
Abstract
Thousands of putative open reading frames (ORFs) are identified via annotation of sequenced plant genomes. Classification of these ORFs into gene families has a crucial importance to understand the evolution, function, and structure of the encoded proteins such as transcription factors, and the non-coding RNAs such as microRNAs (miRNAs). Thus, molecular mechanisms underlying the metabolic processes in plants are uncovered as well. Some members of the gene families are species-specific being more dynamic during evolution whereas others are more conserved, phylogenetically sharing common features. The latter are especially important for the annotation of putative ORFs by revealing known counterparts with high sequence identity via sequence alignment to discover conserved motifs. Various bioinformatic tools are available to find out gene families in plants. The BLAST tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi) is widely used for identification of homologous sequences. Phytozome (http://www.phytozome.net) or GreenPhyl (http://www.greenphyl.org) are the web resources utilized for the functional and comparative genomics in plants to analyze gene families. TRAPID (http://bioinformatics.psb.ugent.be/webtools/trapid) offers a free of charge web source for functional and comparative analyses of transcriptome data sets for identification of gene families, alignment of multiple sequences and phylogenetic tree construction. Some of the databases store specific type of gene families such as plant transcription factor databases PlantTFDB (http://planttfdb.cbi.pku.edu.cn) and PlnTFDB (http://plntfdb.bio.uni-potsdam.de/v3.0), or miRBase (http://www.mirbase.org) for miRNAs. Molecular Evolutionary Genetics Analysis (MEGA) software is an integrated tool for the analyses such as alignment of sequence, construction of phylogenetic trees, and access to online databases. In this chapter, the bioinformatic tools for analyses of genomics and/or transcriptomics data sets to discover gene families as well as sample researches are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The high-throughput next-generation sequencing (NGS) technologies produce a large amount of nucleotide sequences. Complete genome sequences and RNA-seq analysis of many plants utilizing NGS have been published, and a vast quantity of sequences are stored at the publicly available databases such as NCBI GenBank (http://www.ncbi.nlm.nih.gov/nuccore) or Sequence Read Archive (SRA,http://www.ncbi.nlm.nih.gov/sra). Identification of all elements in a genome or transcriptome is highly time-consuming and laborious. Therefore, the databases still contain sequences waiting to be annotated to find out their functions. In silico genome-wide analysis of these sequences using various bioinformatic tools is currently a hot research topic. Among the genome/transcriptome annotation products, one of the main groups is the gene families (Mochida and Shinozaki2011; Martinez2013).
A plant genome includes thousands of protein-coding genes. Although many genes posses distinct features, a large number of genes share homology in terms of sequence structure, which are classified as gene families (Frech and Chen2010). Genome-wide comparative analysis of gene families in plants results in the identification of their distribution pattern, orthology and synteny status, and phylogenic relationships (Chanroj et al.2012; Rawal et al.2013; Hofberger et al.2014; Pan et al.2015). Moreover, comprehensive analysis of gene families may reveal a new function (Wang et al.2015a), a new family (Saito et al.2014), or an evolutionary history among all eukaryotes: plants, animals, and fungi (Li et al.2014b).
Comparative analysis of the transcriptome data produced by the high-throughput sequencing of cDNA libraries (RNA-seq) from a plant exposed to different conditions and/or from differing plant organs gives important clues about the biotic/abiotic stress and hormone responses (Nawaz et al.2014; Okay et al.2014; Kim et al.2015) as well as the plant development (Ha et al.2014; Chettoor et al.2014; Jali et al.2014). The RNA-seq raw data are reconstructed via mapping the reads to a complete reference genome; however, if the reference genome is partial or absent, two strategies can be utilized. In mapping-first approach, the sequence reads are mapped to an unannotated reference genome, and then the overlapping sequences are merged. On the other hand, in assembly-first approach (de novo assembly), the sequence reads are assembled directly, and then the assembly may be mapped to a reference genome, if available (Grabherr et al.2011). For instance, Newbler software assembles the Roche 454 GS reads de novo; however, the lack of a reference genome results in the loss of some data as unassembled.
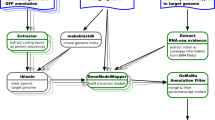
2 Bioinformatic Tools for Identification of Gene Families
Currently, a broad range of bioinformatic tools are present to process the excess amount of data produced by high-throughput sequencing for identification of gene families. First of all, the genome and/or transcriptome sequence data are required for the analyses. If previously produced sequence reads are to be used, the data can be retrieved from a database. Generally, the genome databases store information specific for an organism; however, some of the databases contain data for varying species.
The National Center for Biotechnology Information (NCBI;http://www.ncbi.nlm.nih.gov) portal is one of the most common sources for bioinformatics analyses. NCBI is a division of the U.S. National Library of Medicine (NLM) at the National Institutes of Health (NIH). NCBI server provides diverse resources for storing and analyzing information about genetics, biochemistry, and molecular biology (Fig.1). Among these resources, the Nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore) is a collection of genome, gene, and transcript sequence data. The Sequence Read Archive (SRA;http://www.ncbi.nlm.nih.gov/sra) stores raw sequencing data and alignment data from high-throughput next-generation sequencing platforms. NCBI also provides the Basic Local Alignment Search Tool (BLAST;http://blast.ncbi.nlm.nih.gov/Blast.cgi) for query of nucleotide and amino acid sequences.

The homepage of the NCBI database
The Phytozome database (Goodstein et al.2012;http://phytozome.jgi.doe.gov/pz/portal.html), a joint project of the Center for Integrative Genomics and the Joint Genome Institute (JGI), is also frequently used for retrieval of genome data belonging to green plants (Fig.2). The current version 10.1 of Phytozome includes the genome data of 48 plants and the gene families clustered at 12 phylogenetically important nodes. Additionally, this database contains the information about the genes, gene families, diversity, and expression data for 52 plant genomes. A detailed guide for the utilization of Phytozome can be reached fromhttp://phytozome.jgi.doe.gov/pz/QuickStart.html.

The homepage of the Phytozome database v10.1
The conserved domains representing the protein families should be identified to search the genome/transcriptome sequence. The Protein families (Pfam) database (Finn et al.2014;http://pfam.xfam.org) is widely used for the identification of protein families and clans, the higher-level groupings of related families (Fig.3). In Pfam database, the protein families are represented by multiple sequence alignments and hidden Markov models (HMMs). There are two components of Pfam, Pfam-A and Pfam-B. Pfam-A covers manually curated high quality data whereas Pfam-B includes automatically generated lower quality entries.

The homepage of the Pfam database v27.0
Another database for the domain analysis is the Simple Modular Architecture Research Tool, SMART (Letunic et al.2015;http://smart.embl.de), which contains two modes according to the protein database used (Fig.4). The Normal SMART covers SP-TrEMBL, Swiss-Prot, and stable Ensembl proteomes whereas the Genomic SMART contains Ensembl for metazoans and Swiss-Prot for the rest.

The homepage of the SMART database
Following the determination of gene/protein families, multiple sequence alignment is performed. The Clustal (Sievers et al.2011;http://www.clustal.org) is a widely used tool for multiple sequence alignment (Fig.5). It has two versions: ClustalW2 and Clustal Omega. (1) ClustalW2 (ClustalW/X). ClustalW is the command line version and ClustalX is the graphical version. (2) Clustal Omega is the latest addition offering higher scalability, velocity, and quality. Only command line/web server of Clustal Omega is in use currently. The Clustal can be utilized online from the European Bioinformatics Institute (EMBL-EBI) website (http://www.ebi.ac.uk/Tools/msa).

The homepage of the Clustal tool
The Multiple Sequence Comparison by Log-Expectation (MUSCLE) program (Edgar2004;http://drive5.com/muscle) is also used for multiple sequence alignment. The MUSCLE is faster than Clustal and can be operated using Linux, Windows, Windows with Cygwin, and Mac OSX. Additionally, an online tool is available at the EMBL-EBI website (http://www.ebi.ac.uk/Tools/msa/muscle).
The phylogenetic relationships of the members belonging to a gene family are investigated as well. The Molecular Evolutionary Genetics Analysis (MEGA) software (Tamura et al.2013;http://www.megasoftware.net) is a common tool for phylogenetic analyses. Currently, MEGA v6.0 is accessible (Fig.6), and can be utilized for diverse analyses including sequence alignment, construction of phylogenetic trees, online database search, estimation of divergence time, and molecular evolution rate.

The menu of the MEGA v6.06. The menu options were placed mixed due to the insufficient space
The conserved motifs representing the gene families are also identified. The motif-based sequence analysis tool package MEME Suite (Bailey et al.2009;http://meme.nbcr.net/meme), developed and maintained by seven institutes including National Center for Research Resources and National Biomedical Computation Resource, is widely used for this purpose. Currently, version 4.9.1 of the package includes 13 different tools for discovery, alignment, comparison, enrichment, and annotation of the conserved motifs (Fig.7).

The homepage of the MEME Suite tool
The Expert Protein Analysis System (ExPASy) portal (Artimo et al.2012;http://www.expasy.org), launched by the SIB (Swiss Institute of Bioinformatics), is frequently used to analyze the physicochemical properties such as molecular weight (Mw) and isoelectric point (pI) of the proteins. Although previous version of the portal was utilized only for protein analysis, the current bioinformatics resources portal includes many databases and software tools useful for the different areas of life sciences (Fig.8).

The homepage of the ExPASy bioinformatics resource portal
The most widely used tools are mentioned above; however, there are various additional tools for diverse analyses to identify the gene families. The bioinformatic tools used in the most recent studies on the analysis of gene families in plants are mentioned under the next title.
3 Identification of Gene Families in Plants
3.1 Identification of Gene Families in Monocots
3.1.1 Poaceae (Gramineae)
3.1.1.1 Rice
Gene families in rice (Oryza sativa) have been identified both genome-wide to show their distribution on the chromosomes, and transcriptome-wide to determine their expression patterns related with stress conditions and developmental stages.Nguyen et al. (2014) identified 16Catharanthus roseus receptor-like kinase1-like kinase (CrRLK1L) genes in rice genome using the Greenphyl and Rice Genome Annotation Project (RGAP;http://rice.plantbiology.msu.edu) databases. Moreover, the protein, genomic, and cDNA accession numbers were retrieved from NCBI, the clone name from Knowledge-based Oryza Molecular biological Encyclopedia (KOME;http://cdna01.dna.affrc.go.jp/cDNA), and GO terms from the Rice Oligonucleotide Array Database (http://www.ricearray.org). Additionally, the ortholog names were collected from The Arabidopsis Information Resource database (TAIR;http://www.Arabidopsis.org).
Ma et al. (2014c) identified 21 xylogen-like arabinogalactan protein (XYLP) genes from the rice genome using the RGAP database. The presence of nonspecific lipid transfer protein-like (nsLTP) domains, N-terminal signal peptide, GPI-anchored signal, and N-glycosylation sites were predicted via InterProScan (http://www.ebi.ac.uk/Tools/pfa/iprscan5), SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP), Big-PI Plant Predictor (http://mendel.imp.ac.at/gpi/plant_server.html), and NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc), respectively.
Similarly, the drought-induced 19 (Di19) family of transcription factors in rice genome were analyzed by Wang et al. (2014e) using the RGAP and Rice Annotation Project (RAP-DB;http://rapdb.dna.affrc.go.jp/index.html) databases. The presence of zf-Di19 and Di19_C domains was determined using InterProScan and Pfam servers, the physicochemical parameters of each gene were predicted using ExPASy, and the phylogenetic analyses were performed via MEGA software.
Moreover, de Oliveira et al. (2014) identified the family of wall-associated kinases (WAKs) in rice genomes using RGAP and Gramene (http://www.gramene.org) databases. The protein domains were analyzed via Pfam and SMART, and splicing predictions were performed using GENSCAN (http://genes.mit.edu/GENSCAN.html). The sequence alignment, similarity clustering, and phylogenetic analyses were performed via MUSCLE, Circoletto (Darzentas2010), and MEGA software, respectively.
Zhang et al. (2015a) identified three members of rice CCT [CONSTANS (CO), CO-LIKE, and TIMING OF CAB1 (TOC1)] family, taking role in flowering, through the Gramene, RGAP, NCBI, and DRTF (http://drtf.cbi.pku.edu.cn/index.php) databases. Nucleotide diversity analysis and candidate gene-based association mapping were performed via RiceVarMap (http://ricevarmap.ncpgr.cn).
