
Abstract
Host resistance is the most economical, effective and ecologically sustainable method of controlling diseases in crop plants. In bread wheat, despite the high number of resistance loci that have been cataloged to date, only few have been cloned, underlying the need for genomics-guided investigations capable of providing a prompt and acute knowledge on the identity of effective resistance genes that can be used in breeding programs. Proteins with a nucleotide-binding site (NBS) encoded by the major plant disease resistance (R) genes play an important role in the responses of plants to various pathogens. In this study, a comprehensive analysis of NBS-encoding genes within the whole wheat genome was performed, and the genome scale characterization of this gene family was established. From the recently published wheat genome sequence, we used a data mining and automatic prediction pipeline to identify 580 complete ORF candidate NBS-encoding genes and 1,099 partial-ORF ones. Among complete gene models, 464 were longer than 200 aa, among them 436 had less than 70 % of sequence identity to each other. This gene models set was deeply characterized. (1) First, we have analyzed domain architecture and identified, in addition to typical domain combinations, the presence of particular domains like signal peptides, zinc fingers, kinases, heavy-metal-associated and WRKY DNA-binding domains. (2) Functional and expression annotation via homology searches in protein and transcript databases, based on sufficient criteria, enabled identifying similar proteins for 60 % of the studied gene models and expression evidence for 13 % of them. (3) Shared orthologous groups were defined using NBS-domain proteins of rice and Brachypodium distachyon. (4) Finally, alignment of the 436 NBS-containing gene models to the full set of scaffolds from the IWGSC’s wheat chromosome survey sequence enabled high-stringence anchoring to chromosome arms. The distribution of the R genes was found balanced on the three wheat sub-genomes. In contrast, at chromosome scale, 50 % of members of this gene family were localized on 6 of the 21 wheat chromosomes and ~22 % of them were localized on homeologous group 7. The results of this study provide a detailed analysis of the largest family of plant disease resistance genes in allohexaploid wheat. Some structural traits reported had not been previously identified and the genome-derived data were confronted with those stored in databases outlining the functional specialization of members of this family. The large reservoir of NBS-type genes presented and discussed will, firstly, form an important framework for marker-assisted improvement of resistance in wheat, and, secondly, open up new perspectives for a better understanding of the evolution dynamics of this gene family in grass species and in polyploid systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bread wheat (Triticum aestivum L.) is the most economically important and widely grown crop in the world, which has been farmed and consumed as staple food for thousands of years. This hexaploid (2n = 6x = 42; AABBDD) species originated from hybridization between cultivated tetraploid (2n = 4x = 28; AABB) emmer wheat (Triticum dicoccoides Körn.) and diploid (2n = 14; DD) goat grass (Aegilops tauschii L.) approximately 10,000 years ago. Susceptibility of wheat to multiple pathogens, including fungi, bacteria, viruses and insects, hinders its production increase and quality improvement. An efficient, economical, and environmentally friendly approach for disease opposition is to use resistant cultivars. A prerequisite of this approach is a comprehensive understanding of the genetics, genomics and evolution of the genes controlling disease resistance.
Up to date, thousands of resistance traits have been identified from different plant species since the early years of the twentieth century (Ellis and Jones 2003), and more than 100 genes controlling disease resistance have been cloned according to Plant Resistance Gene Database (PRGdb: http://www.prgdb.org). Most of these cloned resistance genes (R genes) encode nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domains and are thus called NBS–LRR proteins. The NBS domain is homologous to the nucleotide-binding site of ATPases, GTPases and various other nucleotide binding proteins (Traut 1994) and includes several highly conserved and strictly ordered motifs, such as the P-loop, kinase-2 and GLPL motifs (Tan and Wu 2012). The C-terminal LRR domain consists of multiple copies of an imperfect leucine-rich-repeat sequence (Kajava 1998; Bella et al. 2008) and is devoted essentially to protein–protein interactions (Jones and Jones 1997). Two subfamilies of NBS–LRR proteins can be distinguished: TIR–NBS–LRRs (TNLs) containing an N-terminal TIR domain, originally identified as an intracellular part of the Toll receptor in Drosophila and human interleukin 1 receptor; and non-TIR–NBS–LRRs (non-TNLs) containing other domains, the most frequent of which is the coiled-coil (CC) domain that functions in oligomerization processes like the TIR domain (Oakley and Hollenbeck 2001). It should be emphasized that these subfamilies differ not only in terms of the presence or absence of the TIR domain but also in terms of the sequence of the NBS domain (Meyers et al. 1999).
While in dicotyledonous plants, both TNLs and non-TNLs may occur, all so far detected NBS–LRR proteins in monocots are non-TNLs (Marone et al. 2013a). Analyses of different plant genomes (Arabidopsis thaliana, Populus, Oryza sativa) have shown that the non-TNL proteins are characterized by a high structural diversification (Miller et al. 2008) that can be illustrated as follows: (1) first, based on CC, NBS and LRR domains, proteins with two NBS domains (CC–NBS–NBS–LRR), as well as proteins lacking the LRR domain (CC–NBS) and others exclusively composed of a single NBS domain (NBS) have been identified in various plants (Zhou et al. 2004; Krattinger et al. 2011; Sanseverino and Ercolano 2012); (2) secondly, apart from NBS, LRR and CC, the presence of several additional domains has been recently discovered, such as WRKY transcription factors (R gene repressors) which are deactivated by barley MLA proteins following recognition of avirulence (Avr) proteins of Blumeria graminis (Liu and Coaker 2008). These recent findings make the classification of NBS-type R proteins still far from being conclusive and raise new questions concerning the possible involvement of new classes in the resistance process.
With access to the full-genome sequence of many monocot and dicot species, NBS-encoding R genes have been predicted and annotated. For example, it has been the case of Arabidopsis (Meyers et al. 2003), rice (Wang et al. 2004; Monosi et al. 2004), Medicago truncatula (Ameline-Torregrosa et al. 2008) and sorghum (Paterson et al. 2009). The number of NBS-encoding genes that have been identified in several plant species through genome-wide analyses, as extensively reviewed by Marone et al. (2013a), ranged from about 50 in Carica papaya (Porter et al. 2009) and Cucumis sativus (Wan et al. 2013) to 653 in O. sativa L. spp. indica (Shang et al. 2009).
For several years, the very large size and polyploidy complexity of the bread wheat genome have been substantial barriers to genome analysis. However, recently, Brenchley et al. (2012) have reported the sequencing of this large 17 Gigabase-pair hexaploid genome using 454 pyrosequencing technology. Purified nuclear DNA was sequenced to generate 220 million reads (85 Gb of sequence), corresponding to approximately fivefold coverage of genome size. The T. aestivum whole genome shotgun (WGS) sequencing project is accessible for exploration from the NCBI web site (http://www.ncbi.nlm.nih.gov/bioproject/PRJEB217) and consists of two assemblies: (1) The OA genic sub-assembly dataset (EMBL bank accession range CALO01000001–CALO01945079) that consists in the mapping and assembly of wheat raw reads on Orthologous Group (OG) representative grass genes from Brachypodium distachyon, Sorghum bicolor, O. sativa and Hordeum vulgare; and (2): The Low Copy-number Genome assembly (LCG) (EMBL bank accession range CALP010000001–CALP015321847) that was constructed by filtering-out repetitive sequences and assembling the remaining low copy sequences de novo. Both assemblies were submitted to GenBank in January 2013 and are at contiguous sequences (contigs) level. Although fragmentary and lacking functional annotation and sub-genome/chromosome assignments, these assemblies form a powerful framework for identifying genes, accelerating further genome sequencing and facilitating genome scale analyses (Brenchley et al. 2012).
