
Abstract
Polymeric micelles exhibit several features that favor their use for drug delivery applications. Although micellar structures have been studied for about 100 years, they are still considered peculiar objects of physicochemical research.
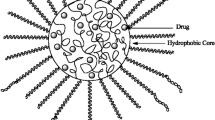
Micelles are nanosized colloidal particles with a hydrophobic inner part (core) and a hydrophilic surface (shell). Drugs and contrast agents can either be embedded in the lipid core of a micelle or covalently bind to its surface. Bifunctional polymeric micelles have been described that are used for simultaneous drug delivery and visualization of damaged tissue. The most important advantage of polymeric micelles is their ability to solubilize poorly water-soluble or hydrophobic drugs within their core, thus enhancing their bioavailability. Based on the type of intermolecular force driving micelle formation, block copolymer micelles can be divided into several categories, including hydrophobically assembled amphiphilic micelles, polyion complex micelles, and micelles stemming from metal complexation. Micelles are advantageous as nanocarriers for drug delivery and treatments owing to their excellent physicochemical properties, drug loading and release capacities, facile preparation methods, biocompatibility, and tumor targetability.
The current line of research in this area is to generate heterogeneous polymeric nanostructures with controlled composition, hydrophilic-hydrophobic balance, and properties. This chapter will consider the existing approaches to preparing polymer micelles for drug delivery applications, the complexity of working with them, and the prospects for their use in therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
In current medicine and pharmacology, targeted drug delivery has gained importance as a method that enables one to increase the concentration of delivered drugs in a certain place and block or strongly limit their accumulation in healthy organs and tissues. Targeted delivery makes it possible not only to increase the duration and efficacy of drug action but also to largely reduce side effects. Nanotechnology and nanoproducts play a special role in this regard. The interest in the development of delivery systems is due to many reasons, above all to the enormous potential benefits, both medical and economic.
After a “normal” drug is injected or administered orally, it enters the bloodstream, which distributes the drug molecules more or less evenly to all organs and tissues of the body. A small portion of the drug (0.001–0.01%) enters the lesion site, whereas the bulk of it is eliminated from the body and also causes toxicity. Targeted delivery implies a different scheme, in which the carrier with the drug enters the bloodstream, circulates in the body, and accumulates only in the lesion site. However, so that this scheme could be implemented, the following requirements must be satisfied (Fig. 1).

Basic requirements for targeted drug delivery
2 Stages in the Preparation of Drug Delivery Systems
The postulate that a drug (agent), for its effect to take place, must first bind to the corresponding receptors in the cells of the pathological focus (target) was formulated by John Langley in 1878 [1]. The idea of targeted delivery of drugs (the so-called magic bullet) belongs to the German scientist Paul Ehrlich, who put it forward in 1906. In his opinion, the “magic bullet” is an ideal medicine “capable of independently finding the source of disease or the focus of disease and striking them without affecting healthy organs and tissues of the body” [2].
The “target agent” concept is based on the fact that a drug, to implement its effect, must first bind to the corresponding receptors located on the target cells. The selectivity of the drug action is determined by its pharmacodynamics and pharmacokinetics and by its metabolism and excretion from the body. There are four levels of drug action:
-
(a)
Molecular level, at which protein molecules are direct targets for most drugs.
-
(b)
Cellular level, at which biochemical and other components of the cell participate in transduction (generating a biological response to a certain external action).
-
(c)
Tissue level, at which changes occur in the functions of the heart, skin, lungs, etc.
-
(d)
Systemic level, at which changes occur in the functions of the cardiovascular and nervous systems, the gastrointestinal tract, etc.
The first drugs based on lipid emulsions were proposed in December 1932 by J. Johnson, when a British patent was registered on behalf of the I.G. Farbenindustrie Aktiengesellschaft. The patent stated that “pharmaceutical preparations for subcutaneous and intramuscular injections may be obtained by combining drugs with liquids such as fats or oils, if necessary together with waxes or wax-like substances, with water or other fluids, and a dispersing agent.” These pharmaceutical preparations can rightfully be considered prototype drug delivery systems [3].
Lipid molecules are capable of forming two types of structures in an aqueous medium: liposomes and micelles. Usually, the terms “liposomes” and “lipid vesicles” are used synonymously. However, liposomes were first described as particles formed by the mechanical dispersion of a suspension of swollen phospholipids in water.
Liposomes are closed bubbles formed by one or more lipid bilayers, inside which there is a space, usually filled with water with substances dissolved in it. In fact, liposomes are spherical vesicles with one or more lipid bilayers. They are formed in mixtures of phospholipids with water. The inside of the liposomes contains water or a solution in which ultrasound treatment was done (Fig. 2).

Lipid bilayer and its closure, forming a liposome (https://commons.wikimedia.org/w/index.php?curid=9035720)
Micelles (a diminutive from Latin mica “particle, grain”) are aggregates of surfactants in a colloidal solution (sol), consisting of a large number of amphiphilic molecules. Figure 3 shows a schematic representation of a phospholipid micelle in an aqueous solution.