Nawaz et al. (2014) analyzed the cyclic nucleotide-gated ion channel (CNGC) gene family and their expression patterns in response to plant hormones and biotic and abiotic stresses in rice. TheArabidopsis CNGC gene sequences were obtained from the TAIR database to find out the rice homologs in RGAP and RAP-DB databases. The non-redundant sequences were collected from Phytozome database, and the domain analysis was performed using the Pfam, SMART, CDD (http://www.ncbi.nlm.nih.gov/cdd), PROSITE (http://prosite.expasy.org), SUPERFAMILY (http://www.supfam.org/SUPERFAMILY), and Gene3D (http://gene3d.biochem.ucl.ac.uk). The chromosomal location and duplication status of the genes were determined using the Rice TOGO Browser (http://agri-trait.dna.affrc.go.jp) and the Plant Genome Duplication Database (PGDD;http://chibba.agtec.uga.edu/duplication), respectively. Exon–intron distribution andcis-acting regulatory elements were analyzed using Gene Structure Display Server (GSDS;http://gsds.cbi.pku.edu.cn) and the PlantCis-acting Regulatory DNA Elements (PLACE;http://www.dna.affrc.go.jp/PLACE) database, respectively. The protein sequences were analyzed via ExPASy, their cellular localizations were determined using PSORT (http://psort.hgc.jp), and the posttranslational modifications were predicted via PROSITE tool. The amino acid sequences were aligned using MUSCLE and MAFFT (http://mafft.cbrc.jp/alignment/software), the phylogenetic analysis was performed via MEGA, and the conserved motifs were determined using MAST (http://meme.nbcr.net/meme/cgi-bin/mast.cgi) search tool.
Additionally, Saha et al. (2015) identified rice ATP-binding cassette (ABC) transporter family, taking role in salt stress, using RGAP and GenBank (http://www.ncbi.nlm.nih.gov/genbank) databases. The protein domains and conserved motifs were predicted via SMART and MEME tools, respectively. The phylogenetic analyses were performed using MEGA software, the genes were mapped to the rice chromosomes via Massively Parallel Signature Sequencing database (MPSS;http://mpss.udel.edu/in9311/mpss_index.php), and the expression patterns of the genes during salinity stress were investigated using Genevestigator (https://genevestigator.com/gv) database.
3.1.1.2 Maize
The gene families in maize have also been analyzed using both genome- and transcriptome-wide methodologies. The protein phosphatase family in maize was identified by Wei and Pan (2014) using Maize Genome Sequence Project (http://ftp.maizesequence.org/release-5b/filtered-set). The hidden Markov model (HMM) profiles and the catalytic domains of the proteins were predicted via HMMER (http://hmmer.janelia.org), Pfam, and SMART. The subcellular localization and pI of the proteins were determined using PSORT and ExPAsy, respectively. The phylogenetic analysis was performed using MEGA. The gene structure, chromosomal location, gene duplication, synteny, and sequence polymorphism analyses were performed via GSDS, MapDraw (Liu and Meng2003), SyMAP (http://www.symapdb.org), and DnaSP (http://www.ub.edu/dnasp), respectively. The putativecis-acting elements in the promoters, and the microRNAs (miRNAs) were identified using PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html) and miRBase (http://www.mirbase.org), respectively.
The expansin gene family in maize genome and their expression in maize endosperm were investigated by Zhang et al. (2014a) using the MaizeSequence (http://ftp.maizesequence.org/ current) database. The protein domains and motifs were determined using Pfam, SMART, and MEME. The pIs, signal peptide cleavage sites, and the gene ontology (GO) terms were predicted using ExPASy, SignalP, and ProtFun (http://www.cbs.dtu.dk/services/ProtFun) servers, respectively. The phylogenetic, gene structure, and chromosomal location analyses were performed via MEGA, GSDS, and MapInspect (http://mapinspect.software.informer.com), respectively. The hormone-responsive elements in promoter regions were predicted using PlantCARE and PLACE.
Shiriga et al. (2014) investigated the genome-wide distribution of NAC family of transcription factors, and their expression pattern under drought stress in maize using the Plant Transcription Factor Database (PlnTFDB;http://plntfdb.bio.uni-potsdam.de). The protein domains were determined using Pfam, SMART, and InterProScan. The phylogenetic analysis was performed using MEGA as well as the membrane-bound proteins and the motifs were predicted via TMHMM (http://www.cbs.dtu.dk/services/TMHMM), and MEME servers, respectively.
Similarly, Fan et al. (2014) identified the NAC transcription factors in maize using the Phytozome database. The Pfam and SMART were used to predict protein domains. The gene and protein structures were analyzed via GSDS, ProtParam (http://web.expasy.org/protparam), and SOPMA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html), respectively. The chromosomal locations and conserved motifs were determined via MapInspect and MEME, respectively. The orthologous sequences were analyzed using OrthoMCL (http://www.orthomcl.org/orthomcl) and the data for NAC gene expression in various maize tissues and developmental stages were obtained from Genevestigator.
Chettoor et al. (2014) analyzed the RNA-seq of different reproductive organs of maize to find out the gametophyte functions, and small signaling proteins and various transcription factor gene families were identified. The data from different samples were determined using the Venny (http://bioinfogp.cnb.csic.es/tools/venny) and the Venn Diagrams were obtained using the BioInfoRx (http://apps.bioinforx.com/bxaf6/tools/app_overlap.php) tools. The GO terms, small peptide genes, and the transcription factors were identified using AgriGO (http://bioinfo.cau.edu.cn/agriGO) toolkit, MaizeSequence (updated link:http://ensembl.gramene.org/Zea_mays/Info/Index) database, and Grassius (http://grassius.org/grasstfdb.html) server. The sequences were aligned using MegAlign (DNASTAR;http://www.dnastar.com), and the phylogenetic analysis was performed using MrBayes (http://mrbayes.sourceforge.net).
Distribution of auxin-responsiveGH3 family genes in maize genome and their expression under abiotic stresses were analyzed by Feng et al. (2014b) using MaizeGDB (http://www.maizegdb.org). The protein domains were predicted using Pfam and InterProScan. The phylogenetic, gene structure, and synteny analyses were performed via MEGA, GSDS, and SyMAP, respectively.
3.1.1.3 Wheat
The distribution of nucleotide binding site–leucine-rich repeats (NBS-LRR) disease resistance genes in wheat (Triticum aestivum) genome was identified by Bouktila et al. (2015) using the NCBI database for wheat (http://www.ncbi.nlm.nih.gov/Traces/wgs/?val=CALP01) and analyzed via European Molecular Biology Open Software Suite (EMBOSS;http://www.ebi.ac.uk/Tools/emboss). The sequences were aligned using MUSCLE, and the HMM profiles were predicted via HMMER. The NBS sequences were obtained from EMBOSS, merged via DNA Baser sequence assembler (http://www.dnabaser.com), and the redundant contigs were checked using CD-HIT Suite (http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi?cmd=cd-hit-est). The candidate wheat NBS-encoding R genes were identified using FGENESH (http://www.softberry.com) and Pfam. The structural domain and orthologous cluster analyses were performed via Geneious (http://www.geneious.com) and InterProScan, and OrthoMCL database, respectively.
Thomas et al. (2014) investigated the Methyltransferase 1 (MET1) gene family in hexaploid wheat using the International Wheat Genome Sequencing Consortium (IWGSC;http://www.wheatgenome.org) and URGI (https://urgi.versailles.inra.fr) databases. The transposable elements were analyzed using TREP (http://wheat.pw.usda.gov/ITMI/Repeats) database. Gene structures and protein domains were identified using SIM4 (http://pbil.univ-lyon1.fr) and FancyGene (http://bio.ieo.eu/fancygene) tools, and Pfam, respectively. The sequences were aligned using MUSCLE, phylogenetic trees were constructed via Interactive Tree of Life (iTOL;http://itol.embl.de), and the exonic sequences were identified using FGENESH. For RNA-seq analysis, the cDNAs were translated using Transeq and Sixpack from the EMBOSS package. The sequence reads were aligned using TopHat (http://ccb.jhu.edu/software/tophat/index.shtml) and Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), and analyzed using Cufflinks (http://cole-trapnell-lab.github.io/cufflinks).
Ariyarathna et al. (2014) characterized the multigene familyTaHKT 2;1 in bread wheat using IWGSS database. The gene structures were predicted using Spidey (http://www.ncbi.nlm.nih.gov/spidey), GeneSeqer (http://www.plantgdb.org/cgi-bin/GeneSeqer/index.cg), and SIM4 software. Sequence and phylogenetic analyses were conducted using Geneious and MEGA, respectively. The structural and functional analyses of the proteins were performed via TMHMM and Membrane Protein Explorer (MPEx;http://blanco.biomol.uci.edu/mpex) tools. 3D structures of the proteins were predicted using PHYRE 2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index). Thecis-acting elements were analyzed via PlantCARE and PLACE databases.
Okay et al. (2014) identified the superfamily of WRKY transcription factors in bread wheat using Plant Transcription Factor Database (PlantTFDB;http://planttfdb.cbi.edu.cn) and NCBI databases. The HMM profiles were predicted using Pfam, the phylogenetic analysis was performed via MEGA, and the conserved motifs were determined using MEME Suite tool. To identify the drought-responsive WRKY members, the RNA-seq data were retrieved from SRA. The proteins were detected using PlantTFDB and analyzed via ExPASy portal.
3.1.1.4 Barley
Genome-wide characterization of the basic leucine zipper (bZIP) family of transcription factors in barley (Hordeum vulgare) and their expression patterns were investigated by Pourabed et al. (2015) using the PlantTFDB and the International Barley Sequencing Consortium (IBSC) IPK BLAST server (http://webblast.ipk-gatersleben.de/barley). The HMM profiles and conserved domains were analyzed using HMMER, Pfam, and SMART. The sequences were aligned and phylogenetic trees were constructed using ClustalX and MEGA, respectively. The bZIP motifs were predicted via MEME, and the gene structures were identified using GSDS. The UTR sequences were retrieved from Ensembl Plants database (http://plants.ensembl.org/index.html), the internal ribosome entry sites (IRES), and sRNA targets were predicted via UTRdb (http://utrdb.ba.itb.cnr.it) and psRNATarget (http://plantgrn.noble.org/psRNATarget) servers, respectively. Thecis-regulatory elements were identified using Plant Promoter Analysis Navigator (PlantPAN;http://plantpan.mbc.nctu.edu.tw) database.
Pandey et al. (2014) identified the heat shock protein 20 (HSP20) gene family in wheat and barley as well as their expression pattern under heat stress using NCBI and Uniprot (http://www.uniprot.org) databases. The HMM profiles were analyzed using Pfam and HMMER. Open reading frames were predicted via ORF finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). The conserved HSP20 motifs and domains were analyzed using MEME and InterProScan, respectively. The physicochemical properties and the subcellular localization of the proteins were predicted using ProtParam, and ESLpred (http://www.imtech.res.in/raghava/eslpred/submit.html) and ProtComp (http://linux1.softberry.com/cgi-bin/programs/proloc/protcomppl.pl) servers, respectively. Simultaneous Alignment and Tree Construction using Hidden Markov Models - Jump Start (SATCHMO-JS (http://phylogenomics.berkeley.edu/q/satchmo) and PROMALS3D (http://prodata.swmed.edu/promals3d/promals3d.php) servers were used for sequence alignment, and the phylogenetic tree was constructed using MEGA.
3.1.1.5 Brachypodium distachyon
The IQ67 domain (IQD) and growth-regulating factor (GRF) gene families found inBrachypodium distachyon genome were investigated by Filiz et al. (2013) and (2014), respectively using NCBI and Phytozome databases. The conserved domains were identified using Pfam and SMART servers. The sequences were aligned via ClustalW, motifs were predicted using MEME, and the phylogenetic tree was constructed using MEGA. The gene structures, ORFs, and physicochemical characteristics of the proteins were identified using GSDS, ORF finder, and ProtParam, respectively. The GO terms were annotated using Gramene resource.
Wen et al. (2014) identified the WRKY family of transcription factors inB. distachyon genome utilizing PlantTFDB, GramineaeTFDB (http://gramineaetfdb.psc.riken.jp),B. distachyon genome database (http://www.brachypodium.org), Phytozome, UniProt, and NCBI databases. The reliability of the results was verified using UniProt, SMART, and the Brachy WRKY Database (http://www.igece.org/WRKY/BrachyWRKY/BrachyWRKYIndex.html). The sequences were aligned using ClustalW and the phylogenetic tree was constructed via MEGA. The conserved motifs andcis-acting elements were analyzed utilizing MEME and PlantCARE, respectively.
Wei et al. (2014a) analyzed the MADS-box gene family inB. distachyon genome usingB. distachyon genome database. The sequences were aligned via ClustalX, and the phylogenetic tree was constructed using MEGA. The Mw and pI of the proteins were predicted using Editseq (DNASTAR), and the gene structures were analyzed utilizing GSDS. The conserved motifs were determined and annotated via MEME and SMART, respectively. The Ka/Ks values were calculated using PAL2NAL (http://www.bork.embl.de/pal2nal) program.
Zhu et al. (2014a) identified the family of protein disulfide isomerase (PDI) genes inB. distachyon genome utilizing NCBI and Phytozome databases. The protein sequences were analyzed using Pfam, CDD, Interpro (http://www.ebi.ac.uk/interpro), and ExPASy databases. The chromosomal locations were mapped using MapInspect software, and the syntenic relationships were investigated using PGDD. The transcription directions and the structures of the genes were analyzed utilizing Gramene and GSDS, respectively. The protein pI/Mw was calculated via ExPASy, and the signal peptides were predicted using SignalP. The transmembrane regions were predicted via TMHMM and SMART databases, phosphorylation and N-/O-glycosylation sites were determined utilizing NetPhos (http://www.cbs. dtu.dk/services/NetPhos) NetNGlyc (http://www.cbs.dtu.dk/services/NetNGlyc), and NetOGlyc (http://www.cbs.dtu.dk/services/NetOGlyc) servers, respectively. The sequences were aligned using ClustalW, and the phylogenetic tree was generated via MEGA software.