The development of a wheat reference genome sequence and complete gene annotation is currently being a major goal of the International Wheat Genome Sequencing Project (http://www.wheatgenome.org/). Although the achievement of this goal at the whole genome scale is currently making progress, it takes considerable time, because of the huge and highly repetitive character of the wheat genome. In anticipation to this milestone, the annotation of single protein families or sets of related protein families making up a single biological subsystem is a challenging goal that could be reached at short-term and could lead to improvements when compared to the genome ‘gene-by-gene’ annotation.
In this paper, in silico search in the wheat genome draft was conducted to identify, nearly exhaustively, members of the NBS-encoding disease resistance gene family. A characterization package of these candidate genes was applied that included the annotation of their structural domains, homology-based classification, expression evidence, analysis of orthology with two model species and genomic localization.
Materials and methods
Retrieval of candidate wheat NBS-encoding loci
The LCG assembly of T. aestivum (5,321,847 contigs; 3,800,325,216 bp) was downloaded in 14 fragment Fasta-format files, from the National Center for Biotechnology Information NCBI database (http://www.ncbi.nlm.nih.gov/Traces/wgs/?val=CALP01). These files were translated in 6 reading frames, using transeq algorithm from the software package of the European Molecular Biology Open Software Suite (EMBOSS version 6.5.0.0, ftp://emboss.open-bio.org/pub/EMBOSS/) and the resulting files were used as a local database in the subsequent search. Aiming to track the NBS domain, we used a set of 422 non-redundant and full length NBS domains belonging to different Triticeae species. This sequence set was identified through PsiBlast search (Altschul et al. 1997) in NCBI GenBank protein database (nr), with the following parameters: e-value cutoff = 10−7, species: taxid: 147389, matrix: BLOSUM-62, initial matrix seed: gb|ACO53397, number of iterations: 6. Following manual cleaning, these 422 Triticeae NBS sequences were aligned with Muscle (http://www.ebi.ac.uk/Tools/msa/muscle/) (Fig. S1). A Hidden Markov Model (HMM) profile was produced from alignment, using hmmbuild tool (HMMER 3.0, http://hmmer.janelia.org/) with default settings. This original HMM profile was applied to the 6-frame translation of wheat contigs, using hmmsearch tool (HMMER 3.0, http://hmmer.janelia.org/) and all nucleotide contigs corresponding to NBS domains were retrieved using seqret tool (EMBOSS version 6.5.0.0, ftp://emboss.open-bio.org/pub/EMBOSS/) and merged into a unique multiFasta file using DNA Baser sequence assembler v. 3.5.4.2 (http://www.dnabaser.com/). This file was checked for any redundant contigs, using cd-hit-est (http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi?cmd=cd-hit-est) and manual removal. Finally, the obtained non-redundant contigs were splitted into individual Fasta files using seqretsplit command of EMBOSS.
Identification of candidate wheat NBS-encoding R genes
All contigs identified were submitted to ab initio gene prediction, using the web version of FGENESH (http://www.softberry.com) (Salamov and Solovyev 2000), with parameters for monocot plants. For gene models displaying a complete ORF structure extending from ATG to Stop codon, and those having a truncated ORF, amino acid sequences were placed in two separate multifasta files. Within both files, gene models were labeled by adding the suffix “−1” to the name of the locus (contig) where they were predicted. In case of multiple predictions in the same contig, suffixes ‘−1’, ‘−2’, ‘−3’, etc… were used. Both multifasta files (each containing a batch of gene models) were subsequently submitted to Pfam v. 27.0 database (http://www.sanger.ac.uk/Software/Pfam/) to select, within each file, only gene models showing at least one significant NBS domain (default e-value = 1). The number of sequences within each file was thus reduced and the sizes of the retained gene models were subsequently determined with sizeseq tool (EMBOSS version 6.5.0.0, ftp://emboss.open-bio.org/pub/EMBOSS/). For complete gene models, only those hypothetical proteins longer than 200 amino acids were retained, based on an average size of the core NBS domain of ~170 aa in Pfam database (http://pfam.sanger.ac.uk/family/NB-ARC#tabview=tab4). Following size-based selection, complete gene models were submitted to cd-hit-est protein clustering algorithm (http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi?cmd=cd-hit-est) calibrated for 70 % similarity cutoff, in order to keep one representative from sequences that may be encoded from allelic or duplicated paralogous genes. The resulting dataset made of complete gene models sized more than 200 aa and non-similar at threshold 70 % was used for the subsequent characterization pipeline.
Structural characterization
For each gene model, size (aa), number of exons, nucleotide span and strand (±) were determined directly from FGENESH output. For further characterization including the NBS domain associations with other domains such as the CC motif (in the N-terminal region) or the LRR one (on the carboxy-terminal region), we used Geneious 6.1.5 (http://www.geneious.com) with added plugins (coiled-coil and transmembrane prediction tools), in order to (1) submit in batch all gene models for protein signature analysis via internet connection to InterProScan with its 13 integrated databases (Gene3D, HAMAP, PANTHER, Pfam, PIR super family, PRINTS, PROFILE, PROSITE, SignalP, SMART, Pro-Dom, SUPERFAMILY, and TIGRFAMs); (2) predict coiled-coils (CC); (3) predict transmembrane domains; and (4) predict cellular topology (protein cytoplasmic/extracellular regions). All structural analyses performed with Geneious were saved in graphical format (jpg).
Multiplex protein homology and expression evidence
Evidence of protein homology was inferred from BlastP (Altschul et al. 1997) against GenBank Non-Redundant protein sequences database (nr) without taxonomic restriction. Evidence of expression was inferred from TBlastN (Altschul et al. 1997) against GanBank Expression Sequence Tags database (dbEST) and Transcriptome Shotgun Assembly Sequence Database (TSA), with restriction to T. aestivum (taxid:4565). For each operation, complete gene models sized more than 200 aa and non-similar at threshold 70 % were submitted in batch to GenBank Blast server (http://www.ncbi.nlm.nih.gov/genbank/). Based on results, a method of classification was developed (Fig. S2) that is similar to “Guidelines for Annotating Wheat Genomic Sequences” (Release 1) published on June 2006 by the “Annotation Working Group” of the “International Wheat Genome Sequencing Consortium IWGSC” (http://wheat.pw.usda.gov/ITMI/Repeats/gene_annotation.pdf).
Proteomic comparison
The predicted whole proteome and domain annotations of O. sativa (version 7.0) and B. distachyon (version 1.2) were retrieved from the ftp server of the Rice Genome Annotation Project (RGAP) (http://rice.plantbiology.msu.edu/) and the ftp server of the Munich Information Center for Protein Sequences (MIPS) (http://ftpmips.helmhottz-muenchen.de/plants/brachypodium/v1.2), respectively. Using domain annotations of these two proteomes, all predicted proteins containing an NBS domain were retrieved from the proteome using seqret (EMBOSS 6.5.0.0) and placed in separate files. These NBS domain-predicted proteins of O. sativa and B. distachyon were used for comparison with T. aestivum complete gene models sized more than 200 aa and non-similar at threshold 70 % that were determined in the present study. NBS proteins from all three species were assigned to orthologous clusters using OrthoMCLdb (http://orthomcl.org/), and a three-way Venn diagram was constructed that shows the distribution of shared orthologous groups among species.