Scheme for a phospholipid micelle in an aqueous solution (https://en.wikipedia.org/wiki/Micelle)
The first idea of the target polymer–drug conjugate was proposed by G. Ringsdorf in 1975 [4]. In 1976, English researcher Gregory Gregoriadis suggested placing drugs inside liposomes to promote their penetration into the body. This can be considered the beginning of the use of nanocontainers.
The widespread use of liposomes with encapsulated drugs led to an increase in their concentration in the pathological focus, increasing the time of drug action and facilitating the release of the drug in a particular location. In the case of local administration of liposomal drugs, no specific delivery of liposomes is required; they act by slowly releasing the drug into the environment. Therefore, these cosmetic products based on liposomes were introduced to the market: in particular, as cosmetic products by Christian Dior, L’Oreal, Procter & Gamble, Johnson & Johnson, Avon, etc. [5].
At the beginning of the twenty-first century, synthesized conjugates are being actively tested in preclinical and clinical trials. Many liposomal preparations successfully pass clinical tests and reach the market [6].
In particular, during the COVID-19 pandemic, Pfizer/BioNTech, and Moderna mRNA vaccines were developed, consisting of nucleoside-modified mRNA encoding the SARS-CoV-2 spike protein, which is encapsulated in lipid nanoparticles [7, 8].
3 Approaches to Making Targeted Drug Delivery Systems
The agent in a targeted drug delivery system can be a drug that binds to a target through a pharmacophore. A target is a specific molecular structure that is the aim of targeted drug delivery. Molecular targets include hormone and neurotransmitter receptors, enzymes, ion channels, transporter molecules, and nucleic acids. The receptor is that part of the target where the binding to the drug occurs, which is followed by activation of specific biochemical processes.
3.1 Active and Passive Transport
It is known that cellular metabolism, bioenergetic processes, the formation of biopotentials, the generation of a nerve impulse, and other processes are related to the transfer of substances through the membranes. In many cases, therapeutic therapy requires drug delivery through the cell membranes. The effectiveness of a drug depends largely on how permeable the membrane is to it.
Targeted delivery can be subdivided into passive and active transport. Transport by simple and facilitated diffusion is called passive; in it, substances are delivered along the concentration gradient. Under nonequilibrium conditions, the directional movement of particles is initiated by various mechanisms. The existence of an artificially maintained concentration gradient or the presence of stationary external forces of various natures leads to the stationary drift of particles. It is this drift that is considered the main mechanism of particle movement through biological membranes and is called passive transport [9, 10].
Passive diffusion across cell membranes plays a large part in the delivery of many pharmaceutical agents to intracellular targets. For measurements, biomimetic systems have been combined with advanced methods; so, attention is paid to time-resolved fluorescence, surface plasmon resonance, light scattering, etc. [11].
Active transport is possible only in conjunction with the hydrolysis of adenosine triphosphoric acid. Concentration, electric potential, pressure, and other gradients, which support life processes, are generated owing to active transport [11, 12].
Advances in drug delivery have prompted researchers to consider two important strategies in the development of new multifunctional liposomal particles: passive and active targeting strategies. In passive targeting, owing to the physical properties of the nanocontainer, the liposome-encapsulated substance accumulates in a certain affected body area and interacts selectively with the anatomical structures of the target tissue vessels while producing its pharmacological effect. Another drug delivery variant is the use of a guiding vector—for example, monoclonal antibodies, receptor ligands, enzymes, or glycoproteins [12,13,14].
3.2 Vector Use in Drug Delivery Systems
A vector is a compound that ensures drug delivery to the pharmacological target. After the vector is attached to the nanocarrier–drug conjugate, the resulting structure must be stable and nontoxic, and the ability to recognize the target and the efficiency of nanocarrier loading must be preserved. Specific proteins (transferrin, the peptide hormone gonadoliberin), radiolabeled monoclonal antibodies, viruses, and folic acid all can be used as vectors.
An experiment using a vector as a delivery system was first done in 1958. For targeting a drug (methotrexate), conjugation to an antibody was done. However, the term “vector” did not exist at that time; it appeared only in the 1970s. In 1975, immunologists S. Milstein and G. Koeller developed a method for generating hybrid somatic cells for antibody production. This opened up the possibility of wide application of antibodies in various fields of biology and medicine.
The first vectors used for targeted delivery were antibodies bound to liposomes. The method of their attachment was relatively simple and allowed the grafting of a sufficient amount of antibodies onto the liposome surface without violating the liposome integrity or changing the affinity and specificity of the antibodies.