3.1.1.6 Foxtail Millet
The AP2/ERF transcription factor family in foxtail millet (Setaria italica) genome and their expression pattern were investigated by Lata et al. (2014). The HMM profiles were obtained from Pfam and searched against Phytozome for foxtail millet. The conserved domains of the proteins were predicted using HMMER. Chromosomal location, segmental duplication, and gene structure analyses were performed using MapChart (https://www.wageningenur.nl/en/ show/Mapchart.htm), Multiple Collinearity Scan (MCScan;http://chibba.agtec.uga.edu/duplication/mcscan) and Circos (http://circos.ca), and GSDS, respectively. Phylogenetic analysis, GO annotation,cis-regulatory element, and miRNA identification were performed using MEGA, Blast2GO (https://www.blast2go.com), PLACE, psRNATarget tools, respectively. For transcriptome analysis, the RNA-seq data were obtained from European Nucleotide Archive (ENA;http://www.ebi.ac.uk/ena), mapped via CLC Genomics Workbench (http://www.clcbio.com/genomics), and the heat map profile was constructed using TIGR Multiexperiment Viewer (MeV;http://www.tm4.org/mev.html).
Likewise, the superfamily of MYB transcription factors in foxtail millet was investigated by Muthamilarasan et al. (2014) using Pfam and Phytozome. The protein domains were also analyzed using CDD and HMMER. The physical map of chromosomal location was generated using MapChart, and the segmental duplications were identified via MCScanX (http://chibba.pgml.uga.edu/mcscan2). The sequence alignment was performed using BioEdit, and the physicochemical properties of the proteins were determined via ExPASy. The phylogenetic tree was constructed using MEGA, and the GO annotation was performed using Blast2GO. Thecis-acting elements and the miRNAs were analyzed using PLACE, Foxtail millet Transcription Factor Database (FmTFDb;http://59.163.192.91/FmTFDb), and psRNATarget, respectively. The RNA-seq data were retrieved from ENA and filtered using the NGS QC Toolkit (http://59.163.192.90:8080/ngsqctoolkit). The heat map for expression profile was generated via TIGR MeV. The orthologous relationships of grass MYBs were visualized and analyzed using Circos and PAL2NAL.
Zhu et al. (2014b) analyzed theNBS-LRR disease resistance genes in foxtail millet using the BioEdit software (http://www.mbio.ncsu.edu/bioedit/bioedit.html) to construct a local platform based on the genome sequence inftp://ftp.jgi-psf.org/pub/JGI_data/Setaria_italica. The protein structures were analyzed using Pfam and COILS (http://ch.embnet.org/software/COILS_form.html) software. The sequences were aligned using ClustalX (http://www.clustal.org/clustal2) and the phylogenetic trees were constructed via MEGA.
The PHT1 family of phosphate transporters in foxtail millet was identified by Ceasar et al. (2014) using Phytozome. The phylogenetic analysis was conducted using MEGA, and thecis-acting elements were identified via PLACE. The conserved regions in the putative promoter regions were predicted using the Evolutionary Analysis of Regulatory Sequences (EARS;http://wsbc.warwick.ac.uk/wsbcToolsWebpage/user_case_form.php).
3.1.1.7 Sorghum
TheNBS-LRR genes in sorghum (Sorghum bicolor) genome were identified by Mace et al. (2014) using Phytozome database. The conserved motifs and coiled coils were detected using Pfam and COILS, respectively. The sequences were aligned via ClustalW and MEGA. The phylogenetic trees were constructed and displayed using TreeBest (http://treesoft.sourceforge.net/treebest.shtml) and Dissimilarity Analysis and Representation for Windows (Darwin;http://darwin.cirad.fr) software, respectively. The synteny analysis was performed utilizing Circos software.
Filiz and Tombuloğlu (2015) investigated the distribution of superoxide dismutase (SOD) gene family in sorghum genome using Phytozome and NCBI databases. The protein domains were detected via Pfam and the physicochemical characteristics were determined using ProtParam tool. The gene structures were identified using GSDS, and transcript levels ofSbSOD genes were determined via the NCBI expressed sequence tag (EST) database (http://www.ncbi.nlm.nih.gov/dbEST). The conserved motifs were analyzed using MEME Suite, the sequences were aligned via ClustalW, and the phylogenetic analysis was conducted using MEGA software. The GO terms were annotated using AmiGO (http://amigo.geneontology.org/amigo), 3D structures of the proteins were predicted utilizing 3DLigandSite server (http://www.sbg.bio.ic.ac.uk/~3dligandsite), and the structural and stereochemical analyses were assessed via RAMPAGE Ramachandran plot analysis (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php).
Panahi et al. (2014) investigated the genes for alternative splicing in sorghum genome using NCBI and Phytozome databases. The transcripts were mapped to the chromosomes using SIM4, and the GTF file was investigated via Alternative Splicing transcriptional landscape visualization tool (Astalavista;http://genome.crg.es/astalavista). For GO analysis, the data were retrieved from Ensemble using BioMart tool (http://www.ensembl.org/info/data/biomart.html) and analyzed via AgriGO.
3.2 Identification of Gene Families in Dicots
3.2.1 Brassicaceae
3.2.1.1 Arabidopsis thaliana
The first plant genome sequence to be completed belongs to the model organismArabidopsis thaliana (TheArabidopsis Genome Initiative2000). Therefore, the genome and transcriptome studies onA. thaliana are numerous, and its genome sequence is used for mapping in many plant genome studies. Here are some of the recent studies on investigation of gene families inA. thaliana utilizing genome and/or transcriptome data:
Jali et al. (2014) investigated the HUA2-LIKE (HULK) gene family inA. thaliana genome using TAIR and Phytozome databases. The sequences were aligned using MUSCLE and the conserved blocks were obtained via Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks.html). The phylogenetic tree was generated and visualized using TREE-PUZZLE (http://www.tree-puzzle.de) and Dendroscope (http://ab.inf.uni-tuebingen.de/software/dendroscope) software, respectively. For transcriptome analysis, the data were retrieved from Gene Expression Omnibus (GEO;http://www.ncbi.nlm.nih.gov/geo). The RNA-seq data were aligned and mapped toArabidopsis genome using TopHat and Bowtie2, and analyzed using Cufflinks.
Ballester et al. (2015) characterized the NGATHA (NGA) clade of transcription factors inA. thaliana using genome sequence database. NGA orthologs from different plant were searched via BLAST, and the pairwise alignments were performed using VISTA (http://genome.lbl.gov/vista/index.shtml). The conserved sequences were aligned using ClustalW tool in MacVector software (http://macvector.com).
Siriwardana et al. (2014) analyzed the NF-YA members of NUCLEAR FACTOR-Y (NF-Y) transcription factor families inA. thaliana genome, and their role in abscisic acid responses during seed germination. The sequence data were retrieved from TAIR and the phylogenetic analyses were performed using MEGA.
Kenzior and Folk (2015) identified a novel family of plant-specific PWWP/RRM (RNA recognition motif) domain proteins inA. thaliana using NCBI and TAIR databases. The sequences were aligned via MultAlin (http://multalin.toulouse.inra.fr/multalin). The Jpred (http://www.compbio.dundee.ac.uk/www-jpred) server was utilized for predicting secondary structures of RRM domain. The phylogenetic analysis was conducted using ClustalW and PHYLogeny Inference Package (PHYLIP;http://evolution.genetics.washington.edu/phylip.html) tools.
3.2.1.2 Brassica spp.
The MADS-box gene family inBrassica rapa (Chinese cabbage) genome was identified by Duan et al. (2014) using theBrassica database (BRAD;http://brassicadb.org/brad). The conserved domains were analyzed using Pfam, SMART, and NCBI databases. The MADS proteins from different plants were retrieved and analyzed using TAIR, Pfam, Phytozome, and PGDD databases. The sequences were aligned using ClustalW and the phylogenetic analysis was conducted using MEGA software. The conserved motifs were analyzed via MEME and SMART, and the gene structures were determined using GSDS. The ortholog groups were identified using OrthoMCL, the genes were linked to chromosomes via Circos, and the network relations were built using Cytoscape (http://www.cytoscape.org) software. The gene duplications (Ka/Ks values) were determined using KaKs_calculator (http://evolution.genomics.org.cn/software.htm).
Duan et al. (2015) investigated the ascorbic acid-related genes inB. rapa genome using BRAD. The sequences were analyzed using FGENESH and verified via NCBI database. The core eukaryotic genes and random genes were retrieved from CEGMA (http://korflab.ucdavis.edu/Datasets/cegma) and the synteny analysis was performed using MCScanX. The homologs were retrieved from Phytozome and Amborella Genome Database (http://www.amborella.org). The sequences were aligned using ClustalW, the phylogenetic trees were generated using MEGA, and the Ka/Ks values were calculated via KaKs_calculator. The conserved motifs were identified using MEME and the GO terms were annotated via InterProScan.
R2R3-MYB transcription factors inB. rapa genome were analyzed by Wang et al. (2015b) using BRAD and PlantTFDB. The conserved protein domains were analyzed using Pfam, SMART, and ExPASy. Mw and pI of the proteins were calculated using the Pepstats (http://www.ebi.ac.uk/Tools/seqstats/emboss_pepstats). The conserved motifs were identified via MEME. Multiple alignments of the sequences were performed using ClustalW and WebLogo (http://weblogo.berkeley.edu/logo.cgi), and the phylogenetic analysis was conducted via MEGA. The orthologous and paralogous genes were analyzed via OrthoMCL and plotted using Circos. The gene duplication analysis was performed using MCScanX.
Wang et al. (2014a) identified theGRF family of genes inB. rapa genome using BRAD. The sequences were aligned using DNAMAN (http://www.lynnon.com) and the phylogenetic analysis was performed utilizing MEGA software. The gene structures were identified via GSDS, the GC content was calculated via DNASTAR, and the physicochemical properties of the proteins were predicted using ProtParam. The Simple Sequence Repeat Identification Tool (SSRIT;http://archive.gramene.org/db/markers/ssrtool) was used to detect the SSR markers. The GO terms were annotated using Gramene, and the conserved motifs were predicted via MEME Suite tool.
The superfamily of WRKY transcription factors inB. rapa genome was investigated by Kayum et al. (2015) utilizing BRAD. The conserved domains and the properties of the proteins were analyzed using SMART and ExPASy, respectively. The chromosomal locations were identified via MapChart.
Ma et al. (2014b) analyzed the NAC transcription factor family inB. rapa genome using BRAD. The conserved motifs were predicted via MEME, the sequences were aligned utilizing ClustalW, and the phylogenetic tree was generated using MEGA software.
Arya et al. (2014a) investigated the family of heterotrimeric G-protein subunit genes inB. rapa genome using Phytozome database. The alignment of sequences and phylogenetic analysis were conducted using ClustalW and MEGA, respectively. The Ka/Ks values were calculated using DnaSP.
The family ofNBS-LRR resistance genes inB. oleracea was analyzed by Kim et al. (2015). The RNA-seq reads were de novo assembled via Velvet (https://www.ebi.ac.uk/~zerbino/velvet) and Oases (https://www.ebi.ac.uk/~zerbino/oases) programs, validated using Phytozome, and mapped to the assembled unigenes using Bowtie. The number of mapped reads was normalized utilizing DESeq (http://bioconductor.org/packages/release/bioc/html/DESeq.html) software. Functional enrichment and annotation analysis was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID;http://david.abcc.ncifcrf.gov). The RNA-seq data were deposited in the National Agricultural Biotechnology Information Center (NABIChttp://nabic.rda.go.kr) database. NBS-encoding genes in the other plant genomes were retrieved from BRAD and PlantTFDB, and the conserved domains were searched in theBrassica oleracea Genome Database (Bolbase;http://www.ocri-genomics.org/bolbase). The protein domains were analyzed using SMART, EMBOSS, and myHits (http://myhits.isb-sib.ch/cgi-bin/motif_scan). The conserved motifs were predicted via MEME, and the genes were linked to chromosomes using MapChart.
Yao et al. (2015) identified the genome-wide distribution ofWRKY gene family inB. oleracea var.capitata using Bolbase database. The HMM profiles were analyzed via Pfam and HMMER, the sequences were aligned using MUSCLE, and the protein domains were identified via SMART. For RNA-seq analyses, the data were retrieved from SRA, the sequence reads were mapped using Bowtie and TopHat, and the transcript reconstruction was performed using Cufflinks. The conserved motifs were predicted via MEME, the sequences were aligned using MUSCLE and BioEdit, and the phylogenetic tree was generated using MEGA software.