Localization in T. aestivum genome
While no complete physical map has yet been developed for T. aestivum, chromosome and chromosome arm-specific scaffolds are available at the IWGSC Survey Sequence repository (http://wheat-urgi.versailles.inra.fr/Seq-Repository/) with access to blasting and download. Thus, it was interesting to determine the genomic distribution of T. aestivum NBS-encoding gene family members, at chromosomes, sub-genomes and arms levels. To this aim, DNA sequences of gene models identified in our study were individually used to perform BlastN against the full set of scaffolds from the IWGSC’s wheat chromosome survey sequence (CSS), including repeats. Access to IWGSC wheat genome data was done through The Genome Analysis Center (TGAC) bread wheat Blast server (http://tgac-browser.tgac.ac.uk/iwgsc_css/blast.jsp), providing the advantage of simultaneous queuing of 3 Blast jobs.
Results
Identification of NBS genes
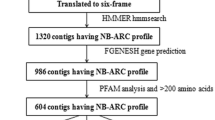
Screening the wheat genome translations with the Triticeae-specific HMM profile of the NBS domain and the subsequent removal of redundancies have resulted in 3,010 contigs. Among these, 1,032 did not produce any gene prediction, while 1,978 produced at least one gene model per contig, leading to a total of 2,081 gene models (Table S1). These gene models were divided into two distinct groups: (1) a first group including 883 complete ORF models and (2) a second group including 1,198 models that show a truncated ORF (Table 1). The number of sequences within both groups was, later, reduced to 580 and 1,099, respectively, following checking for the significance of the NBS domain by Pfam analysis (default e-value = 1.0). In a first step, from 580 complete ORF gene models we have retained only 464 sequences that were longer than 200 aa, and in a second step, only 436 sequences that showed less than 70 % of similarity. The different steps, tools and outputs of the data mining procedure are summarized in Fig. 1. Considered as representative of the diversity of wheat NBS protein family, the final 436-gene models set (Table S2) was, therefore, used for structural and homology-based classifications, proteomic comparison with model Poaceae species and anchoring to the wheat genome.

Data mining pipeline developed in this study for the identification of members of the NBS-encoding R genes. A primary pipeline starts from 5,321,847 T. aestivum genome contigs (GanBank accession CALP000000000) that were translated in all frames and used as template for HMM-based searching. 3,010 non-redundant contigs resulting from HMM investigation were used for creating 580 complete ORF models and 1,099 incomplete ORF ones. A secondary pipeline consisted in the filtration of complete models based on size and similarity criteria. Text on the right of arrows explains the software and databases used for each operation of the pipeline. Rectangles contain the output of each step
Structure
Among the 436 gene models obtained from the previous filtration steps, the shortest (CALP011095362.1-1) was of 201 aa, while the longest (CALP010000002.1-1) was of 1,721 aa (Table S2). This wide size range was not accompanied by a homogeneous distribution, as most (75 %) of gene models were shorter than 508 aa, as illustrated by the box plot analysis (Fig. 2a). The number of exons varied between 1 and 11 with an average of 3 (Table S2). Three cell arrangements were identified with respect to cell topology: proteins with only a cytoplasmic domain (92 %), those with a cytoplasmic domain and a transmembrane anchor (1 %), and those with cytoplasmic, transmembrane and extracellular domains (7 %) (Fig. 2b; Table S2). Depending on the presence or absence of CC and LRR domains, we could assign the 436 gene models identified to 4 distinct groups; CC–NBS–LRR (334/436; 76.61 %), NBS–LRR (96/436; 22.02 %), CC–NBS (5/436; 1.15 %) and NBS (1/436; 0.23 %) (Fig. 2c; Table S2). In addition to the CC and LRR domains, other structural features were optionally present in the identified models. These characteristics included the presence of signal peptides, BED (PF02892) and RVT (PF13966) zinc fingers, two types of kinases (serine/threonine and tyrosine), DNA-binding, heavy metal-associated (HMA) (PF00403) and WRKY (PF03106) domains, and NB–ARC domains over two distinct intervals (Fig. 3). Percentage representation of each feature is illustrated in Table 2.

Classification of 436 complete gene models representing the NBS protein family of T. aestivum, based on structural features. a Box plot analysis revealing unequal distribution of wheat NBS-type proteins based on their sizes: although the size range was 201–1,721 aa, only 25 % had sizes above 508 aa. b Classes of cellular topology showing that the majority of identified proteins were composed of a unique cytoplasmic domain (C), followed by proteins with cytoplasmic, transmembrane and extracellular domains (C + T + E), and those with a cytoplasmic and a transmembrane domains (C + T). c Classes of domain architecture, based on coiled-coil (CC), nucleotide-binding site (NBS) and leucine-rich repeats (LRR) domains

Examples illustrating variability in conserved domain associations within the identified wheat NBS proteins. a Gene model CALP010016798.1-1 (669 aa); b gene model CALP010011135.1-1 (339 aa); c gene model CALP01056883.1-1 (285 aa); d gene model CALP010955910.1-1 (271 aa); e gene model CALP010004716.1-1 (997 aa); f gene model CALP010000049.1-1 (690 aa). CC–NBS–LRR: a, b, e, f; NBS–LRR: c; NBS: d; Cytoplasmic domain only protein: a, c, d and e; Cytoplasmic, transmembrane and extracellular domains protein: b; Zinc finger in reverse transcriptase (Zf-RVT; Pfam: PF13966): a; NB–ARC domain (Pfam: PF00931) over two intervals: b; WRKY domain (Pfam: PF03106): e; HMA domain (Pfam: PF00403): f
Homology-based classification
Homology searches with proteins or transcripts already available in public databases have enabled assigning the collection of gene models identified in the present study to 4 out of the 6 likely statuses explained in Figure S2. Among these statuses, the first corresponds to proteins with evidence of their existence based on homology at similar predicted-protein and transcript levels (Expressed R protein similar to a predicted one; ~8.0 %), the second includes proteins with evidence of existence only at transcript level (Expressed R protein, ~5.0 %), the third comprises proteins with evidence of existence at only similar predicted-protein level (Hypothetical R protein similar to a predicted one, ~52.0 %), and the last is represented by proteins whose existence remains hypothetical because they were inferred only from structure, without any homology support (Hypothetical R protein, ~35.0 %) (Fig. 4).

Classification of 436 candidate NBS-encoding genes, based on homologies at peptide (GenBank nr) and transcript (GenBank dbEST and TSA) levels. H Hypothetical resistance protein, H/P hypothetical resistance protein similar to a predicted one, E expressed resistance protein, E/P expressed resistance protein similar to a predicted one. Numbers of gene models of each class are shown inside histograms and percentages to the right
The principal protein homologies were detected from T. tauschii (126), followed by T. urartu (59), while a single match was produced from each of T. durum, Phyllostachys edulis, Thinopyrum ponticum and Nicotiana tabacum (dicot) (Fig. 5; Table S3). All these species are monocots except N. tabacum, for which a homology was detected between a part of I2 protein (ABB00570.1) and wheat gene model CALP010125674.1-1 (265 aa).