Viruses such as the adenovirus, the vesicular stomatitis virus, the cytomegalovirus, the lentivirus, and the retrovirus are widely used vectors because they ensure highly contagious, efficient delivery [15,16,17]. However, viral vectors have several limitations, such as toxicity, immunogenicity, carcinogenicity, high cost, and difficulty of large-scale production in clinical practice [18,19,20]. Consequently, increasing scientific attention has been given to the development of nonviral vectors and carriers [21,22,23]. Recent studies have shown that nonviral vectors have the following advantages: low immunogenicity, biodegradability, easy synthesis, low production cost, and absence of restrictions on the size of injected molecules [24,25,26,27,28]. The most widely studied nonviral vectors are polymers, liposomes, and inorganic nanoparticles [29,30,31,32,33]. The main vectors used as drug delivery systems are listed in Table 1.
Nonviral vectors can prevent premature degradation of nucleic acids, proteins, or drugs and can prolong the therapeutic effect and reduce side effects. Biomedical applications place high demands on the physicochemical properties of vectors. Meanwhile, the residual toxic effects of catalysts, solvents, and other substances in the synthetic process cannot be ignored. Therefore, it is necessary to standardize toxicological tests and determine safe exposure limits. Despite these difficulties, the widespread use of nonviral vectors to improve the efficiency of drug delivery and gene therapy is expected in the near future.
The key role of vectors can be understood as increasing drug pharmacokinetics to improve therapy. The growing number of gene therapy and vaccine vectors and drug delivery carriers has been extensively investigated because of their ease of use, targeting ability, high bioavailability, and good biocompatibility [35,36,37].
For example, a system was described that delivers doxorubicin (DOX) to the lungs and is based on the macrophages of active [38] and inactive cells [39]. Efficient and minimally invasive drug delivery systems have been developed for the treatment of persistent human diseases. These are based on chimeric vector systems combining at least two different vector systems. For example, chimeric drug delivery systems combining viral and nonviral features have been developed. Fusigenic nonviral particles have been constructed by conferring viral fusion proteins onto nonviral vectors. HVJ (hemagglutinating virus of Japan; Sendai virus) liposomes were constructed by combining DNA-loaded liposomes with a fusogenic envelope derived from the HVJ. The resulting HVJ envelope vector efficiently and rapidly introduced plasmid DNA into both cultured cells in vitro and organs in vivo. In addition, proteins, synthetic oligonucleotides, and drugs were also efficiently introduced into cells with the HVJ envelope vector. The authors have shown that the HVJ envelope vector is a promising tool for gene therapy experiments both ex vivo and in vivo [40].
3.3 Cellular Penetration of Delivery Systems and Release of Drugs
Separate attention should be paid to the release of the drug and its penetration into the cell when the delivery system enters the pathological focus. This process depends largely on the nature of the interaction between the carrier (e.g., liposomes) and the cell membrane. The following kinds of the interaction of the carrier with the cell membrane can be distinguished (Fig. 4) [41]:
-
(a)
Liposomes are adsorbed (attached) on the cell surface.
-
(b)
The drug goes directly into the cell through the uptake of liposomes by the cell (endocytosis).
-
(c)
Liposome membranes may fuse with cell membranes and become part of them, in which case the properties of the cell membranes may change. This includes an increase in the cell membrane permeability owing to the formation of additional membrane channels.
-
(d)
Sometimes the cell membrane and the liposome exchange lipids.
-
(e)
Liposomes may fuse with cell membranes and become part of them. The properties of the cell membranes, such as their viscosity and permeability and the amount of electric charge, may change. The number of channels crossing the membranes may also increase or decrease.

Ways of penetration of the liposome contents into the cell. (Reprinted under the Creative Commons Attribution (CC BY SA 3.0) [41])
Thus, liposomes have paved the way for a new method of cell targeting, which can be called “membrane engineering.” A multiscale diffusion model was developed incorporating chemical properties of material and geometry of microstructure, which is especially useful in predicting mass release from drug vectors. Owing to this model, it is possible to predict mass distribution in a flow similar to the one found in capillaries [42].
3.4 Development of Liposomal Drugs for Targeted Delivery
Liposomal drug delivery systems have proven breakthroughs and are innovative in the treatment of many diseases [43]. The main factors behind the development of liposome-based drug delivery systems were the use of three key methods.
The first method is the coating of liposomes with polyethylene glycol (PEG). This coating makes liposomes better protected from the reticuloendothelial system by reducing their recognition by macrophages, which allows liposome elimination from the body to be slowed down.
PEG also generates increased osmotic pressure around the liposome, preventing the approach of other cells. As a result, PEGylated liposomes circulate longer in blood and accumulate in tumor tissues in larger amounts than conventional liposomes.
The second method is the use of antibodies as vectors, which ensures the possibility of targeted drug delivery owing to the interaction of the antibody attached to the particle with the cell membrane receptor.
The third method is related to the fact that the endothelial cells of tumor vessels proliferate 30–40 times faster than do the endothelial cells of normal tissue vessels, so tumor capillaries are characterized by larger pores. These pores are used for the passive delivery of liposomal drugs to the tumor.