3.2.2 Fabaceae (Leguminosae)
3.2.2.1 Chickpea
Distribution of Ethylene Responsive Factor (ERF) gene family in chickpea (Cicer arietinum) genome was identified by Deokar et al. (2015) utilizing PlnTFDB, PlantTFDB, and International Chickpea Genetics and Genomics Consortium (ICGGC;http://www.icrisat.org/gt-bt/ICGGC/GenomeManuscript.htm) databases. The protein domains were analyzed using CDD, SMART, and Pfam. The phylogenetic tree was constructed using MEGA, and the gene duplication was predicted via MCScanX. The sequence homology was analyzed using BLAST, and the protein sequence analyses were performed via ExPASy and Membrane protein IdeNtificatioN withOUt explicit use of hydropathy profiles and alignments (MINNOU;http://minnou.cchmc.org) tools. Secondary structures were predicted using YASPIN (http://www.ibi.vu.nl/programs/yaspinwww) and Advanced Protein Secondary Structure Prediction Server (APSSP;http://imtech.res.in/raghava/apssp). Subcellular localization of the proteins was analyzed using PredictProtein (https://www.predictprotein.org) and WoLF PSORT (http://wolfpsort.org) servers. Thecis-acting elements were predicted via PLACE and PlantCARE. The DNA and amino acid analysis were performed using BioEdit and DNASTAR.
Sharma et al. (2014b) investigated the uridine diphosphate glycosyltransferase (UGT) family of genes in chickpea genome utilizing chickpea browser of Legume Information System (LIS;http://cicar.comparative-legumes.org). The family of UGTs and the conserved motifs were identified using SUPERFAMILY and MEME tools, respectively. The HMM profiles were analyzed using Pfam and HMMER. The dendrogram for evolutionary analysis was drawn via PhyML (http://atgc.lirmm.fr/phyml). To determine the functional specificity of UGTs, characterized proteins with known substrate specificity from different plants were retrieved from Swiss-Prot (http://www.uniprot.org) database. The molecular modeling of the proteins was performed using the Protein Data Bank (PDB;http://www.rcsb.org/pdb/home/home.do). The stereochemical properties of the 3D models were analyzed via PROCHECK (http://www.ebi.ac.uk/thornton-srv/software/PROCHECK), Verify3D (http://services.mbi.ucla.edu/Verify_3D), ProSA (https://prosa.services.came.sbg.ac.at/prosa.php), and ERRAT (http://services.mbi.ucla.edu/ERRAT) tools. The UGT orthologs were predicted using Blast2GO, and the gene structures were identified via GSDS.
Sharma and Suresh (2015) analyzed the proteases and protease inhibitors in chickpea genome using LIS database. The HMM profiles were identified using Pfam and HMMER. The sequences were aligned, bootstrapped, and analyzed via ClustalX, PHYLIP, and ProtDist (http://evolution.genetics.washington.edu/phylip/doc/protdist.html), respectively, and the dendrograms were obtained utilizing the FigTree (http://tree.bio.ed.ac.uk/software/figtree). The conserved domains were analyzed using SMART and DomainGraph (http://domaingraph.bioinf.mpi-inf.mpg.de), and the signal peptides were predicted via SignalP. The orthologs were identified using Blast2GO, and the codon composition was analyzed via MEGA. The gene structure was identified using GSDS. For gene expression analyses, the RNA-seq data were retrieved from SRA, mapped to the chickpea genome using TopHat, and the abundance of reads were estimated via Cufflinks.
Ha et al. (2014) investigated the NAC family of transcription factors in chickpea genome utilizing the Chickpea Transcriptome Database (CTDB;http://www.nipgr.res.in/ctdb.html) PlantTFDB and iTAK (http://bioinfo.bti.cornell.edu/cgi-bin/itak/index.cgi) program. Additionally, the genome sequence of chickpea cultivar “desi” (CGAP;http://nipgr.res.in/CGAP/home.php) was used, and the HMM profiles were identified via TMHHM. The phylogenetic analyses were conducted using MEGA, and the sequence alignment was visualized via GeneDoc (http://www.nrbsc.org/gfx/genedoc).
Jain et al. (2014) performed the genome-wide identification of the miRNAs in chickpea. The RNA-seq data were pre-processed using miRTools (http://centre.bioinformatics.zj.cn/mirtools) software, and annotated for small nucleolar RNAs using Plant SnoRNAbase (http://bioinf.scri.sari.ac.uk/cgi-bin/plant_snorna/home), for tRNAs using Genomic tRNA Database (http://gtrnadb.ucsc.edu/download.html), and for rRNAs using RFAM (http://rfam.xfam.org) database. The rest of the sequence was screened via RepBase (http://www.girinst.org/server/RepBase). The chickpea miRNAs were identified in miRBase and the filtered reads were mapped to these miRNAs using Bowtie. The secondary structures of the genomic sequences were determined using RNAfold (http://www.tbi.univie.ac.at/RNA/RNAfold.html) software, and processed utilizing miRDeep-P core algorithm (http://faculty.virginia.edu/lilab/miRDP). The mature miRNA candidates were clustered into families via CD-HIT server, and the putative targets of the miRNAs were predicted using psRNATarget server. The conserved domains of the targets were identified via Pfam and HMMER program. The GO terms were annotated using BİNGO (http://www.psb.ugent.be/cbd/papers/BiNGO/Home.html) software, and the eukaryotic orthologous groups (KOGs) were identified utilizing NCBI KOG server (ftp://ftp.ncbi.nih.gov/pub/COG/KOG).
3.2.2.2 Soybean
The cupin gene family in soybean (Glycine max) genome was investigated by Wang et al. (2014f) utilizing Phytozome. The conserved domains were identified via InterProScan. Sequence alignment and phylogenetic analysis were conducted using ClustalX and MEGA, respectively. The logos for amino acid residues in conserved domains were generated using WebLogo. The conserved motifs were analyzed via MEME, and annotated using SMART, Pfam, and NCBI database. The exon/intron organizations were identified via GSDS, and the chromosomal locations were mapped using Chromosome Visualization Tool (CViT) at the LIS database (http://cvit.comparative-legumes.org). The Ka/Ks values were calculated using DnaSp. For gene expression analysis, the data were obtained from the SoyBase database (http://soybase.org), analyzed using Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm), and the heat map was visualized via Java Treeview (http://jtreeview.sourceforge.net). For evolutionary analysis, the SNP data were retrieved from the Soybean Knowledge Base (SoyKB;http://soykb.org).
Mainali et al (2014) identified the cyclophilin (CYP) gene family in soybean genome utilizing Phytozome database. The Mw and subcellular localization of the proteins were predicted using ProtParam program, and TargetP (http://www.cbs.dtu.dk/services/TargetP) and WoLF PSORT servers, respectively. The transcribedCYPs were analyzed using soybean gene index (ftp://occams.dfci.harvard.edu/pub/bio/tgi/data/Glycine_max). The sequences were aligned and analyzed using ClustalX, MEGA, and iTOL. For transcriptome analysis, the data were retrieved from GEO, and the heat map was generated using the gplots CRAN library (http://cran.r-project.org/web/packages/gplots/index.html).
Li et al. (2014a) investigated the family of heat shock transcription factors (Hsfs) in soybean genome utilizing Phytozome and SoyBase. The conserved domains were analyzed using SMART, Pfam, Predict Nuclear Localization Signals (PredictNLS;https://rostlab.org/owiki/index.php/PredictNLS), and Nuclear Export Signals (NetNES;http://www.cbs.dtu.dk/services/NetNES) servers, and the genes were mapped to the chromosomes via MapDraw. The exon-intron substructures and thecis-acting elements were analyzed using GSDS and PLACE tools, respectively. The phylogenetic tree was constructed using MEGA software.
The family of R2R3-MYB transcription factors in soybean was identified by Aoyagi et al. (2014) utilizing PlantTFDB, SoyDB, and Phytozome. The conserved domains and motifs were predicted using Pfam and SMART, and MEME, respectively. For transcriptome analysis, RNA-seq data were retrieved from LGE Soybean Genome Project (http://bioinfo03.ibi.unicamp.br/soja) and Genevestigator databases. The C-terminal amino acid sequences of the proteins were analyzed via MEME, and the phylogenetic tree was generated using MEGA.
Belamkar et al. (2014) analyzed the distribution of homeodomain leucine zipper (HD-Zip) transcription factor family in soybean using Phytozome database. Following BLAST search, the sequences were aligned using MUSCLE and processed via SeaView (http://doua.prabi.fr/software/seaview). The phylogenetic trees were generated using CLUSTAL, PhyML, and FigTree. The HMM profiles were analyzed using HMMER. For RNA-seq analysis, the data were obtained from SoyBase and SoyKB. The GO terms were annotated using Blast2GO and SoyDB (http://casp.rnet.missouri.edu/soydb), and the heat map was obtained via the gplots CRAN library.
Bencke-Malato et al. (2014) investigated the family of WRKY transcription factors in soybean genome utilizing Phytozome, PlantTFDB, PLAZA (http://bioinformatics.psb.ugent.be/plaza), and SoyBase. The protein domains were identified using SMART. The coding sequences were analyzed via GENSCAN and FGENESH, and the conserved motifs were predicted using MEME. The functional analysis of the proteins was performed via FancyGene. The sequences were aligned using MUSCLE option in MEGA, and the phylogenetic analyses were conducted via the Bayesian Evolutionary Analysis Sampling Trees (BEAST;http://beast.bio.ed.ac.uk) software. The best-fit model of amino acid replacement was analyzed using ProtTest (https://code.google.com/p/prottest3), and the phylogenetic trees were visualized via FigTree.
Feng et al. (2014a) investigated the family ofIQD genes in soybean using Phytozome. The HMM profiles were determined utilizing Pfam and SMART. The Mw and pI of the proteins were predicted using ExPASy, and the subcellular localizations were determined via TargetP and WoLF PSORT. The sequence alignment and phylogenetic tree construction were performed using ClustalX and MEGA, respectively. The gene structures were identified using GSDS, and the conserved motifs were determined via MEME. The putative calmodulin-binding sites were predicted using the Calmodulin Target Database (http://calcium.uhnres.utoronto.ca/ctdb/ctdb/home.html). The chromosomal locations of the genes were mapped via MapInspect, and the segmental duplications were analyzed using SoyBase. The Smith-Waterman algorithm (http://www.ebi.ac.uk/Tools/psa) was utilized to calculate the local alignment of two protein sequences. The amino acid sequences were aligned via ClustalX, the codon alignments were generated using PAL2NAL, and Ka/Ks values were calculated using CODEML program of Phylogenetic Analysis by Maximum Likelihood (PAML;http://abacus.gene.ucl.ac.uk/software/paml.html) package. The microsynteny analysis was conducted using PGDD. For transcriptome analysis, the RNA-seq data were retrieved from SoyBase.
The soybean glutamate decarboxylase (GAD) gene family was analyzed by Hyun et al. (2014) utilizing the Phylogeny.fr server (http://phylogeny.lirmm.fr/phylo_cgi/index.cgi) and PGDD. The sequence alignment was performed via PAL2NAL, and the Ka/Ks calculation was conducted using PAML. The start codon forGAD genes was obtained from Phytozome, and thecis-acting elements were analyzed using PLACE.
3.2.2.3 Medicago truncatula
The family of auxin/indoleacetic acid (Aux/IAA) genes inMedicago truncatula genome was investigated by Shen et al. (2014) using Phytozome. The conserved domains and the synteny blocks were analyzed via InterProScan and SyMAP, respectively. Multiple sequence alignment was performed via ClustalW, and the phylogenetic tree was constructed using MEGA and visualized via TreeView. The chromosomal locations of the genes were mapped using Circos, and the motif analysis was conducted via MEME.
The distribution of Gretchen Hagen 3 (GH3) gene family inM. truncatula genome was identified by Yang et al. (2015) usingM. truncatula Genome Database (MtGDB;http://www.plantgdb.org/MtGDB). The conserved domains were analyzed via InterProScan. The sequence alignment was performed via ClustalW and visualized using GeneDoc. The phylogenetic tree was generated using MEGA software. The synteny blocks were analyzed using SyMAP, and the conserved motifs were predicted via MEME. Thecis-regulating elements were determined using PLACE.
The LEED..PEED (LP) gene family, unique to theMedicago lineage, was identified by Trujillo et al. (2014) inM. truncatula genome using NCBI, Phytozome, LIS, J. Craig Venter Institute (http://www.jcvi.org/medicago), Kazusa DNA Research Institute (http://www.kazusa.or.jp/lotus), and Dana Farber Cancer Institute—Gene Indices (http://compbio.dfci.harvard.edu/tgi) databases. The small peptides were analyzed using SPADA program (Zhou et al.2013), and the conserved domains were identified via InterProScan. The synteny analysis was conducted using MUMmer (http://mummer.sourceforge.net). The gene homology patterns were analyzed via Genome Evolution Analysis (GEvo;https://genomevolution.org/CoGe/GEvo.pl) and visualized using Multi-Genome Synteny Viewer (mGSV;http://cas-bioinfo.cas.unt.edu/mgsv) tools. The sequences were aligned using ClustalW, the phylogenetic analysis was conducted using MEGA and MrBayes, and the tree was visualized via FigTree. Gene duplications were analyzed and displayed using DILTAG (http://www-lbit.iro.umontreal.ca/DILTAG) program.