Histogram of species ranking based on the number of hits matching T. aestivum gene models, using BlastP against GenBank Non-Redundant protein sequences database (nr)
Even though the two statuses of “Resistance protein” and “Putative resistance protein” (Fig. S2) were not represented, some gene models of our set did show similarities to four T. aestivum characterized functional disease resistance proteins, namely Lr1, Lr10, Lr21 and Pm3, with similarity ranges covering only in part the subject resistance proteins. For instance, our gene models CALP010000738.1-1, CALP010121935.1-1, CALP010049524.1 and CALP010709654.1 (all of them CC–NBS–LRRs of 330, 332, 218 and 209 aa, respectively; Table S2) showed local BlastP similarity with Lr1, Lr10, Lr21 and Pm3, respectively. This shows, at least to some degree, that our analysis was deep enough to identify well-characterized gene models.
Orthology
Based on domain annotations, 593 and 231 NBS-containing predicted proteins were retrieved from the predicted whole proteomes of O. sativa and B. distachyon, respectively. Using these proteins, in addition to the set of 436 T. aestivum NBS proteins, allowed identifying a total of 56 gene sub-families (sequence clusters). Wheat proteins belonged to 31 sub-families, among which 25 were common to all three species, thus representing ancestral gene clusters, 3 were shared between T. aestivum and rice, and 3 others specific to T. aestivum (Fig. 6; Table S4). None of B. distachyon clusters (30) was specific to this species, while 20 clusters proved specific to rice.

Three-way Venn diagram showing the distribution of species-specific and shared NBS gene clusters among Triticum aestivum, Brachypodium distachyon and Oryza sativa NBS-encoding predicted proteins. The first number under the species name indicates the total number of NBS genes annotated for a particular species, and the second indicates the number of genes in groups for that organism. The difference between the two accounts for singleton genes that were not present in any cluster. Numbers of clusters are provided in each intersection of the Venn diagram. For wheat, out of 436 genes identified, 416 were successfully clustered that belonged to 31 gene families: three specific to T. aestivum, three shared with O. sativa only; and 25 shared with both O. sativa and B. distachyon
Genomic distribution
Using the survey sequence assemblies of the IWGSC (http://wheat-urgi.versailles.inra.fr/Seq-Repository) together with the Blast server of TGAC (http://tgac-browser.tgac.ac.uk/iwgsc_css/blast.jsp), we could assign 373 out of the total 436 studied complete NBS-type gene models to their chromosome arms. Sixty-three gene models could not be assigned either because they resulted in no hits or less significance ones (identity to subject scaffold <90 % and blast score <1,000). Five gene models were assigned to duplicate loci because they matched scaffolds located on different chromosome arms with an identical blast score. This made the number of loci slightly higher (378) than the number of gene models (373). Chromosome 4A had the highest number of R-genes (41; 10.85 %), followed by chromosome 2B (35; 9.26 %), whereas the most underrepresented chromosome was chromosome 4D, with just 1 gene (Fig. 7; Table S5). Out of 378 loci, 189 (50 %) were located on 6 out of 21 chromosomes, namely chromosomes 1B, 2B, 4A, 7A, 7B and 7D. Comparing wheat homeologous groups for NBS loci showed that chromosome group 7 had the greatest number: 84 of the 378 (22.22 %). At sub-genomes level, the distribution of NBS loci was almost balanced, with sub-genomes A, B and D containing 118, 160 and 100 NBS loci, respectively (Fig. 7).

Distribution of 378 loci of 373 wheat NBS-encoding candidate genes across T. aestivum subgenomes, chromosomes and chromosome arms
Discussion
Worldwide, wheat is cultivated over an area of 225 Mha, with production of 681 Mt (Sharma 2012). The world’s population is increasing every year and is expected to reach 8.9 billion people by 2050. To meet the food requirement of this alarmingly increasing population, wheat research programs have to be oriented towards genetic improvement for high-yield potential and resistance to biotic and abiotic stresses. Wheat crop is attacked by a variety of pests, including bacteria, fungi, oomycetes, viruses, nematodes and insects. These pests cause substantial yield losses worldwide, highlighting the threat of starvation to Human populations in several regions of the world. Genetic transfer of resistance (R) genes is an efficient, economical, and environment-friendly method of controlling wheat pests. Despite the increasing numbers of disease resistance genes identified by segregation methods, and deployed in wheat breeding programs, only few genes have been cloned and characterized and most of them were found to encode an NBS domain. Among these genes, Pm3 (Yahiaoui et al. 2004), Lr1 (Cloutier et al. 2007), Lr10 (Feuillet et al. 2003), Lr21 (Huang et al. 2009) and Lr34 (Krattinger et al. 2011) were identified in T. aestivum, whereas other genes such as Sr33 (Periyannan et al. 2013) and Sr35 (Saintenac et al. 2013) were derived from wheat relatives and transferred into bread wheat.
NBS-containing proteins are the largest R-proteins family and have a major effect on the defense of the plant against its pathogens, operating mainly as cytoplasmic receptors, directly or indirectly recognizing pathogen effectors introduced into the host cells, or (in some cases) acting in signal transduction pathways (Glowacki et al. 2011). With the advent of high throughput molecular tools, genomic DNA sequence analyses revealed variable numbers of putative NBS-containing genes in different crop and model species, even though the number of functional or expressed R genes remained unknown. Analyses of NBS–LRR-containing genomic sequences from various crop plants suggested that only a small fraction of the R genes may be functional (Chin et al. 2001; Sun et al. 2001; Shen et al. 2002).
In our study, we have used a data mining and filtering pipeline starting from more than 5 million raw contigs. Based on a Triticeae-specific HMM model, a set comprising 3,010 genome contigs was constructed and the subsequent gene prediction and filtration have allowed the creation of 1,679 hypothetical NBS-containing genes in the wheat genome. Because gene prediction algorithms are not flawless and could not identify some genes (Mathé et al. 2002), we listed all potentially interesting contigs and gene models identified on them (Table S1), which will facilitate further analyses based on different prediction software or the simultaneous integration of multiple software using different algorithms (D’Hont et al. 2012). The total number of genes we have found in wheat is higher than the 600 already reported in rice (Goff et al. 2002; Shang et al. 2009), 126 in B. distachyon (Tan and Wu 2012) and 191 in H. vulgare (International Barley Genome Sequencing Consortium 2012). This fact seems proportional to the hexaploid nature of T. aestivum and corresponds to almost three times the number of genes found in T. urartu, the hypothetical A genome progenitor (593; Ling et al. 2013). Beyond the fact of polyploidy, this high number certainly reflects the important size and complexity of NBS gene family in wheat.