The drug inclusion in liposomes enhances patient tolerability of the encapsulated drug and enables the drug therapeutic index (ratio between therapeutic effect and toxicity) to be increased. The reduction in systemic toxicity is based on the preferential accumulation of medium-sized (50–200-nm) particles in tumor tissue owing to enhanced permeability and retention (increased capillary permeability and impaired lymphatic drainage in tumor tissue) [44, 45]. To be well transported to tumor tissue, liposomes can be equipped with carbohydrate ligands of the sialyl Lewis family, which are specific to selectins—carbohydrate-binding lectins involved in the primary interaction of blood leukocytes with endothelial cells and thus involved in various inflammatory and metastatic processes. Selectins are promising targets for the delivery of therapeutic agents to the vessels of tumor tissue [46].
The liposome membrane is usually formed from the same phospholipids that are part of the biological membrane: phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine. Depending on the method of liposome preparation, the liposome size may range from a few microns to tens of nanometers (nanosomes). If an aqueous drug solution is used in the production of liposomes, part of this solution is closed inside the liposomal container and is introduced into the human body in just such form of dosage. This is important when a toxic compound, such as an anticancer agent, is used or when the drug substance needs to be protected from degradation until it is delivered to the target. The attachment of PEG molecules protects the liposomes themselves from capture by the immune system cells and thus increases the time for which the liposomes remain in the bloodstream. Liposomes deliver the drug to the cells either by fusion with their membrane or by endocytosis. On the basis of different targeting strategies, liposomes are classified into passive, active, physicochemically targeting, and multifunctional. The manufacturing technology of drug liposomes is mature and consists of film dispersion, reverse-phase evaporation, chemical gradient loading, and other methods such as the following:
-
(a)
Heating to produce empty liposomes by hydration of phospholipids in an aqueous solution containing 3% glycerol by increasing the temperature to 60 °C or 120 °C
-
(b)
Lyophilization of a monophasic solution for the encapsulation of heat-sensitive drugs such as DNA [47]
Liposomal drug delivery systems are believed promising in the treatment of cancer and other diseases. Owing to the development of pharmaceutical technologies, a new type of liposome, the double functional liposome, has been proposed. The advantage of these new liposomes is their expanded function, which enables the elimination of drug-resistant cancer, destruction of cancer stem cells and mitochondria, induction of apoptosis, regulation of autophagy by using the microenvironment, and suppression of resistant cancer genes [48].
A targeted liposome is a drug delivery system that selectively localizes the drug in the target tissue, organ, cell, or intracellular structure through local or systemic administration. Homobifunctional and heterobifunctional cross-linking approaches are used to prepare antibody–drug conjugates and antibody-mediated targeted liposomes. For the preparation of antibody-mediated liposomes, antibodies are conjugated directly to the distal end of PEG, which is already bound to the liposome membrane [47, 49].
For the preparation of ligand-mediated liposomes, a chemical targeting ligand is often used. Such a ligand forms a complex with a specific protein on cells or organelles to reach a target; for example, dequalinium is used to deliver drugs to mitochondria [49, 50].
3.5 Drug Release from the Delivery System
Targeted drug delivery leads to the preferential accumulation of the drug in the target area, which does not depend on the method and route of drug administration. On the other hand, targeted therapy or targeted medicine means a specific interaction between a drug and its receptor at the molecular level. Effective targeted drug delivery systems imply four basic requirements: preservation of the therapeutic dose, avoidance of prior drug degradation, targeting, and release of the encapsulated drug.
Three controlled release technologies are currently available including (a) periodic release (a constant amount of the drug is released at a constant interval); (b) feedback release (the drug is released on command from a physical signal); and (c) continuous release (the drug is released at a constant rate) [51,52,53,54,55,56].
The methods of drug release from the polymer are divided into physical and chemical. In the physical method, the release is controlled by diffusion of the drug/solvent or penetration of the drug through the membrane [57]. The drug is placed in a container with a membrane wall. The rate of its release from diffusion-controlled systems depends strongly on the physical properties and size of the drug molecules and the loading level. The surface area of the membrane and the length of the diffusion pathway are also important. The chemical method consists in the hydrolytic or enzymatic cleavage of the main chain or the detachment of a side chain from a biodegradable polymer. The drug release from the delivery system is controlled by diffusion control. Functionalization of the surface of the drug delivery system and selection of the correct membrane material is crucial for the kinetics of drug release.
The controlled-release frame/membrane material should be nonimmunogenic, nontoxic, and biocompatible, have reduced tensile strength, retain large drug amounts, and, most importantly, allow controlled release of the drug. Typically, nanoparticles with a specific drug are loaded into the reservoir, and then the reservoir surface is coated with a programmable-rate membrane, which can be further engineered to control the drug release behavior.