3.2.3 Solanaceae
3.2.3.1 Tomato
The family of CLAVATA3/EMBRYO-SURROUNDING REGION-RELATED (CLV3/ESR,CLE) genes in tomato (Solanum lycopersicum) genome was identified by Zhang et al. (2014c) using Phytozome and the tomato resource in SOL Genomics Network (SGN;http://solgenomics.net/organism/Solanum_lycopersicum/genome). The sequence alignment and phylogenetic analysis were conducted using ClustalX and MEGA, respectively. The conserved motifs andcis-acting elements were analyzed via MEME and PLACE, respectively.
Zhang et al. (2014d) identified the family of HD-Zip transcription factors in tomato using SGN. The protein domains were analyzed via Pfam and SMART. Mw and pI of the proteins were determined using ProtParam, and the subcellular localizations were predicted via CELLO (http://cello.life.nctu.edu.tw). The multiple alignment of the sequences was performed using ClustalX, and the phylogenetic tree was constructed via MEGA. The gene structures were identified using GSDS. The conserved motifs were analyzed via MEME. Thecis-acting elements were predicted using PLACE.
Cao and Li (2014) analyzed the family of late embryogenesis abundant (LEA) genes in tomato using Phytozome. The physicochemical properties of the proteins were investigated via ProtParam, and intrinsically disordered proteins were analyzed using IUPred (http://iupred.enzim.hu) server. The subcellular localizations were predicted via CELLO server and PSORT. Multiple sequence alignment and phylogenetic analysis were performed via MUSCLE and MEGA, respectively. The gene duplication/lost analysis was conducted using NOTUNG (http://www.cs.cmu.edu/~durand/Notung) software. The K-Estimator program (https://bioweb.biology.uiowa.edu/labs/comeron/software) was used for Ka/Ks calculation. Thecis-elements were analyzed via PLACE, and the recombination events were predicted using the Recombination Detection Program (RDP;http://web.cbio.uct.ac.za/~darren/rdp.html). Additionally, the site-specific positive selection and purifying selection was analyzed using the Selecton Server (http://selecton.tau.ac.il). For transcriptome-wide expression analysis, the data were retrieved from GEO and processed via Genesis program (http://genome.tugraz.at).
Wu et al. (2014) investigated the mitogen-activated protein kinase (MAPK) kinase (MAPKK) and MAPKKK family in tomato genome using SGN and the Kazusa Full-length Tomato cDNA Database (KafTom;http://www.pgb.kazusa.or.jp/kaftom/blast.html). The protein domains were analyzed using Pfam and SMART. The Mw and pI of the proteins were determined using ExPASy, and the subcellular localizations were predicted via CELLO. The sequences were aligned via ClustalX, and the phylogenetic analysis was performed using MEGA. The Plant Phosphorylation (PlantsP;http://plantsp.genomics.purdue.edu/index.html) was used for motif and domain analysis, and thecis-elements were analyzed using PLACE. The chromosomal locations were identified via SGN, and the synteny analysis was conducted using PGDD.
Chen et al. (2014) identified the mildew resistance locus o (MLO) gene family in tomato genome utilizing SGN and the Plant Genome and Systems Biology (PGSB;http://pgsb.helmholtz-muenchen.de/plant/tomato/searchjsp/index.jsp) database. The conserved domains were identified via Pfam, the sequences were aligned using ClustalX, and the phylogenetic tree was generated using MEGA. The gene structure was determined via GSDS, and the genes were mapped to the chromosomes using MapDraw. The MEME tool was utilized for prediction of conserved motifs.
The basic helix-loop-helix (bHLH) family of transcription factors in tomato genome was investigated by Sun et al. (2015) via SGN. The conserved domains were analyzed using Pfam, SMART, and HMMER. The motif analysis was performed using MEME. The sequences were aligned using MultAlin and Clustal Omega (http://www.clustal.org/omega) and visualized via WebLogo. The phylogenetic analysis was performed using MEGA and FigTree. The gene duplications were analyzed via MUMmer and mapped to the chromosomes using MapChart. Thecis-acting elements were identified using PLACE.
3.2.3.2 Potato
The family of ERF transcription factors in potato (Solanum tuberosum) genome was investigated by Charfeddine et al. (2014) using Phytozome. The conserved domains were searched using Pfam, and the gene structures were corrected using FGENESH. The sequence alignment was conducted via ClustalW and the phylogenetic analysis was performed using MEGA. The genes were mapped to the chromosomes using MapChart, and the Pamilo–Bianchi–Li substitution model in MEGA was utilized for a codon-based Z-test for each block. The pI of the proteins was calculated using ProtParam, and the FoldIndex (http://bioportal.weizmann.ac.il/fldbin/findex) program was used for prediction of protein folding. The subcellular localization of the protein was analyzed via TargetP. For RNA-seq analysis, the data were retrieved from SRA and clustered using MeV. The conserved motifs were predicted via MEME, and the gene structures were analyzed using GSDS.
Charfeddine et al. (2015) investigated theLEA gene family in potato genome using Phytozome. The conserved domains were analyzed via Pfam and FGENESH. The sequences were aligned using ClustalW and the phylogenetic tree was generated via MEGA. The signal peptides, transmembrane regions, and subcellular localization of the proteins were predicted using SignalP, TMHHM, and TargetP, respectively. The gene structures were predicted via GSDS, and the conserved motifs were analyzed using MEME. The chromosomal locations of the genes were mapped using MapChart, and a codon-based Z-test was applied using MEGA. The physicochemical and folding properties of the proteins were predicted via ProtParam and FoldIndex, respectively.
Yang et al. (2014) identified the miR159 family and MYB transcription factors as their targets in potato using mirBase, NCBI, PlantTFDB, the Potato Genome Sequencing Consortium (PGSC;http://potatogenome.net/index.php/Main_Page) database, and the Unified Nucleic Acid Folding and hybridization package (UNAFold;http://www.bioinfo.rpi.edu/applications/mfold). The miRNA targets were predicted via psRNATarget, the sequences were aligned using ClustalX, and the phylogenetic tree was generated using MEGA. The conserved domains of the MYBs were analyzed using Pfam, the physicochemical properties were determined via ExPASy, and the gene structures were identified using Splign (http://www.ncbi.nlm.nih.gov/sutils/splign).
Sharma et al. (2014a) investigated the BEL1-like (BELL) family of transcription factors in potato using PGSC server. The sequences were aligned via ClustalW algorithm in BioEdit, and the phylogenetic tree was generated using MEGA. The ORFs were predicted using FGENESH.
3.2.4 Trees
3.2.4.1 Apple
TheNBS-LRR gene family in apple (Malus x domestica) genome was investigated by Arya et al. (2014b) using Phytozome. The conserved domains were identified via Pfam and HMMER. The sequence alignment was performed via ClustalW, and the coil-coiled motif in proteins was identified using COILS program. The conserved NBS–leucine-rich repeat (LRR) motifs were analyzed via MEME. The genes were mapped to the chromosomes using MapInspect, and the duplication events were analyzed using MCScanX. The phylogenetic analysis was conducted via the Randomized Axelerated Maximum Likelihood tool (RAxML;http://sco.h-its.org/exelixis/web/software/raxml/index.html).
The cystatin gene family in apple genome was identified by Tan et al. (2014) using NCBI andM. x domestica genome data in Genome Database for Rosaceae (GDR;http://www.rosaceae.org/species/malus/malus_x_domestica). The protein domains were analyzed using Pfam and SMART. The MapDraw was utilized to map the genes to the chromosomes. The signal peptides were predicted via SignalP, and the Mw and pI of the proteins were calculated using ExPASY. The multiple alignment of the sequences was performed using CLC Combined Workbench, and the phylogenetic tree was generated using MEGA software. The gene structures were predicted via GSDS, and the cis-acting elements were analyzed utilizing PLACE and PlantCARE databases.
Wei et al. (2014b) investigated the sugar transporter (SUT) gene family in apple genome using GDR and the apple genome database in the Istituto Agrario San Michele all'Adige (IASMA;http://genomics.research.iasma.it). The sequences were aligned via DNAMAN and MUSCLE, and the phylogenetic tree was obtained using MEGA. The subcellular localizations were predicted via TargetP and WoLF PSORT. The chromosomal locations of the genes were mapped using MapDraw.
The distribution of teosinte branched1/cycloidea/proliferating cell factor1 (TCP) family of transcription factors in apple genome was identified by Xu et al. (2014) using the Apple Gene Function and Gene Family DataBase (AppleGFDB;http://www.applegene.org), NCBI, and GDR databases. The conserved domains were analyzed using Pfam and SMART. The Mw and pI of the proteins were calculated via ExPASy. The sequence alignment was conducted using ClustalX and MUSCLE, and the phylogenetic tree was generated via MEGA. The genes were mapped to the chromosomes using MapDraw, and the gene structures were analyzed using GSDS.
Tian et al. (2015) analyzed the MADS-box gene family in apple genome using the IASMA database. The conserved domains were determined using Pfam and CDD. The sequence logo was generated via WebLogo. The protein structure homology models were predicted using SWISS-MODEL (http://swissmodel.expasy.org). The 3D structure models were presented using RasTop (http://www.geneinfinity.org/rastop). The sequence alignment was conducted using CLC Combined Workbench, and the phylogenetic tree was generated via MEGA. The conserved motifs were analyzed using MEME. Chromosomal location of the genes was mapped via MapInspect. The intron–exon structures were identified and visualized via PLAZA and SigmaPlot (http://www.sigmaplot.com), respectively.
Shao et al. (2014) investigated the sucrose non-fermenting-1-related protein kinase 2 (SnRK2) gene family inM. prunifolia (Chinese apple) genome utilizing GDR and IASMA databases. The conserved domains were analyzed using CDD, Pfam, SMART, and PROSITE. The sequences were aligned via ClustalW, MUSCLE, and DNAMAN, and the synteny analysis was performed using PGDD. The gene structure and conserved motif analyses were conducted via GSDS and MEME, respectively. The phylogenetic tree was constructed using MEGA software.
Pessina et al. (2014) identifiedMLO gene family in apple using GDR. The conserved protein motifs were analyzed using HMMER. The membrane spanning helices were identified using InterPro. The sequence alignment was conducted via CLC Sequence Viewer. The orthology and synteny analyses were performed using GBrowse-Syn tool at GDR (http://www.rosaceae.org/gb/gbrowse_syn/peach_apple_strawberry) and Mercator (https://www.biostat.wisc.edu/~cdewey/mercator), respectively.
3.2.4.2 Citrus spp.
The MYB transcription factor family in sweet orange (Citrus sinensis) genome was identified by Hou et al. (2014) using the Orange Genome Annotation Project (OGAP;http://citrus.hzau.edu.cn/orange/index.php) database. The conserved domains were analyzed using Pfam, and the sequences were aligned via ClustalW. The gene structures were analyzed using GSDS. The physicochemical properties and the subcellular localizations of the proteins were analyzed using ProtParam and the Protein Localization Server (PLOC;http://www.genome.jp/SIT/plocdir). The conserved motifs were predicted using MEME, and the phylogenetic tree was constructed via MEGA. The GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations were conducted using Blast2GO. The chromosomal locations of the genes were mapped via MapChart, and the Ka/Ks values were determined using CODEML module of the PAML. The repetitive elements were analyzed using Tandem Repeats Finder (TRF;http://tandem.bu.edu/trf/trf.html) and Inverted Repeats Finder (IRF;http://tandem.bu.edu/irf/irf.download.html). The low copy repeats (LCRs) and transposable elements (TEs) were identified using RepeatMasker (http://www.repeatmasker.org) and the simple sequence repeats (SSRs) were determined via the Simple Sequence Repeat Identification Tool (SSRIT;http://archive.gramene.org/db/markers/ssrtool).
Ito et al. (2014) identified the AP2/ERF superfamily of transcription factors in sweet orange genome using the Citrus Genome Database (http://www.citrusgenomedb.org/species/sinensis). The ORFs were detected using ORF finder, the sequences were aligned, and the phylogenetic tree was generated using ClustalW and MEGA, respectively. The conserved motifs of the proteins were predicted via MEME, and the gene structures were identified using GSDS tool.
Xie et al. (2014) investigated theR2R3-MYB gene family in the genomes of sweet orange and clementine (C. clementina) utilizing Phytozome and PlantTFDB. The conserved domains of the proteins were predicted via PROSITE and SMART. Multiple sequence alignment was conducted using ClustalX and adjusted via BioEdit. The gene structures were analyzed using GSDS, and the phylogenetic analysis was conducted via MEGA.
Lin et al. (2015) analyzed the heat shock transcription factors in Ponkan (C. reticulata Blanco cv. Ponkan) using the Citrus Genome Database. The CAP3 sequence assembly program was used to eliminate redundant sequences and alignment was performed via ClustalX. The phylogenetic tree was generated using TreeView. The conserved motifs were identified using MEME Suite tool.