Nearly a third (580) of the total number of genes is likely to encode functional products, as they are characterized by a complete ORF structure, while two-thirds (1,099) would correspond to pseudogenes, just waiting to be eliminated from the genome, or standing as reservoirs of genetic diversity that could be reached under recombination or gene conversion (Meyers et al. 1999). In case of their expression, the truncated R polypeptides could play a role in promoting disease resistance by acting as adaptator molecules (Lozano et al. 2012; Kohler et al. 2008, reviewed in Marone et al. 2013a). The proportion of these incomplete, probably inactive genes in wheat was much higher than that reported in all identified types of NBS-encoding genes (CC–NBS–LRR, NBS–LRR, CC–NBS and NBS) in T. urartu, where 511 complete ORF and only 82 partial genes were reported (Ling et al. 2013). Therefore, we think that events of genome duplication through polyploidization have significantly influenced the structural gene rearrangements in T. aestivum, leading to the non-functionalization of many genes (Akhunov et al. 2013).
An overview of domain architecture within a representative subset comprising 436 complete ORF, non-redundant (<70 % similarity) and longer than 200 aa gene models shows that the wheat NBS-containing protein family displays the conserved standard architecture of NBS proteins of monocotyledons, characterized by the presence of coiled-coil (CC) and LRR domains. We have demonstrated the possible presence of some atypical domains such as zinc fingers (Zf-RVT and Zf-BED), WRKY and HMA domains, together with the possible presence of a second NB–ARC domain, a signal peptide and two types of kinases (serine/threonine and tyrosine). The inventory and analysis of these additional domains should have an importance in refining the classification of NBS-type proteins into subclasses and, especially, for the examination of their localization in plant cells and the role they play in signal transduction during the plant resistance response to biotic stress factors. For example, plant WRKY that we have identified in C-terminal position of gene model CALP010004716.1-1 (997 aa) (Fig. 3e; Table S2) has been so far reported in RRS-1R receptor, an A. thaliana TIR–NBS–LRR–WRKY protein, that recognizes the PopP2 effector of the bacterium Ralstonia solanacearum (Deslandes et al. 2002). In this protein, the C-terminal WRKY domain was predicted to act as an inhibitor of signaling pathways responsible for resistance to this pathogen (Noutoshi et al. 2005). Up to our knowledge, this study is the first to report the presence of a WRKY domain in the C-terminus of a non-TIR–NBS–LRR protein from a Poaceae species, and we suggest that it would act in a manner similar to that described in RRS-1R receptor of A. thaliana. It is worth signaling that the WRKY (WRKYGQK) domain has been already described in barley, at the amino-terminus of NBS-type MLA proteins, as a component of an HvWRKY transcription factor (Ulker and Somssich 2004; Eulgem and Somssich 2007). This transcription factor is a repressor of the resistance genes that is deactivated following the recognition of an appropriate Avr elicitor of Blumeria graminis (Liu and Coaker 2008).
Apart from WRKY, the HMA (heavy metal-associated) domain that we have identified in N-terminal position of gene model CALP010000049.1-1 (690 aa) (Fig. 3f; Table S2) is a conserved protein domain found in a number of heavy metal transport or detoxification proteins (Bull and Cox 1994). It contains two conserved Cysteines that are probably involved in metal binding. This is the first report of this domain in N-terminal position of an NBS–LRR protein from a Triticeae species. Yet, in rice (Poaceae but non-Triticeae), Okuyama et al. (2011) reported cloning of resistance gene Pia conferring resistance to the blast fungus Magnaporthe oryzae with an HMA domain located in the C-terminus. The role this domain might play in the rice resistance to the blast fungal pathogen M. oryzae was hypothesized, later, by Kanzaki et al. (2013) who have found that the HMA domains of several rice proteins belonging to Heavy Metal-Associated domain protein family (sHMAs) interact with the avirulence effector Avr-Pik of M. oryzae. The high similarity of this protein family (sHMAs) with the CC domain of the rice R protein Pik-1, together with the role of coiled-coil (CC) domain of Pik-1 in the interaction with the pathogen, suggested that rice sHMA proteins could be involved in generating sHMA RNAi and overexpressing lines in rice (Kanzaki et al. 2013). In addition to its possible involvement in regulating expression activity of NBS genes, as hypothesized by Kanzaki et al. (2013), we suggest that NBS genes with HMA domain may play a role in resistance to pathogenic bacteria in heavy metals-exposed plants (Fones et al. 2010), in a way functionally analogous to the resistance conferred by normal (without HMA) NBS genes in non-accumulating plants. Though wheat plants are not accumulators, transfer of heavy metals to different organs of the wheat plant, including edible ones, was demonstrated in several varieties (Karatas et al. 2006; Jamali et al. 2009).
The existence of two distinct intervals of the NBS domain that are separated by an interdomain seems to be quite frequent in wheat, as this phenomenon was identified in 10.55 % (46/436) of our gene models. We could explain this fact by a probable breaking of the intramolecular interaction between NB and ARC subdomains, which is predicted to increase the rate of nucleotide exchange (Takken et al. 2006; van Ooijen et al. 2007). Such intramolecular interactions between NBS–LRR functional domains have been demonstrated (Moffett et al. 2002; Leister et al. 2005), and progress has been made toward delimiting the regions involved (Rairdan and Moffett 2006), although the precise nature and role of interdomain contacts are still incompletely understood (Ashfield et al. 2012).
In silico functional assignment is always approximate, since understanding the relationship between an organism’s predicted gene and its phenotype often requires wet-lab experimentation, such as gene knocking-out or transcriptome profiling using microarrays. Yet, it is always very interesting for sequences identified as putative genes to be confirmed by further evidence, such as similarity to cDNA or EST sequences from the same organism, similarity of the predicted protein sequence to known proteins, or association with promoter sequences. In this context, we have performed comparisons of our complete gene models set, against proteins (BlastP), ESTs (TBlastN) and Full-Length cDNAs (TBlastN). We could assign the 436 models to four functional and expression statuses, namely “expressed resistance proteins” (E) (~5.0 %), “expressed resistance proteins similar to predicted ones” (E/P) (~8.0 %), “hypothetical resistance proteins” (H) (~35.0 %) and “hypothetical resistance proteins similar to predicted ones” (H/P) (~52.0 %). The recent availability of genome annotations of two wheat relatives, namely the hypothetical A genome progenitor, T. urartu (Ling et al. 2013; Genbank accession AOTI000000000), and the D genome progenitor, A. tauschii (Jia et al. 2013; Genbank accession AOCO010000000), has provided numerous hits matching our wheat gene models, among predicted NBS proteins of these two species. This explains to a certain extent the relatively high proportion (~60 %) of our gene models falling within the statuses E/P and H/P. For these proteins matching similar ones in “nr” database, hits were different, reflecting functional diversity of our R-proteins dataset. Among 56 genes characterized as expressed, 18 had homologous sequences within wheat ESTs, while the remaining (38) only had a match within full length cDNAs of TSA. As TSA is an archive of computationally assembled sequences from primary data, expression evidence inferred from ESTs should be considered stronger.
Studying the genomic distribution of the analyzed 436 predicted wheat NBS genes revealed the presence of at least one predicted gene by chromosome. Unlike this ubiquity, the distribution of NBS-encoding loci across the 21 chromosomes was very unequal, varying from 1 on chromosome 4D to 41 on chromosome 4A. This unequal distribution was expected, as it was previously reported in several plant genomes (Yang et al. 2008; Mun et al. 2009; Ameline-Torregrosa et al. 2008; Monosi et al. 2004; Porter et al. 2009; Lozano et al. 2012). This fact is due to the clustered nature of NBS–LRR genes, which facilitates their evolution through sequence exchange via gene duplication (Friedman and Baker 2007).