The release of a particular drug from the drug delivery system (DDS) can be modulated by external or internal stimuli. The rationale behind activation-modulated drug delivery systems is that different organs have different biological environments (physical, chemical, electrical, and biochemical). For example, different organs/parts of the body have different pH values, such as blood (pH 7.4), tumor tissues (pH 6.5–7.2), lysosomes (pH 4.5–5.0), and the gastrointestinal tract (pH 6.2–7.9). Therefore, a pH-sensitive delivery system releases the drug only in its target area. Thus, the pH becomes an intrinsic stimulus for the targeted delivery of therapeutic agents [58,59,60].
The reduced cell cytosolic environment, as compared to body fluids, becomes a stimulus for redox-sensitive drug delivery systems to release active agents only in the cytosol and not in body fluids [61]. Tumor tissue suffers from hypoxia owing to impaired metabolism, and hypoxia-sensitive drug delivery systems serve as excellent modulated activation (in this case, hypoxia) of targeted drug delivery systems [62]. Magnetic nanoparticles loaded with therapeutic agents can be directed by an external magnetic field to a specific organ and can be stimulated to release the drug only at that location—an ideal example of an externally controlled/modulated drug delivery system [63].
Recent advances in microfabrication have made it possible to develop controlled-release systems for drug delivery. Two types of delivery devices are most popular: microvessels and micro-/nanofluidic ones. Much promise is held by controlled-release chip-based drug delivery systems [64].
4 Selection of Carriers for Delivery Systems
Most low-molecular-weight drugs used in clinical practice have high hydrophobicity and low bioavailability. Therefore, nanotechnology development can increase drug bioavailability.
Nanoscale systems, depending on the material from which they are made, can be divided into lipid, polymeric, inorganic, peptide, and viruslike [65]. Lipid-based formulations include liposomes, self-assembled colloidal structures consisting of lipid bilayers surrounding an aqueous core, and solid lipid nanoparticles. Among polymeric nanosystems designed by using natural or synthetic polymers, one can find several structures used in pharmaceuticals and medicine (e.g., nanoparticles, nanocapsules, micelles, and dendrimers), whereas inorganic nanocarriers such as gold, silica, and silver are used in both imaging and therapy [66, 67]. Figure 5 shows structures composed of a core material containing both a hydrophobic and a hydrophilic region, surface modifiers (biocompatibility modifiers and targeting moieties), and a therapeutic payload [68].

Generalized nanomedicine carrier. Structure composed of a core material containing both a hydrophobic and a hydrophilic region, surface modifiers (biocompatibility modifiers and targeting moieties), and a therapeutic payload [68]
Nanocomposites include polymers or lipid-based systems to form bubbles and physically or chemically trap the drug in them. Nanocomposites solubilize drugs and impart stability to them, increasing their circulation in the bloodstream and accumulation in the target organs [65].
In the selection of nanocarriers, a key part is played by drug optimization through the improvement of the water solubility of hydrophobic drugs [69, 70] and the stabilization of easily degradable compounds [71, 72]. In addition, one should consider the role of nanosystems in ensuring drug retention in tissues, protection of drugs from enzymatic degradation, and enhancement of cellular absorption and high-precision delivery [73]. All aspects have a substantial impact on treatment efficacy [74].
4.1 Carriers: Their Advantages and Disadvantages
The use of a carrier is extremely important to ensure drug efficacy and safety. Some substances cannot withstand the journey through the digestive or circulatory system and will be inactive by the time they reach the disease area. Others can be hazardous to healthy tissues, so their potential must be unlocked at the disease site. Solid dosage forms (tablets, capsules, etc.) account for up to 90% of medications used as research objects and are the most popular and convenient dosage forms. The choice of a carrier for dosage forms containing high-molecular-weight components as an active pharmaceutical substance or an additive requires a comprehensive approach, with account taken of the effect of the carrier on the physicochemical and adsorptive properties of the active material. The following requirements are imposed on carriers:
-
(a)
They should not interact with the drugs.
-
(b)
They should ensure drug stability for the required period and contain the required amount of the dispersed phase in the dispersion medium.
-
(c)
They should be nontoxic.
-
(d)
They should ensure the optimal therapeutic effect of the drug [75].
All carriers can be divided into three types: artificial, natural (biological), and hybrid. They can also be divided into three generations on the basis of their chronological appearance and physical size:
-
(a)
First-generation drug carriers (microcapsules, microspheres; size, 1–2 microns), produced as various dosage forms: powders, tablets, capsules, suspensions, emulsions, and so on. In pharmaceutical technology, microencapsulation began to be used in the late 1950s–early 1960s.
-
(b)
Second-generation drug carriers (nanocapsules, nanoparticles, nanotubes, dendimers, liposomes, polymer conjugates, etc.; size, <1 micron), which are collectively called colloidal carriers. Nanocapsules are intended for parenteral administration near a specific organ or tissue. The size is less than 100 nm [76].