3.2.4.3 Poplar
The WRKY transcription factor family in poplar (Populus trichocarpa) genome was investigated by Jiang et al. (2014) using Phytozome and PlantTFDB. The sequence alignment was performed using ClustalX, and the phylogenetic tree was constructed via MEGA. Thecis-regulatory elements were analyzed using PlantCARE. For gene expression analysis, the data were retrieved from Phytozome and the sequence reads were mapped to the poplar genome using Short Oligonucleotide Analysis Package (SOAP;http://soap.genomics.org.cn).
Ma et al. (2014a) identified theIQD gene family in poplar genome using Phytozome. The conserved domains were analyzed using Pfam and SMART. The physicochemical properties of the proteins were determined via ExPASy, and the subcellular localization of the proteins was identified using WoLF PSORT. The calmodulin-binding sites were predicted via the Calmodulin Target Database. The gene structures and the conserved motifs were identified using GSDS and MEME, respectively. The genes were mapped to the chromosomes using MapInspect, and the synteny analysis was performed using the Vista Synteny browser (http://pipeline.lbl.gov/cgi-bin/gateway2). The sequences were aligned using ClustalX, and the phylogenetic analysis was conducted via MEGA. The Ka/Ks values were calculated using CODEML program in PAML after multiple alignment via PAL2NAL. For gene expression analysis, the data were obtained from Gene Indices (http://compbio.dfci.harvard.edu/tgi) and GEO. The heat map was visualized via Heatmapper Plus (http://bar.utoronto.ca/ntools/cgi-bin/ntools heatmapper plus.cgi) and Cluster tools.
Li and Lu (2014) analyzed the SQUAMOSA PROMOTER BINDING PROTEIN LIKE (SPL) gene family in poplar using Phytozome. The conserved domains and motifs were identified using Pfam and CDD, and MEME, respectively. Sequence logos were generated via WebLogo. The paralogs were identified via PGDD, and the Ka/Ks calculation was performed using DnaSP. The Mw and pI of the proteins were predicted using ExPASy, and the gene structure was analyzed via GSDS. The sequence alignment was constructed using ClustalW, and the phylogenetic tree was generated via MEGA software.
Chai et al. (2014) identified theR2R3-MYB gene family in poplar utilizing Phytozome. The conserved domains were analyzed using Pfam, the sequences were aligned via ClustalX, and phylogenetic analysis was conducted using MEGA. The gene structures and the conserved motifs were identified using GSDS and MEME, respectively. Detected motifs were searched in databases using MAST. The gene expression was analyzed using the data retrieved from GEO, and normalization was conducted via Genesis.
3.2.4.4 Eucalyptus grandis
The R2R3-MYB transcription factor family inEucalyptus grandis was identified by Soler et al. (2014) using Phytozome. The protein domains were analyzed via InterProScan, the sequences were aligned using MAFFT, and the phylogenetic tree was generated using MEGA. The gene structures were retrieved from Phytozome and represented via FancyGene. The physical positions of the genes on the corresponding chromosomes were mapped using MapChart. The gene duplication was analyzed using a Z-test in MEGA. The conserved motifs were predicted using MEME. For transcriptome analysis, the RNA-seq data were obtained from EucGenIE (http://www.eucgenie.org) and normalized via the EXpression Analyzer and DisplayER (EXPANDER;http://acgt.cs.tau.ac.il/expander).
TheAux/IAA gene family inE. grandis genome was investigated by Yu et al. (2015) utilizing Phytozome. The conserved domains were identified via Pfam and CDD. The gene models were processed using FGENESH and mapped to the chromosomes via MapChart. The physicochemical features of the proteins were predicted using ProtParam, and the conserved motifs were analyzed using MEME. The exon–intron structures were retrieved from Phytozome and visualized using FancyGene. The sequences were aligned via ClustalX and the phylogenetic tree was constructed using MEGA.
Yu et al. (2014) analyzed the family of AUXIN RESPONSE FACTOR (ARF) genes inE. grandis genome using Phytozome. The protein domains were analyzed using CDD and Pfam, and the gene models were processed via FGENESH and mapped to the related chromosomes using MapChart. The sequences were aligned via ClustalX, and the phylogenetic analysis was conducted using MEGA. The gene structures were represented using FancyGene. The small RNA target sites were predicted via psRNATarget. The stem-loop structures of the RNAs were analyzed and visualized via RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) and RNAstructure (http://rna.urmc.rochester.edu/RNAstructure.html) servers, respectively.
Hussey et al. (2014) identified the NAC family of transcription factors inE. grandis genome using Phytozome, PlantTFDB, Eucspresso (http://eucspresso.bi.up.ac.za), and EucGenIE. The protein domains were analyzed using Pfam and HMMER, and the transmembrane helix structures were predicted via TMHMM. The gene models were processed using FGENESH, and the gene structures were predicted via GSDS. The multiple sequence alignment was generated via MUSCLE and trimmed using Gblocks. The phylogenetic tree was constructed and visualized using PhyML and MEGA, respectively. The conserved motifs were predicted via MEME; overrepresented motifs were annotated using Pfam-A and Pfam-B, and schematically represented via DomainDraw (http://domaindraw.imb.uq.edu.au). The genes were mapped to the chromosomes using MapChart. The sequences were aligned via MUSCLE, and the phylogenetic analysis was conducted using MEGA. For transcriptome analysis, the data were retrieved from EucGenIE and analyzed using TopHat and Cufflinks. The expression values were clustered using the QT clustering tool in the MeV.
3.2.5 Other Plants
3.2.5.1 Cotton
The family of WRKY transcription factors in the genomes ofGossypium raimondii andG. arboreum was investigated by Ding et al. (2015) using Phytozome and Cotton Genome Project database (CGP;http://cgp.genomics.org.cn/page/species/index.jsp), respectively. The FGENESH was utilized for gene and protein prediction. The conserved domains were analyzed using HMMER, Pfam, and SMART, and revised using PlantTFDB. Tandem duplications were detected using MCScanX. Ka/Ks calculation was performed via Ka_Ks Calculator. The gene conversion events were identified via GENECONV (http://www.math.wustl.edu/~sawyer/geneconv). The sequences were aligned and the phylogenetic tree was generated using MUSCLE and MEGA, respectively. The gene structures were evaluated via the Plant Intron Exon Comparison and Evolution (PIECE;http://wheat.pw.usda.gov/piece) database. The functional divergence of the subgroups was identified using DIVERGE (http://xungulab.com/software/diverge2/diverge2.html). The selective pressures on codons were analyzed using CODEML package of PAML.
TheSPL gene family inG. hirsutum genome was investigated by Zhang et al. (2015b) using the CGP and CottonGen (http://www.cottongen.org). The genes were mapped to the corresponding chromosomes using MapChart. The phylogenetic analysis was conducted via MEGA. The conserved motifs were predicted using MEME. The miRNAs targeting theGhSPLs were identified using miRBase and psRNATarget.
Zhang et al. (2014b) investigated the MAPK family inG. raimondii genome using Phytozome and NCBI EST database. The conserved domains were analyzed via HMMER, Pfam, InterProScan, SMART, PlantsP, and MOTIF (http://www.genome.jp/tools/motif) tools. The chromosomal locations of the genes were mapped using MapInspect. The subcellular localization of the proteins was detected using CELLO. The sequences were aligned via ClustalX and the phylogenetic tree was obtained using MEGA.
Yurchenko et al. (2014) identified the omega-3 fatty acid desaturase (FAD) gene family inG. hirsutum genome using the databases NCBI, Phytozome, and Cotton Genome Database (CottonDB;http://www.cottondb.org/wwwroot/cdbhome.php). The intron/exon structures were determined using Softberry package (http://www.softberry.com). The sequences were aligned using T-Coffee (http://tcoffee.crg.cat) and cleaned via Gblocks. The phylogenetic tree was generated using PhyML and visualized via FigTree.
Wang et al. (2014c) analyzed the family of heat shock transcription factors inG. hirsutum genome using the NCBI EST database. The conserved domains and motifs were analyzed via SMART and MEME, and visualized using ProSite. The sequences were aligned using DNAMAN and ClustalX. The phylogenetic analyses including gene duplication were performed using MEGA. Physicochemical features of the proteins were analyzed via ExPASy.
The aldehyde dehydrogenase (ALDH) gene superfamily inG. raimondii genome was investigated by He et al. (2014) utilizing NCBI and Phytozome. The protein domains were determined using Pfam. Multiple sequences were aligned and edited using ClustalW and BioEdit, respectively. The phylogenetic analysis was performed via MEGA. The intron–exon structures were identified using FancyGene, and the sequence repeats were determined using RepeatMasker. The synteny analysis was conducted using MCScanX. For gene expression analysis, the microarray and RNA-seq data were retrieved from the GEO and Plant Expression Database (PLEXdb;http://www.plexdb.org), and SRA, respectively. RNA-seq reads were mapped to the gene models via TopHat, and differentially regulated genes at the transcriptional or post-transcriptional level were estimated using Cuffdiff (http://cole-trapnell-lab.github.io/cufflinks/cuffdiff).
3.2.5.2 Grapevine
The MADS-box transcription factors in grapevine (Vitis vinifera) were identified by Wang et al. (2014d) using the GENOSCOPE database (http://www.genoscope.cns.fr/spip/Vitis-vinifera.html). The conserved domains were identified via Pfam, SMART, and Domain Graph software (DOG;http://dog.biocuckoo.org). Gene duplication events were analyzed using PGDD. Multiple sequence alignment was conducted using ClustalW, and the phylogenetic analysis was performed via MEGA. The gene structures were identified using GSDS.
The MAPKKK family in grapevine was analyzed by Wang et al. (2014b) using Grape Genome Database (http://genomes.cribi.unipd.it/grape), Vitis-URGI (http://urgi.versailles.inra.fr/Species/Vitis), and NCBI. The conserved domains were identified via Pfam, HMMER, and SMART. The sequence alignment was performed using ClustalX and GeneDoc, and the phylogenetic tree was generated using Phylogeny.fr. The gene structures were analyzed using GSDS.
The subtilase gene family in grapevine was investigated by Cao et al. (2014). The protease-associated subtilisin-like domain (PA_subtilisin_like domain) was searched using CDD. The primary structural analyses were performed using TargetP and PredoTar (https://urgi.versailles.inra.fr/Tools/Predotar). The sequences were aligned via MUSCLE and ClustalW, and the phylogenetic analyses were performed using PhyML and PHYLIP. The protein substitution model and rate heterogeneity were evaluated using ModelGenerator (http://bioinf.nuim.ie/modelgenerator). The chromosomal locations were identified using GENOSCOPE, and the gene structure information was gathered from NCBI and Phytozome. Ka/Ks analysis was performed using K-Estimator, and the conserved motifs were identified via MEME. The functional divergence was analyzed using DIVERGE. Site-specific selection analyses were conducted via SLAC, REL, and FEL methods in Datamonkey web interface (http://www.datamonkey.org/dataupload.php).
Matus et al. (2014) identified the BURP superfamily in grapevine using Grape Genome Database and GENOSCOPE database. The sequences were aligned via MUSCLE and the phylogenetic trees were obtained using MEGA and FigTree. The conserved motifs were identified using MEME.
4 Conclusion and Future Perspective
Identification of gene families in the genome/transcriptome of a plant gives important clues about the organism’s phylogenetic position, genome mobility, stress tolerance, gene expression profiles, and so on. Hence, the number of articles on this subject is getting higher day by day as more plant genome sequences are released.
Currently, various bioinformatic tools are present to analyze a vast amount of data provided by the next-generation sequencing technologies, and new tools are still produced depending on the need of analysts. In this chapter, mainly the methodologies used in the recent studies analyzing the plant gene families were mentioned. The majority of these bioinformatic tools are accessible on the internet platforms; therefore, their website addresses were provided for ease of use. However, it should be noted that sometimes the links for these addresses might be broken or they might be moved to another address.
As the number of sequenced plant genomes/transcriptomes increased, more studies will be performed on the analysis of gene families. New analysis methods will give rise to the discovery of novel bioinformatic tools. Hereby, our understanding on the roles of these gene families will broaden, especially having an impact on the molecular breeding of stress-tolerant cultivars. Additionally, the knowledge obtained via in silico analyses will be used for the functional gene expression and/or gene silencing studies in plants.