Chromosome arm 4AL represented the richest one (38/378) in potentially functional NBS loci producing diversified (<70 % of similarity) products. This fact looks in perfect agreement with the diversity of resistance traits mapped to this chromosome arm, including resistance genes to powdery mildew, leaf rust, Hessian fly, yellow rust, stem rust, wheat streak mosaic and Septoria tritici blotch.
Wheat homeologous group 7 was characterized by a high number of loci (84/378). Within this group, chromosome 7A was highlighted as the richest wheat chromosome in powdery mildew (Pm) genes (Marone et al. 2013b). Chromosome 7B is known to harbor powdery mildew resistance genes Pm5e (Huang et al. 2003), Pm5d (Nematollahi et al. 2008), Pm47 (Xiao et al. 2013), yellow rust resistance genes Yr2 (Lin et al. 2005) and Yr6 (Li and Niu 2007), stem rust resistance gene Sr17 and leaf rust resistance gene Lr14 (McIntosh et al. 1967). The long arm of 7D is known to carry several greenbug (Schizaphis graminum) resistance genes (Zhu et al. 2005; Weng et al. 2005), while the short arm of 7D comprises genes Stb4 and Stb5 underlying resistance to S. tritici blotch (Arraiano et al. 2001; Adhikari et al. 2004), powdery mildew resistance gene Pm15 (Tosa and Sakai 1990) and leaf rust resistance Lr34 (Spielmeyer et al. 2005; Krattinger et al. 2011). For most of these genes, the molecular structure has not been characterized yet, while Lr34 is known to encode an ABC transporter and has an NBS, but not an LRR domain (Krattinger et al. 2011; PRGdb, http://www.prgdb.org). From our investigation, it appears that the most significant enrichment areas of NBS-type resistance genes (NBS-encoding hot spots) are represented by chromosome arms 1BS, 2BS, 3B, 4AL, 7AS, 7BL and 7DS.
Five gene models (namely: CALP010036014.1-1, CALP010075277.1-1, CALP010317351.1-1, CALP010000017.1-2 and CALP010001533.1-2) (Table S5) were assigned to duplicate loci because they matched scaffolds located on different chromosome arms with an identical blast score. This fact is expected as hexaploid wheat often has gene functional redundancy because of tripled genomes. Redundant genes tend to be lost or become pseudogenes to avoid a fitness cost to the host species (Tian et al. 2003).
This study provides an exhaustive report of the NBS-containing gene family in the cultivated hexaploid wheat T. aestivum, with rich and multifaceted genome-wide information, hence offering a genomics-guided framework for molecular isolation, cloning, and elucidation of the functions of all R genes. The information generated in this study will be of great use on two levels: the applied one with the aim to improve disease resistance in wheat, and the fundamental one through comparative genomics approaches across grass genomes, to reveal the evolutionary significance of some wheat genes.
References
Adhikari TB, Cavaletto JR, Dubcovsky J, Gieco JO, Schlatter AR, Goodwin SB (2004) Molecular mapping of the Stb4 gene for resistance to Septoria tritici blotch in wheat. Phytopathol 94:1198–1206
Akhunov ED, Sehgal S, Liang H, Wang S, Akhunova AR, Kaur G, Li W, Forrest KL, See D, Šimková H, Ma Y, Hayden MJ, Luo M, Faris JD, Dolezel J, Gill BS (2013) Comparative analysis of syntenic genes in grass genomes reveals accelerated rates of gene structure and coding sequence evolution in polyploid wheat. Plant Physiol 161:252–265
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Ameline-Torregrosa C, Wang BB, O’Bleness MS, Deshpande S, Zhu H, Roe B, Young ND, Cannon SB (2008) Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant medicago truncatula. Plant Physiol 146:5–21
Arraiano LS, Worland AJ, Ellerbrook C, Brown JKM (2001) Chromosomal location of a gene for resistance to septoria tritici blotch (Mycosphaerella graminicola) in the hexaploid wheat ‘Synthetic 6x’. Theor Appl Genet 103:758–764
Ashfield T, Ashley NE, Pfeil BE, Chen NWG, Podicheti R, Ratnaparkhe MB, Ameline-Torregrosa C, Denny R, Cannon S, Doyle JJ, Geffroy V, Roe BA, Saghai Maroof MA, Young ND, Innes RW (2012) Evolution of a complex disease resistance gene cluster in diploid Phaseolus and tetraploid Glycine. Plant Physiol 159(1):336–354
Bella J, Hindle KL, McEwan PA, Lovell SC (2008) The leucine-rich repeat structure. Cmls-cell Mol Life Sci 277:519–527
Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D’Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D, Kay S, Waite D, Trick M, Bancroft I, Gu Y, Huo N, Luo MC, Sehga S, Gill B, Kianian S, Anderson O, Kersey P, Dvorak J, McCombie WR, Hall A, Mayer KFX, Edwards KJ, Bevan MW, Hall N (2012) Analysis of the breadwheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Bull PC, Cox DW (1994) Wilson disease and Menkes disease: new handles on heavy-metal transport. Trends Genet 10(7):246–252
Chin DB, Arroya-Garcia R, Ochoa OE, Keselli RV, Lavelle DO, Michelmore RW (2001) Recombination and spontaneous mutation at the major cluster of resistance genes in lettuce (Lactuca sativa). Genetics 157:831–849
Cloutier S, McCallum BD, Loutre C, Banks TW, Wicker T, Feuillet C, Keller B, Jordan MC (2007) Leaf rust resistance gene Lr1, isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol Biol 65:93–106
Deslandes L, Olivier J, Theulieres F, Hirsch J, Feng DX, Bittner-Eddy PD, Beynon J, Marco Y (2002) Resistance to Ralstonia solanacearum in Arabidopsis thaliana is conferred by the recessive RRS1-R gene, a member of a novel family of resistance genes. Proc Natl Acad Sci USA 99:2404–2409
D’Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M, Da Silva C, Jabbari K, Cardi C, Poulain J, Souquet M, Labadie K, Jourda C, Lengellé J, Rodier-Goud M, Alberti A, Bernard M, Correa M, Ayyampalayam S, Mckain MR, Leebens-Mack J, Burgess D, Freeling M, Mbéguié-A-Mbéguié D, Chabannes M, Wicker T, Panaud O, Barbosa J, Hribova E, Heslop-Harrison P, Habas R, Rivallan R, Francois P, Poiron C, Kilian A, Burthia D, Jenny C, Bakry F, Brown S, Guignon V, Kema G, Dita M, Waalwijk C, Joseph S, Dievart A, Jaillon O, Leclercq J, Argout X, Lyons E, Almeida A, Jeridi M, Dolezel J, Roux N, Risterucci AM, Weissenbach J, Ruiz M, Glaszmann JC, Quétier F, Yahiaoui N, Wincker P (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488:213–217. doi:10.1038/nature11241
Ellis J, Jones D (2003) Plant disease resistance genes. In: Ezekowitz A, Hoffman J (eds) Innate immunity. Humana Press Inc., New Jersey, pp 27–44
Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10:366–371
Feuillet C, Travella S, Stein N, Albar L, Nublat A, Keller B (2003) Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc Natl Acad Sci USA 100:15253–15258
Fones H, Davis CAR, Rico A, Fang F, Smith JAC, Preston GM (2010) Metal hyperaccumulation armors plants against disease. PLoS Pathog 6(9):e1001093. doi:10.1371/journal.ppat.1001093
Friedman AR, Baker BJ (2007) The evolution of resistance genes in multiprotein plant resistance systems. Curr Opin Genet Dev 17:493–499
Glowacki S, Macioszek VK, Kononowicz AK (2011) R proteins as fundamentals of plant innate immunity. Cell Mol Biol Lett 16:1–24
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296:92–100
Huang X, Wang L, Xu M, Röder M (2003) Microsatellite mapping of the powdery mildew resistance gene Pm5e in common wheat (Triticum aestivum L.). Theor Appl Genet 106:858–865
Huang L, Brooks S, Li W, Fellers J, Nelson JC, Gill B (2009) Evolution of new disease specificity at a simple resistance locus in a crop-weed complex: reconstitution of the Lr21 gene in wheat. Genetics 182(2):595–602
International Barley Genome Sequencing Consortium (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716
Jamali MK, Kazi TG, Arain MB, Afridi HI, Jalbani N, Kandhro GA, Shah AQ, Baig JA (2009) Heavy metal accumulation in different varieties of wheat (Triticum aestivum L.) grown in soil amended with domestic sewage sludge. J Hazardous Materials 164:1386–1391
Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X, Jing R, Zhang C, Ma Y, Gao L, Gao C, Spannagl M, Mayer KFX, Li D, Pan S, Zheng F, Hu Q, Xia X, Li J, Liang Q, Chen J, Wicker T, Gou C, Kuang H, He G, Luo Y, Keller B, Xia Q, Lu P, Wang J, Zou H, Zhang R, Xu J, Gao J, Middleton C, Quan Z, Liu G, Wang J, International Wheat Genome Sequencing Consortium, Yang H, Liu X, He Z, Mao L, Wang J (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Jones DA, Jones JDG (1997) The role of leucine-rich repeat proteins in plant defences. Adv Bot Res 24:90–167
Kajava AV (1998) Structural diversity of leucine-rich repeat proteins. J Mol Biol 277:519–527
Kanzaki K, Saitoh H, Fujisaki K, Kobayashi M, Ito K, Kanzaki E, Mitsuoka C, Banfield M, Kamoun S, Terauchi R (2013) Rice proteins that interact with the magnaporthe oryzae avirulence effector AVR-Pik (A-22). In: International rice blast conference, Jeju, Korea, August 20–24, 2013, Proceedings book, p 98. http://irbc2013.riceblast.snu.ac.kr/attach/IRBC2013_proceeding_book.pdf
Karatas M, Dursun S, Guler E, Ozdemir C, Argun ME (2006) Heavy metal accumulation in wheat plants irrigated by waste water. Cellulose Chem Technol 40:575–579
Kohler A, Rinaldi C, Duplessis S, Baucher M, Geelen D, Duchaussoy F, Meyers BC, Boerjan W, Martin F (2008) Genome-wide identification of NBS resistance genes in Populus trichocarpa. Plant Mol Biol 66:619–636
Krattinger SG, Lagudah ES, Wicker T, Risk JM, Ashton AR, Selter LL, Matsumoto T, Keller B (2011) Lr34 multi-pathogen resistance ABC transporter: molecular analysis of homoeologous and orthologous genes in hexaploid wheat and other grass species. Plant J 65:392–403
Leister RT, Dahlbeck D, Day B, Li Y, Chesnokova O, Staskawicz BJ (2005) Molecular genetic evidence for the role of SGT1 in the intramolecular complementation of Bs2 protein activity in Nicotiana benthamiana. Plant Cell 17:1268–1278
Li Y, Niu YC (2007) Identification of molecular markers for wheat stripe rust resistance gene Yr6. Acta Agr Boreali-Sinica 22:189–192
Lin F, Xu SC, Zhang LJ, Miao Q, Zhai Q, Li L (2005) SSR marker of wheat stripe rust resistance gene Yr2. J Tritical Crops 25:17–19
Ling HQ, Zhao S, Liu D, Wang J, Sun H, Zhang C, Fan H, Li D, Dong L, Tao Y, Gao C, Wu H, Li Y, Cui Y, Guo X, Zheng S, Wang B, Yu K, Liang Q, Yang W, Lou X, Chen J, Feng M, Jian J, Zhang X, Luo G, Jiang Y, Liu J, Wang Z, Sha Y, Zhang B, Wu H, Tang D, Shen Q, Xue P, Zou S, Wang X, Liu X, Wang F, Yang Y, An X, Dong Z, Zhang K, Zhang X, Luo MC, Dvorak J, Tong Y, Wang J, Yang H, Li Z, Wang D, Zhang A, Wang J (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90
Liu J, Coaker G (2008) Nuclear trafficking during plant innate immunity. Mol Plant 1(3):411–422
Lozano R, Ponce O, Ramirez M, Mostajo N, Orjeda G (2012) Genome-wide identification and mapping of NBS-encoding resistance genes in solanum tuberosum group Phureja. PLoS ONE 7(4):e34775. doi:10.1371/journal.pone.0034775
Marone D, Russo MA, Laidò G, De Leonardis AM, Mastrangelo AM (2013a) Plant nucleotide binding site–leucine-rich repeat (NBS–LRR) genes: active guardians in host defense responses. Int J Mol Sci 14:7302–7326
Marone D, Russo MA, Laidò G, De Vita P, Papa R, Blanco A, Gadaleta A, Rubiales D, Mastrangelo AM (2013b) Genetic basis of qualitative and quantitative resistance to powdery mildew in wheat: from consensus regions to candidate genes. BMC Genom 14:562. doi:10.1186/1471-2164-14-562
Mathé C, Sagot M-F, Schiex T, Rouzé P (2002) Current methods of gene prediction, their strengths and weaknesses. Nucl Acids Res 30:4103–4117. doi:10.1093/nar/gkf543
McIntosh R, Luig N, Baker E (1967) Genetic and cytogenetic studies of stem rust, leaf rust, and powdery mildew resistances in Hope and related wheat cultivars. Aust J Biol Sci 20:1181–1192
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotidebinding superfamily. Plant J 20:317–332
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS–LRR–encoding genes in Arabidopsis. Plant Cell Online 15:809–834
Miller RN, Bertioli DJ, Baurens FC, Santos CM, Alves PC, Martins NF, Togawa RC, Souza MT, Pappas GJ (2008) Analysis of non-TIR–NBS–LRR resistance gene analogs in Musa acuminata Colla: isolation, RFLP marker development, and physical mapping. BMC Plant Biol 8:15. doi:10.1186/1471-2229-8-15
Moffett P, Farnham G, Peart J, Baulcombe DC (2002) Interaction between domains of a plant NBS–LRR protein in disease resistance-related cell death. EMBO J 21:4511–4519
Monosi B, Wisser RJ, Pennill L, Hulbert SH (2004) Full-genome analysis of resistance gene homologues in rice. Theor Appl Genet 109:1434–1447
Mun JH, Yu HJ, Park S, Park BS (2009) Genome-wide identification of NBS encoding resistance genes in Brassica rapa. Mol Genet Genomics 282:617–631
Nematollahi G, Mohler V, Wenzel G, Zeller FJ, Hsam SLK (2008) Microsatellite mapping of powdery mildew resistance allele Pm5d from common wheat line IGV1-455. Euphytica 159:307–313