-
(c)
Third-generation drug carriers, involving the use of nanotechnology, biotechnology, genetic engineering, and other fields. These include antibodies [77,78,79,80], glycoproteins [81, 82], cellular markers and receptors [3, 83,84,85,86], viruses and oncolytic viruses [87, 88], and other materials. Third-generation carriers open new possibilities for high levels of selective action and targeted drug delivery. Different types of stimuli-responsive nanocarriers are shown in Fig. 6 [89].

Different types of stimuli-responsive nanocaries [89]
Nanoscale drug delivery agents (1–250 nm) can change the therapy of various diseases, owing first of all to the increased ability to overcome biological barriers, increased half-life, and targeted drug delivery. Currently, several nanotechnological platforms are used for targeted drug delivery that differ in their physical and chemical structure. These include polymersomes, nanoshells, dendrimers, polymer micelles, and polymer–drug conjugates [55, 90, 91]. Liposomes as a dosage form have numerous advantages. The most significant of them include (a) the unique ability to deliver drugs into cells, biocompatibility; (b) the absence or the minimal possibility of allergic reactions (invisible liposomes for the immune system—stealth liposomes); (c) protection of drugs from degradation in the body; (d) improving the pharmacokinetic profile of drugs and increasing their therapeutic efficacy; and (e) reduction of the general toxic effect on the body, versatility and the ability to modify the liposome structure so as to achieve specific properties [92, 93]. The main difficulty with liposome design is related to their in vitro and in vivo stability, which varies with nanoparticle size, surface charge, lipid composition, number of lamellae, and surface modifications (with ligands or polymers). All these factors make liposomes difficult to manufacture and use [94]. Liposomes can hardly penetrate tissues with severe microcirculation disorders; they can block pulmonary capillaries (leading to microvascular embolism), can cause an increase in blood glucose levels, and lead to impaired blood clotting and cholesterol metabolism. Nanosized polymer particles (nanocontainers, nanospheres, dendrimers) are loaded with drugs either by drug absorption or by conjugation with side acid groups and end of groups. For example, OH groups of polyethylene glycol are associated with vector molecules.
Among various drug nanocarriers, carbon carriers such as fullerenes, graphene and its oxide, carbon nanotubes, and detonation nanodiamonds are considered very promising. The prospects for the use of these carriers are due to their physicochemical characteristics, the possibility of targeted modification of the surface, and variation in particle size. Two main approaches have been adopted for modifying the surface of carriers: adsorption and covalent grafting.
For DDS as a carrier, it was proposed to use not graphene itself but its oxide. This is due to the possibility of chemical modification of its surface. Because anticancer drugs are usually water-insoluble, graphene oxide overcomes this disadvantage. It has been proposed as a platform for doxorubicin, camptothecin, cisplatin, etc. Graphene oxide can be used to track tumor angiogenesis. However, when graphene toxicity studies were carried out, it turned out that graphene and its derivatives can be dangerous to biological systems. Therefore, the use of graphene and its oxide as a carrier for delivery systems requires additional toxicological studies. There are two known ways of using carbon nanotubes as a carrier including (a) the attachment of drug molecules to the outer surface of the tube; (b) the placement of drug molecules inside the tube, after which it acts as a container.
Carbon nanotubes, like other pure carbon materials, have an inert hydrophobic surface, as a result of which they are poorly dispersed in water, stick together, and form very large aggregates. However, data on the toxicity of carbon nanotubes themselves have been obtained, and their further application requires additional studies. Inorganic materials such as gold, iron, and silicon dioxide are used in nanoparticle synthesis that has various applications in biology and medicine. The synthesis of inorganic nanoparticles makes it possible to control the size, structure, and geometric shape of the final product. Colloidal gold nanoparticles are also considered a promising platform for making drug delivery systems. They are easily synthesized and biocompatible, and their surface can be functionalized. For example, gold nanoparticles (AuNPs), which are the most studied, are used in various forms such as nanospheres, nanorods, nanostars, nanoshells, and nanocells [95]. In addition, inorganic nanoparticles have unique physical, electrical, magnetic, and optical properties owing to the properties of the base material itself. For example, AuNPs have free electrons on their surface that continuously vibrate at a frequency that depends on their size and shape, which gives them photothermal properties [96]. AuNPs are also easily functionalized, which makes it possible to give them additional properties and delivery capabilities [95]. Iron oxide is another widely studied material for the synthesis of inorganic nanoparticles, and the resulting nanoparticles based on it make up the majority of FDA-approved inorganic nanomedicines [97]. Other common inorganic nanoparticles include calcium phosphate and mesoporous silica nanoparticles, which have been successfully used for gene and drug delivery [98, 99]. Owing to their unique properties, inorganic nanoparticles have good biocompatibility and stability and fill a niche where properties are required that are unattainable for organic materials. However, their clinical use is limited because of their low solubility and toxicity, especially in formulations containing heavy metals [100].