References
Aoyagi LN, Lopes-Caitar VS, de Carvalho MCCG, Darben LM, Polizel-Podanosqui A, Kuwahara MK, Nepomuceno AL, Abdelnoor RV, Marcelino-Guimarães FC (2014) Genomic and transcriptomic characterization of the transcription factor family R2R3-MYB in soybean and its involvement in the resistance responses toPhakopsora pachyrhizi. Plant Sci 229:32–42
Ariyarathna HA, Ul-Haq T, Colmer TD, Francki MG (2014) Characterization of the multigene familyTaHKT 2;1 in bread wheat and the role of gene members in plant Na+ and K+ status. BMC Plant Biol 14:159
Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40:W597–W603
Arya GC, Kumar R, Bisht NC (2014a) Evolution, expression differentiation and interaction specificity of heterotrimeric G-protein subunit gene family in the mesohexaploidBrassica rapa. PLoS One 9(9):e105771
Arya P, Kumar G, Acharya V, Singh AK (2014b) Genome-wide identification and expression analysis of NBS-encoding genes inMalus x domestica and expansion of NBS genes family in Rosaceae. PLoS One 9(9):e107987
Bailey TL, Bodén M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208
Ballester P, Navarrete-Gómez M, Carbonero P, Oñate-Sánchez L, Ferrándiz C (2015) Leaf expansion inArabidopsis is controlled by a TCP-NGA regulatory module likely conserved in distantly related species. Physiol Plant doi. doi:10.1111/ppl.12327
Belamkar V, Weeks NT, Bharti AK, Farmer AD, Graham MA, Cannon SB (2014) Comprehensive characterization and RNA-Seq profiling of the HD-Zip transcription factor family in soybean (Glycine max) during dehydration and salt stress. BMC Genomics 15:950
Bencke-Malato M, Cabreira C, Wiebke-Strohm B, Bücker-Neto L, Mancini E, Osorio MB, Homrich MS, Turchetto-Zolet AC, De Carvalho MC, Stolf R, Weber RL, Westergaard G, Castagnaro AP, Abdelnoor RV, Marcelino-Guimarães FC, Margis-Pinheiro M, Bodanese-Zanettini MH (2014) Genome-wide annotation of the soybean WRKY family and functional characterization of genes involved in response toPhakopsora pachyrhizi infection. BMC Plant Biol 14:236
Bouktila D, Khalfallah Y, Habachi-Houimli Y, Mezghani-Khemakhem M, Makni M, Makni H (2015) Full-genome identification and characterization of NBS-encoding disease resistance genes in wheat. Mol Genet Genomics 290(1):257–271
Cao J, Li X (2014) Identification and phylogenetic analysis of late embryogenesis abundant proteins family in tomato (Solanum lycopersicum). Planta. doi:10.1007/s00425-014-2215-y
Cao J, Han X, Zhang T, Yang Y, Huang J, Hu X (2014) Genome-wide and molecular evolution analysis of the subtilase gene family inVitis vinifera. BMC Genomics 15:1116
Ceasar SA, Hodge A, Baker A, Baldwin SA (2014) Phosphate concentration and arbuscular mycorrhizal colonisation influence the growth, yield and expression of twelvePHT1 family phosphate transporters in foxtail millet (Setaria italica). PLoS One 9(9):e108459
Chai G, Wang Z, Tang X, Yu L, Qi G, Wang D, Yan X, Kong Y, Zhou G (2014)R2R3-MYB gene pairs inPopulus: evolution and contribution to secondary wall formation and flowering time. J Exp Bot 65(15):4255–4269
Chanroj S, Wang G, Venema K, Zhang MW, Delwiche CF, Sze H (2012) Conserved and diversified gene families of monovalent cation/H+ antiporters from algae to flowering plants. Front Plant Sci 3:25
Charfeddine M, Saïdi MN, Charfeddine S, Hammami A, Gargouri Bouzid R (2014) Genome-wide analysis and expression profiling of the ERF transcription factor family in potato (Solanum tuberosum L.). Mol Biotechnol doi:10.1007/s12033-014-9828-z
Charfeddine S, Saïdi MN, Charfeddine M, Gargouri-Bouzid R (2015) Genome-wide identification and expression profiling of the late embryogenesis abundant genes in potato with emphasis on dehydrins. Mol Biol Rep. doi:10.1007/s11033-015-3853-2
Chen Y, Wang Y, Zhang H (2014) Genome-wide analysis of the mildew resistance locus o (MLO) gene family in tomato (Solanum lycopersicum L.). Plant Omics J 7(2):87–93
Chettoor AM, Givan SA, Cole RA, Coker CT, Unger-Wallace E, Vejlupkova Z, Vollbrecht E, Fowler JE, Evans MM (2014) Discovery of novel transcripts and gametophytic functions via RNA-seq analysis of maize gametophytic transcriptomes. Genome Biol 15(7):414
Darzentas N (2010) Circoletto: visualizing sequence similarity with Circos. Bioinformatics 26(20):2620–2621
de Oliveira LF, Christoff AP, de Lima JC, de Ross BC, Sachetto-Martins G, Margis-Pinheiro M, Margis R (2014) The Wall-associated Kinase gene family in rice genomes. Plant Sci 229:181–192
Deokar AA, Kondawar V, Kohli D, Aslam M, Jain PK, Karuppayil SM, Varshney RK, Srinivasan R (2015) Funct Integr Genomics 15(1):27–46
Ding M, Chen J, Jiang Y, Lin L, Cao Y, Wang M, Zhang Y, Rong J, Ye W (2015) Genome-wide investigation and transcriptome analysis of the WRKY gene family inGossypium. Mol Genet Genomics 290(1):151–171
Duan W, Song X, Liu T, Huang Z, Ren J, Hou X, Du J, Li Y (2014) Patterns of evolutionary conservation of ascorbic acid-related genes following whole-genome triplication inBrassica rapa. Genome Biol Evol 7(1):299–313
Duan W, Song X, Liu T, Huang Z, Ren J, Hou X, Li Y (2015) Genome-wide analysis of the MADS-box gene family inBrassica rapa (Chinese cabbage). Mol Genet Genomics 290(1):239–255
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Fan K, Wang M, Miao Y, Ni M, Bibi N, Yuan S, Li F, Wang X (2014) Molecular evolution and expansion analysis of the NAC transcription factor inZea mays. PLoS One 9(11):e111837
Feng L, Chen Z, Ma H, Chen X, Li Y, Wang Y, Xiang Y (2014a) The IQD gene family in soybean: structure, phylogeny, evolution and expression. PLoS One 9(10):e110896
Feng S, Yue R, Tao S, Yang Y, Zhang L, Xu M, Wang H, Shen C (2014b) Genome-wide identification, expression analysis of auxin-responsiveGH3 family genes in maize (Zea mays L.) under abiotic stresses. J Integr Plant Biol doi:10.1111/jipb.12327
Filiz E, Tombuloğlu H (2015) Genome-wide distribution of superoxide dismutase (SOD) gene families inSorghum bicolor. Turk J Biol 39:49–59
Filiz E, Tombuloglu H, Ozyigit II (2013) Genome-wide analysis of IQ67 domain (IQD) gene families inBrachypodium distachyon. Plant Omics J 6(6):425–432
Filiz E, Koç İ, Tombuloğlu H (2014) Genome-wide identification and analysis of growth regulating factor genes inBrachypodium distachyon: in silico approaches. Turk J Biol 38:296–306
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230
Frech C, Chen N (2010) Genome-wide comparative gene family classification. PLoS One 5(10):e13409
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:D1178–D1186
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652
Ha CV, Esfahani MN, Watanabe Y, Tran UT, Sulieman S, Mochida K, Nguyen DV, Tran LS (2014) Genome-wide identification and expression analysis of the CaNAC family members in chickpea during development, dehydration and ABA treatments. PLoS One 9(12):e114107
He D, Lei Z, Xing H, Tang B (2014) Genome-wide identification and analysis of thealdehyde dehydrogenase (ALDH) gene superfamily ofGossypium raimondii. Gene 549(1):123–133
Hofberger JA, Zhou B, Tang H, Jones JD, Schranz ME (2014) A novel approach for multi-domain and multi-gene family identification provides insights into evolutionary dynamics of disease resistance genes in core eudicot plants. BMC Genomics 15:966
Hou XJ, Li SB, Liu SR, Hu CG, Zhang JZ (2014) Genome-wide classification and evolutionary and expression analyses of citrus MYB transcription factor families in sweet orange. PLoS One 9(11):e112375
Hussey SG, Saïdi MN, Hefer CA, Myburg AA, Grima-Pettenati J (2014) Structural, evolutionary and functional analysis of the NAC domain protein family inEucalyptus. New Phytol. doi:10.1111/nph.13139
Hyun TK, Eom SH, Han X, Kim JS (2014) Evolution and expression analysis of the soybean glutamate decarboxylase gene family. J Biosci 39(5):899–907
Ito TM, Polido PB, Rampim MC, Kaschuk G, Souza SG (2014) Genome-wide identification and phylogenetic analysis of the AP2/ERF gene superfamily in sweet orange (Citrus sinensis). Genet Mol Res 13(3):7839–7851
Jain M, Chevala VV, Garg R (2014) Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J Exp Bot 65(20):5945–5958
Jali SS, Rosloski SM, Janakirama P, Steffen JG, Zhurov V, Berleth T, Clark RM, Grbic V (2014) A plant-specificHUA2-LIKE (HULK) gene family inArabidopsis thaliana is essential for development. Plant J 80(2):242–254
Jiang Y, Duan Y, Yin J, Ye S, Zhu J, Zhang F, Lu W, Fan D, Luo K (2014) Genome-wide identification and characterization of thePopulus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J Exp Bot 65(22):6629–6644
Kayum MA, Jung HJ, Park JI, Ahmed NU, Saha G, Yang TJ, Nou IS (2015) Identification and expression analysis ofWRKY family genes under biotic and abiotic stresses inBrassica rapa. Mol Genet Genomics 290(1):79–95
Kenzior A, Folk WR (2015)Arabidopsis thaliana MSI4/FVE associates with members of a novel family of plant specific PWWP/RRM domain proteins. Plant Mol Biol doi. doi:10.1007/s11103-014-0280-z
Kim Y-W, Jung H-J, Park J-I, Hur Y, Nou I-S (2015) Response of NBS encoding resistance genes linked to both heat and fungal stress inBrassica oleracea. Plant Physiol Biochem 86:130–136
Lata C, Mishra AK, Muthamilarasan M, Bonthala VS, Khan Y, Prasad M (2014) Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS One 9(11):e113092
Letunic I, Doerks T, Bork P (2015) SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43:D257–D260
Li C, Lu S (2014) Molecular characterization of the SPL gene family inPopulus trichocarpa. BMC Plant Biol 14:131
Li PS, Yu TF, He GH, Chen M, Zhou YB, Chai SC, Xu ZS, Ma YZ (2014a) Genome-wide analysis of the Hsf family in soybean and functional identification ofGmHsf-34 involvement in drought and heat stresses. BMC Genomics 15:1009
Li Q, Zhang N, Zhang L, Ma H (2014b) Differential evolution of members of the rhomboid gene family with conservative and divergent patterns. New Phytol. doi:10.1111/nph.13174
Lin Q, Jiang Q, Lin J, Wang D, Li S, Liu C, Sun C, Chen K (2015) Heat shock transcription factors expression during fruit development and under hot air stress in Ponkan (Citrus reticulata Blanco cv. Ponkan) fruit. Gene pii: S0378-1119(15)00040-2
Liu RH, Meng JL (2003) MapDraw: a microsoft excel macro for drawing genetic linkage maps based on given genetic linkage data. Yi Chuan 25(3):317–321
Ma H, Feng L, Chen Z, Chen X, Zhao H, Xiang Y (2014a) Genome-wide identification and expression analysis of theIQD gene family inPopulus trichocarpa. Plant Sci 229:96–110
Ma J, Wang F, Li M-Y, Jiang Q, Tan G-F, Xiong A-S (2014b) Genome wide analysis of the NAC transcription factor family in Chinese cabbage to elucidate responses to temperature stress. Scientia Horticulturae 165:82–90
Ma T, Ma H, Zhao H, Qi H, Zhao J (2014c) Identification, characterization, and transcription analysis of xylogen-like arabinogalactan proteins in rice (Oryza sativa L.). BMC Plant Biol 14:299
Mace E, Tai S, Innes D, Godwin I, Hu W, Campbell B, Gilding E, Cruickshank A, Prentis P, Wang J, Jordan D (2014) The plasticity of NBS resistance genes in sorghum is driven by multiple evolutionary processes. BMC Plant Biol 14:253
Mainali HR, Chapman P, Dhaubhadel S (2014) Genome-wide analysis ofCyclophilin gene family in soybean (Glycine max). BMC Plant Biol 14(1):282
Martinez M (2013) From plant genomes to protein families: computational tools. Comput Struct Biotechnol J 8:e201307001
Matus JT, Aquea F, Espinoza C, Vega A, Cavallini E, Dal Santo S, Cañón P, Rodríguez-Hoces de la Guardia A, Serrano J, Tornielli GB, Arce-Johnson P (2014) Inspection of the grapevine BURP superfamily highlights an expansion ofRD22 genes with distinctive expression features in berry development and ABA-mediated stress responses. PLoS One 9(10):e110372
Mochida K, Shinozaki K (2011) Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant Cell Physiol 52(12):2017–2038
Muthamilarasan M, Khandelwal R, Yadav CB, Bonthala VS, Khan Y, Prasad M (2014) Identification and molecular characterization of MYB transcription factor superfamily in C4 model plant foxtail millet (Setaria italica L.). PLoS One 9(10):e109920