Noutoshi Y, Ito T, Seki M, Nakashita H, Yoshida S, Marco Y, Shirasu K, Shinozaki KA (2005) Single amino acid insertion in the WRKY domain of the Arabidopsis TIR–NBS–LRR–WRKY-type disease resistance protein SLH1 (sensitive to low humidity 1) causes activation of defense responses and hypersensitive cell death. Plant J 43:873–888
Oakley MG, Hollenbeck JJ (2001) The design of antiparallel coiled coils. Curr Opin Struct Biol 11:450–457
Okuyama Y, Kanzaki H, Abe A, Yoshida K, Tamiru M, Saitoh H, Fujibe T, Matsumura H, Shenton M, Clark DG, Undan J, Ito A, Sone T, Terauchi R (2011) A multifaceted genomics approach allows the isolation of the rice Pia-blast resistance gene consisting of two adjacent NBS–LRR protein genes. Plant J 66:467–479
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob-ur-Rahman, Ware D, Westhoff P, Mayer KF, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Periyannan S, Moore J, Ayliffe M, Bansal U, Wang X, Huang L, Deal K, Luo M, Kong X, Bariana H, Mago R, McIntosh R, Dodds P, Dvorak J, Lagudah E (2013) The gene Sr33, an ortholog of barley Mla genes, encodes resistance to wheat stem rust race Ug99. Science 341:786–788. doi:10.1126/science.1239028
Porter BW, Paidi M, Ming R, Alam M, Nishijima WT, Zhu YJ (2009) Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Mol Genet Genomics 281:609–626
Rairdan GJ, Moffett P (2006) Distinct domains in the ARC region of the potato resistance protein Rx mediate LRR binding and inhibition of activation. Plant Cell 18:2082–2093
Saintenac C, Zhang W, Salcedo A, Rouse MN, Trick HN, Akhunov E, Dubcovsky J (2013) Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341:783–786. doi:10.1126/science.1239022
Salamov A, Solovyev V (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res 10:516–522
Sanseverino W, Ercolano MR (2012) In silico approach to predict candidate R proteins and to define their domain architecture. BMC Res Notes 5:678. doi:10.1186/1756-0500-5-678
Shang J, Tao Y, Chen X, Zou Y, Lei C, Wang J, Li X, Zhao X, Zhang M, Lu Z, Xu J, Cheng Z, Wan J, Zhu L (2009) Identification of a new rice blast resistance gene, Pid3, by genome wide comparison of paired nucleotide-binding site leucine-rich repeat genes and their pseudogene alleles between the two sequenced rice genomes. Genetics 182:1303–1311
Sharma I (2012) Diseases in wheat crops, an introduction. In: Sharma I (ed) Disease resistance in wheat. CABI, Oxfordshire, pp 1–17
Shen KA, Chin DB, Arroyo-Garcia R, Ochoa OE, Lavelle DO, Wroblewski T, Meyers BC, Michelmore RW (2002) Dm3 is one member of a large constitutively expressed family of nucleotide binding site-leucine-rich repeat encoding genes. Mol Plant-Microbe Interact 15:251–261
Spielmeyer W, McIntosh RA, Kolmer J, Lagudah ES (2005) Powdery mildew resistance and Lr34/Yr18 genes for durable resistance to leaf and stripe rust cosegregate at a locus on the short arm of chromosome 7D of wheat. Theor Appl Genet 111:731–735
Sun Q, Collins NC, Ayliffe M, Smith SM, Drake J, Pryor T, Hulbert SH (2001) Recombination between paralogues at the Rp1 rust resistance locus in maize. Genetics 158:423–438
Takken FL, Albrecht M, Tameling WI (2006) Resistance proteins: molecular switches of plant defence. Curr Opin Plant Biol 9:383–390
Tan S, Wu S (2012) Genome wide analysis of nucleotide-binding site disease resistance genes in Brachypodium distachyon. Comp Funct Genomics. doi:10.1155/2012/418208
Tian D, Traw M, Chen J, Kreitman M, Bergelson J (2003) Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423:74–77
Tosa Y, Sakai K (1990) The genetics of resistance of hexaploid wheat to the wheatgrass powdery mildew fungus. Genome 33:225–230
Traut TW (1994) The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide binding sites. Eur J Biochem 222:9–19
Ulker B, Somssich IE (2004) WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol 7:491–498
van Ooijen G, van den Burg HA, Cornelissen BJC, Takken FLW (2007) Structure and function of resistance proteins in solanaceous plants. Ann Rev Phytopathol 45:43–72
Wan H, Yuan W, Bo K, Shen J, Pang X, Chen J (2013) Genome-wide analysis of NBS-encoding disease resistance in Cucumis sativus and phylogenetic study of NBS-encoding genes in Cucurbitaceae crops. BMC Genom 14:109
Wang Y, Chen J-Q, Araki H, Jing Z, Jiang K, Shen J, Tian D (2004) Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NBS-LRR genes. Mol Genet Genomics 271:402–415
Weng Y, Li W, Devkota RN, Rudd JC (2005) Microsatellite markers associated with two Aegilops tauschii-derived greenbug resistance loci in wheat. Theor Appl Genet 110:462–469
Xiao M, Song F, Jiao J, Wang X, Xu H, Li H (2013) Identification of the gene Pm47 on chromosome 7BS conferring resistance to powdery mildew in the Chinese wheat landrace Hongyanglazi. Theor Appl Genet 126:1397–1403
Yahiaoui N, Srichumpa P, Dudler R, Keller B (2004) Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J 37(4):528–538
Yang S, Zhang X, Yue JX, Tian D, Chen JQ (2008) Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol Genet Genomics 280:187–198
Zhou T, Wang Y, Chen JQ, Araki H, Jing Z, Jiang K, Shen J, Tian D (2004) Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR–NBS–LRR genes. Mol Genet Genomics 271:402–415
Zhu LC, Smith CM, Fritz A, Boyko E, Voothuluru P, Gill BS (2005) Inheritance and molecular mapping of new greenbug resistance genes in wheat germplasms derived from Aegilops tauschii. Theor Appl Genet 111:831–837
Acknowledgments
We gratefully acknowledge Jacques-Déric Rouault (Laboratoire Evolution, Génome et Spéciation, CNRS, Gif sur Yvette, France) for providing training; Abdelkader Aïnouche and Malika-Lily Aïnouche (UMR-CNRS Ecobio, Université de Rennes-1, France) for useful discussion and manuscript revision, and Rafika Challouf (Institut National des Sciences et Technologies de la Mer, INSTM, Monastir, Tunisia) for providing help in computational analyses. This study was financially supported by the Tunisian Ministry of Higher Education and Scientific Research.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bouktila, D., Khalfallah, Y., Habachi-Houimli, Y. et al. Full-genome identification and characterization of NBS-encoding disease resistance genes in wheat. Mol Genet Genomics 290, 257–271 (2015). https://doi.org/10.1007/s00438-014-0909-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-014-0909-2