Polymeric nanoparticles can be synthesized from natural or synthetic materials, as well as from monomers or preformed polymers, providing a wide range of possible structures and characteristics. When choosing polymers as drug carriers, investigators rely on their physical properties, which determine the release rate of drugs. Thus, it is preferable to use hydrophobic polymers that decompose to small water-soluble molecules, which ensures their rapid clearance. When hydrophilic biodegradable polymers are used, owing to their high affinity for water, when chemical bonds are broken, rather large molecules pass into the environment, which complicates their participation in metabolism. The most common polymers used as platforms for building delivery systems are polyesters. They are biodegradable, biocompatible, and easily degraded owing to the hydrolysis of the ester bond. Typically, polyglycolic, polylactic acids, and copolymers of lactic and glycolic acids are used.
Despite being biocompatible and biodegradable in vivo, polymers have disadvantages such as lack of stability, sterilization difficulties, and scaling up problems. One of the modern variants of polymeric nanocarriers is dendrimers. The most commonly used are polyamidoamine dendrimers. The unique properties of dendrimers are due to the radial symmetry of their molecules, highly ordered structure, and tree-like branching. They have sizes of 1.1 nm and above, depending on the number of stages of synthesis, and a strictly fixed molecular weight.
The drug is encapsulated inside the dendrimer molecules (Fig. 7), which increases its solubility and stability under physiological conditions. In addition, drugs can be grafted to the surface of dendrimers. This makes the use of dendrimers as carriers for targeted transport promising.

Schematic representation of the internal cavities of a drug-filled dendrimer (https://en.wikipedia.org/wiki/Dendrimer)
From many types of nanosized particles and materials studied, cyclodextrins (CDs), natural cyclic oligosaccharides, and molecular nanocontainers have attracted researchers’ attention. CDs were discovered in 1891 by M.-A. Villiers, and the first detailed description of their preparation from starch was given in 1903. F. Kramer in 1954 was the first to show that CDs can form molecular inclusion complexes with a wide range of substrates of the “guest-host” type, in which CD molecules with their internal hydrophobic cavity play the role of hosts. The formation of inclusion complexes by CDs can radically change the physicochemical and biological properties of the included molecules, which has led to their demand as an object of modern chemical and pharmaceutical technologies. CDs can increase the solubility of poorly soluble drugs in water, as well as enhance the penetration of drugs through biological membranes [101]. CDs are of great interest because they are non-toxic [102, 103].
The disadvantage of many drugs based on CDs is a rapid decrease in their concentration in plasma blood owing to metabolic destruction in the body itself, which necessitates an increase in dose loads and, accordingly, increases the likelihood of side effects. Cases of damage to the auditory nerve have been described [104], and nephrotoxic effects have been observed after CD application [105,106,107].
Special attention should be paid to the direction in which natural containers are used as carriers—blood cells of humans or animals. They have the following advantages: a wide range of delivery targets, a decrease in immunogenicity and an increase in drug circulation time in vivo, biocompatibility, and controlled release of drugs. In addition, owing to the specific receptors on the membrane surface, they can be used in targeted delivery. Delivery systems based on blood cells are divided into systems using erythrocytes, platelets, and leukocytes.
The disadvantage of erythrocytes as carriers in delivery systems is their large volume (90 μm3). Because of this, the erythrocyte cannot penetrate the tissues, and the area of its delivery is limited only to the affected foci accessible to the bloodstream.
Platelets have a short lifespan (7–10 days), but despite this, they are also considered promising drug carriers. Platelets specifically target the sites of damage and contain many biologically active proteins, and during pathological processes, these proteins are released from platelets by exocytosis. Owing to this feature, drugs are released locally at the sites of platelet activation.
Leukocytes, unlike other blood cells, have adhesive properties. In inflammatory conditions, leukocytes can adhere to the endothelium, which contains the protein E-selectin, which is synthesized in response to inflammation. Thus, leukocytes can be used in antitumor therapy owing to their similarity in adhesive properties to tumor cells. Leukocytes are used as carriers for the targeted transport of antibiotics to the site of inflammation owing to the slow release of the drug from cells in the vascular bed.
The most common forms of polymer nanoparticles are nanocapsules (cavities surrounded by a polymer membrane or shell) and nanospheres (solid matrix systems). Within these two broad categories, nanoparticles are divided into polymersomes, micelles, and dendrimers.
In general, polymeric nanoparticles are ideal candidates for drug delivery, because they are biodegradable, water-soluble, biocompatible, biomimetic, and storage stable. Their surfaces can easily be modified for additional targeting [104], which allows them to deliver drugs, proteins, and genetic material to target tissues, making them useful in oncological medicine, gene therapy, and diagnostics. However, the disadvantages of polymer nanoparticles include an increased risk of particle aggregation and toxicity [108].
