampson JH, McLendon RE, Herndon JE, Bulusu A, Threatt S, Friedman AH, Vredenburgh JJ, Friedman HS (2012) Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naive, recurrent glioblastoma. J Neurooncol 107:155–164
Shirai K, Siedow MR, Chakravarti A (2012) Antiangiogenic therapy for patients with recurrent and newly diagnosed malignant gliomas. J Oncol 2012:193436
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8:592–603
Wick W, Weller M, Weiler M, Batchelor T, Yung AW, Platten M (2011) Pathway inhibition: emerging molecular targets for treating glioblastoma. Neuro Oncol 13:566–579
Acknowledgments
Medical writing support, funded by Boehringer Ingelheim, was provided by Duncan Campbell of GeoMed in the production of this manuscript. This investigator-initiated study was supported by a research grant from Boehringer Ingelheim, Germany, who also provided the study medication.
Conflicts of interest
No authors have conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Muhic, A., Poulsen, H.S., Sorensen, M. et al. Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. J Neurooncol 111, 205–212 (2013). https://doi.org/10.1007/s11060-012-1009-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-012-1009-y