Nawaz Z, Kakar KU, Saand MA, Shu QY (2014) Cyclic nucleotide-gated ion channel gene family in rice, identification, characterization and experimental analysis of expression response to plant hormones, biotic and abiotic stresses. BMC Genomics 15:853
Nguyen QN, Lee YS, Cho LH, Jeong HJ, An G, Jung KH Genome-wide identification and analysis ofCatharanthus roseus RLK1-like kinases in rice. Planta doi:10.1007/s00425-014-2203-2
Okay S, Derelli E, Unver T (2014) Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Mol Genet Genomics 289(5):765–781
Pan X, Peng FY, Weselake R (2015) Genome-wide analysis ofPHOSPHOLIPID:DIACYLGLYCEROL ACYLTRANSFERASE genes in plants reveals the eudicot-widePDAT gene expansion and altered selective pressures acting on the core eudicotPDAT paralogs. Plant Physiol pii: pp.114.253658
Panahi B, Abbaszadeh B, Taghizadeghan M, Ebrahimie E (2014) Genome-wide survey of alternative splicing inSorghum bicolor. Physiol Mol Biol Plants 20(3):323–329
Pandey B, Kaur A, Gupta OP, Sharma I, Sharma P (2014) Identification ofHSP20 gene family in wheat and barley and their differential expression profiling under heat stress. Appl Biochem Biotechnol. doi:10.1007/s12010-014-1420-2
Pessina S, Pavan S, Catalano D, Gallotta A, Visser RG, Bai Y, Malnoy M, Schouten HJ (2014) Characterization of theMLO gene family in Rosaceae and gene expression analysis inMalus domestica. BMC Genomics 15:618
Pourabed E, Ghane Golmohamadi F, Soleymani Monfared P, Razavi SM, Shobbar ZS (2015) Basic leucine zipper family in barley: genome-wide characterization of members and expression analysis. Mol Biotechnol 57(1):12–26
Rawal HC, Singh NK, Sharma TR (2013) Conservation, divergence, and genome-wide distribution ofPAL andPOX A gene families in plants. Int J Genomics 2013:678969
Saha J, Sengupta A, Gupta K, Gupta B (2015) Molecular phylogenetic study and expression analysis of ATP-binding cassette transporter gene family inOryza sativa in response to salt stress. Comput Biol Chem 54:18–32
Saito F, Suyama A, Oka T, Yoko-O T, Matsuoka K, Jigami Y, Shimma YI (2014) Identification of novel peptidyl serine α-galactosyltransferase gene family in plants. J Biol Chem 289:20405–20420
Shao Y, Qin Y, Zou Y, Ma F (2014) Genome-wide identification and expression profiling of the SnRK2 gene family inMalus prunifolia. Gene 552(1):87–97
Sharma R, Suresh CG (2015) Genome-wide identification and structure-function studies of proteases and protease inhibitors inCicer arietinum (chickpea). Comput Biol Med 56:67–81
Sharma P, Lin T, Grandellis C, Yu M, Hannapel DJ (2014a) The BEL1-like family of transcription factors in potato. J Exp Bot 65(2):709–723
Sharma R, Rawat V, Suresh CG (2014b) Genome-wide identification and tissue-specific expression analysis of UDP-glycosyltransferases genes confirm their abundance inCicer arietinum (chickpea) genome. PLoS One 9(10):e109715
Shen C, Yue R, Yang Y, Zhang L, Sun T, Xu L, Tie S, Wang H (2014) Genome-wide identification and expression profiling analysis of theAux/IAA gene family inMedicago truncatula during the early phase ofSinorhizobium meliloti infection. PLoS One 9(9):e107495
Shiriga K, Sharma R, Kumar K, Yadav SK, Hossain F, Thirunavukkarasu N (2014) Genome-wide identification and expression pattern of drought-responsive members of the NAC family in maize. Meta Gene 2:407–417
Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Siriwardana CL, Kumimoto RW, Jones DS, Holt BF III (2014) Gene family analysis of theArabidopsis NF-YA transcription factors reveals opposing abscisic acid responses during seed germination. Plant Mol Biol Rep 32(5):971–986
Soler M, Camargo EL, Carocha V, Cassan-Wang H, San Clemente H, Savelli B, Hefer CA, Paiva JA, Myburg AA, Grima-Pettenati J (2014) TheEucalyptus grandis R2R3-MYB transcription factor family: evidence for woody growth-related evolution and function. New Phytol. doi:10.1111/nph.13039
Sun H, Fan H-J, Ling H-Q (2015) Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics 16:9
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tan Y, Wang S, Liang D, Li M, Ma F (2014) Genome-wide identification and expression profiling of the cystatin gene family in apple (Malus × domestica Borkh.). Plant Physiol Biochem 79:88–97
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plantArabidopsis thaliana. Nature 408(6814):796–815
Thomas M, Pingault L, Poulet A, Duarte J, Throude M, Faure S, Pichon JP, Paux E, Probst AV, Tatout C (2014) Evolutionary history of Methyltransferase 1 genes in hexaploid wheat. BMC Genomics 15:922
Tian Y, Dong Q, Ji Z, Chi F, Cong P, Zhou Z (2015) Genome-wide identification and analysis of the MADS-box gene family in apple. Gene 555(2):277–290
Trujillo DI, Silverstein KA, Young ND (2014) Genomic characterization of the LEED.PEEDs, a gene family unique to the Medicago lineage. G3 (Bethesda) 4(10):2003–2012
Wang F, Qiu N, Ding Q, Li J, Zhang Y, Li H, Gao J (2014a) Genome-wide identification and analysis of the growth-regulating factor family in Chinese cabbage (Brassica rapa L. ssp.pekinensis). BMC Genomics 15:807
Wang G, Lovato A, Polverari A, Wang M, Liang YH, Ma YC, Cheng ZM (2014b) Genome-wide identification and analysis of mitogen activated protein kinase kinase kinase gene family in grapevine (Vitis vinifera). BMC Plant Biol 14:219
Wang J, Sun N, Deng T, Zhang L, Zuo K (2014c) Genome-wide cloning, identification, classification and functional analysis of cotton heat shock transcription factors in cotton (Gossypium hirsutum). BMC Genomics 15:961
Wang L, Yin X, Cheng C, Wang H, Guo R, Xu X, Zhao J, Zheng Y, Wang X (2014d) Evolutionary and expression analysis of a MADS-box gene superfamily involved in ovule development of seeded and seedless grapevines. Mol Genet Genomics. doi:10.1007/s00438-014-0961-y
Wang L, Yu C, Chen C, He C, Zhu Y, Huang W (2014e) Identification of rice Di19 family revealsOsDi19-4 involved in drought resistance. Plant Cell Rep 33(12):2047–2062
Wang X, Zhang H, Gao Y, Sun G, Zhang W, Qiu L (2014f) A comprehensive analysis of the Cupin gene family in soybean (Glycine max). PLoS One 9(10):e110092
Wang J, Chu S, Zhu Y, Cheng H, Yu D (2015a) Positive selection drives neofunctionalization of the UbiA prenyltransferase gene family. Plant Mol Biol. doi:10.1007/s11103-015-0285-2
Wang Z, Tang J, Hu R, Wu P, Hou XL, Song XM, Xiong AS (2015b) Genome-wide analysis of the R2R3-MYB transcription factor genes in Chinese cabbage (Brassica rapa ssp.pekinensis) reveals their stress and hormone responsive patterns. BMC Genomics 16(1):17
Wei K, Pan S (2014) Maize protein phosphatase gene family: identification and molecular characterization. BMC Genomics 15:773
Wei B, Zhang RZ, Guo JJ, Liu DM, Li AL, Fan RC, Mao L, Zhang XQ (2014a) Genome-wide analysis of the MADS-box gene family inBrachypodium distachyon. PLoS One 9(1):e84781
Wei X, Liu F, Chen C, Ma F, Li M (2014b) TheMalus domestica sugar transporter gene family: identifications based on genome and expression profiling related to the accumulation of fruit sugars. Front Plant Sci 5:569
Wen F, Zhu H, Li P, Jiang M, Mao W, Ong C, Chu Z (2014) Genome-wide evolutionary characterization and expression analyses of WRKY family genes inBrachypodium distachyon. DNA Res 21(3):327–339
Wu J, Wang J, Pan C, Guan X, Wang Y, Liu S, He Y, Chen J, Chen L, Lu G (2014) Genome-wide identification of MAPKK and MAPKKK gene families in tomato and transcriptional profiling analysis during development and stress response. PLoS One 9(7):e103032
Xie R, Li Y, He S, Zheng Y, Yi S, Lv Q, Deng L (2014) Genome-wide analysis of citrusR2R3MYB genes and their spatiotemporal expression under stresses and hormone treatments. PLoS One 9(12):e113971
Xu R, Sun P, Jia F, Lu L, Li Y, Zhang S, Huang J (2014) Genomewide analysis ofTCP transcription factor gene family inMalus domestica. J Genet 93(3):733–746
Yang J, Zhang N, Mi X, Wu L, Ma R, Zhu X, Yao L, Jin X, Si H, Wang D (2014) Identification of miR159s and their target genes and expression analysis under drought stress in potato. Comput Biol Chem 53PB:204–213
Yang Y, Yue R, Sun T, Zhang L, Chen W, Zeng H, Wang H, Shen C (2015) Genome-wide identification, expression analysis ofGH3 family genes inMedicago truncatula under stress-related hormones andSinorhizobium meliloti infection. Appl Microbiol Biotechnol 99(2):841–854
Yao QY, Xia EH, Liu FH, Gao LZ (2015) Genome-wide identification and comparative expression analysis reveal a rapid expansion and functional divergence of duplicated genes in the WRKY gene family of cabbage. Brassica oleracea var. capitata. Gene 557(1):35–42
Yu H, Soler M, Mila I, San Clemente H, Savelli B, Dunand C, Paiva JA, Myburg AA, Bouzayen M, Grima-Pettenati J, Cassan-Wang H (2014) Genome-wide characterization and expression profiling of theAUXIN RESPONSE FACTOR (ARF) gene family inEucalyptus grandis. PLoS One 9(9):e108906
Yu H, Soler M, San Clemente H, Mila I, Paiva JA, Myburg AA, Bouzayen M, Grima-Pettenati J, Cassan-Wang H (2015) Comprehensive genome-wide analysis of theAux/IAA gene family inEucalyptus: evidence for the role ofEgrIAA4 in wood formation. Plant Cell Physiol pii:pcu215
Yurchenko OP, Park S, Ilut DC, Inmon JJ, Millhollon JC, Liechty Z, Page JT, Jenks MA, Chapman KD, Udall JA, Gore MA, Dyer JM (2014) Genome-wide analysis of the omega-3 fatty acid desaturase gene family inGossypium. BMC Plant Biol 14:312
Zhang W, Yan H, Chen W, Liu J, Jiang C, Jiang H, Zhu S, Cheng B (2014a) Genome-wide identification and characterization of maize expansin genes expressed in endosperm. Mol Genet Genomics 289(6):1061–1074
Zhang X, Wang L, Xu X, Cai C, Guo W (2014b) Genome-wide identification of mitogen-activated protein kinase gene family inGossypium raimondii and the function of their corresponding orthologs in tetraploid cultivated cotton. BMC Plant Biol 14(1):345
Zhang Y, Yang S, Song Y, Wang J (2014c) Genome-wide characterization, expression and functional analysis ofCLV3/ESR gene family in tomato. BMC Genomics 15:827
Zhang Z, Chen X, Guan X, Liu Y, Chen H, Wang T, Mouekouba LD, Li J, Wang A (2014d) A genome-wide survey of homeodomain-leucine zipper genes and analysis of cold-responsive HD-Zip I members’ expression in tomato. Biosci Biotechnol Biochem 78(8):1337–1349
Zhang L, Li Q, Dong H, He Q, Liang L, Tan C, Han Z, Yao W, Li G, Zhao H, Xie W, Xing Y (2015a) Three CCT domain-containing genes were identified to regulate heading date by candidate gene-based association mapping and transformation in rice. Sci Rep 5:7663
Zhang X, Dou L, Pang C, Song M, Wei H, Fan S, Wang C, Yu S (2015b) Genomic organization, differential expression, and functional analysis of theSPL gene family inGossypium hirsutum. Mol Genet Genomics 290(1):115–126
Zhou P, Silverstein KAT, Gao L, Walton JD, Nallu S, Guhlin J, Young ND (2013) Detecting small plant peptides using SPADA (Small Peptide Alignment Discovery Application). BMC Bioinformatics 14:335
Zhu C, Luo N, He M, Chen G, Zhu J, Yin G, Li X, Hu Y, Li J, Yan Y (2014a) Molecular characterization and expression profiling of the protein disulfide isomerase gene family inBrachypodium distachyon L. PLoS One 9(4):e94704
Zhu YB, Xie XQ, Li ZY, Bai H, Dong L, Dong ZP, Dong JG (2014b) Bioinformatic analysis of the nucleotide binding site-encoding disease-resistance genes in foxtail millet (Setaria italica (L.) Beauv.). Genet Mol Res 13(3):6602–6609
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Okay, S. (2016). Identification of Gene Families Using Genomics and/or Transcriptomics Data. In: Hakeem, K., Tombuloğlu, H., Tombuloğlu, G. (eds) Plant Omics: Trends and Applications. Springer, Cham. https://doi.org/10.1007/978-3-319-31703-8_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-31703-8_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31701-4
Online ISBN: 978-3-319-31703-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)