5 Polymer Carrier Micelles in Delivery Systems
Polymeric nanoparticles are nanosized drug delivery systems characterized by a core–shell structure that results from the self-assembly of amphiphilic block copolymers in an aqueous solution. In a dilute aqueous solution, the amphiphilic molecules exist separately, and the amphiphiles act as surfactants, lowering the surface tension at the air–water interface. If more chains are added to the system, adsorption at the interface becomes higher until unimer aggregation occurs owing to the saturation of the solution volume. At this stage, the critical micelle concentration (CMC) is reached. Thus, this variable is defined as the minimum concentration of polymers in solution leading to the formation of micelles. According to this, micelles are stable at a concentration of polymer chains higher than the CMC, whereas disassembly of the system is observed after dilution below the CMC [109, 110].
Owing to the presence of various functional groups (hydroxyl, carboxyl, and amino groups) in their molecular chains, micelles can be chemically altered and modified with side chains. The incorporation of hydrophobic drugs into the micellar core can further enhance the stability of the micelles. This is an important feature for injectable biomedical applications.
Polymeric micelles are of interest for drug delivery, because the hydrophobic drug loaded into the micelle core is protected by an external hydrophilic corona. This corona hinders micelle removal by the mononuclear phagocytic system. Because of this system, the stability of the active substance is increased, and its circulation time in vivo is extended.
The driving force behind the formation of micelles is a decrease in the free energy of the system owing to the removal of hydrophobic segments from the aqueous medium with the formation of a micellar core. An important factor in drug delivery is the relative thermodynamic (possibility of disassembly) and kinetic (disassembly rate) stability of the substance. The relatively small size of micelles allows them to accumulate passively in neovascularized or poorly vascularized tumors, which may lead to reduced systemic toxicity [111, 112].
Structural features of polymeric micelles (hydrophilic shell) help to avoid unexpected loss of drugs from serum components and prevent rapid elimination of drugs from systemic circulation [113, 114]. Ideal polymeric micelles are expected to reduce the toxicity of therapeutic compounds.
5.1 Brief History of Obtaining Polymer Micelles
The term “micelle” was first introduced by McBain in 1913. According to modern concepts, micelles are aggregates of long-chain amphiphilic molecules or surfactant ions that spontaneously appear in their solutions at a certain concentration. The last property essentially depends on the nature of the polar group and, especially, on the length of the molecular chain [115].
In the past decade, polymer micelles have been studied in nanotechnologies, biotechnologies [116], biomedical engineering [117], and environmental technologies [118]. In biomedicine, especially in the detection and treatment of cancer, polymer-based micellar systems have been widely studied owing to their success at the clinical level. In 1984, polymer micelles (~200 nm) were first used by Bader et al. [119] to deliver anti-cancer molecules.
The term “micellar nanoparticles” has been mentioned in publications since the mid-1990s [120], especially in transdermal therapy [121]. Micellar nanoparticles are used in veterinary medicine; for example, Scott-Moncrieff et al. [122] showed that whereas insulin in combination with mixed micelles is completely absorbed in dogs, its bioavailability is much lower than in similar studies in rats. It has been shown [123] that the rate of insulin release from micelles can be controlled by changing the concentration of glucose.
Comparative studies of the biodynamic parameters of the aqueous form of diminazene and diminazene enclosed in water-dispersed micelles in ram erythrocytes and sheep blood plasma showed that surfactants improve the intracellular penetration of the active substance owing to the interaction with the cell membrane [124, 125].
Vail et al. [126] found the efficacy and safety of water-soluble micellar paclitaxel (Pascal Vet), as compared with free lomustine, for the treatment of inoperable grade 2 and 3 mast cell tumors in dogs.
Oral intake of natural vitamin E, contained in micelles in racehorses, effectively increased the concentration of α-tocopherol in blood plasma, as compared with the control [127]. Another study by the same authors in adult and weaned piglets showed that oral administration of micellized natural vitamin E to sows (75 mg/day) and piglets (1.7 mg/day) altered the fatty acid profile in piglet tissues and improved their oxidative status [128]. Micelles were also used for the oral delivery of vitamin B12 [129].
The bioavailability and pharmacokinetic parameters of tilmicosin (a semi-synthetic antimicrobial agent) were studied in broiler chickens by oral administration by using various micellar nanoparticles (solid lipid nanoparticles, nanostructured lipid carriers, and lipid core nanocapsules). Al-Qushawi et al. [130] showed that lipid nanoparticles improved the bioavailability and pharmacokinetic parameters of tilmicosin in broiler chickens. Troncarelli et al. [131] described the importance of various nanoparticles as antimicrobial agents in veterinary medicine.
Micellar nanoparticles have a greater loading capacity and excellent stability and can be considered safer for parenteral administration.
5.2 Basic Principles for the Preparation of Polymer Micelles
Drugs may be encapsulated depending on the preparation method and the drug physicochemical characteristics. The simplest preparation method is direct dissolution. Other methods include dialysis, evaporation of the emulsion with a solvent (or co-solvent), and pouring of the solution, followed by hydration of the film. The choice of method depends on the polymer characteristics and the drug, as described in [132, 133].
Because the properties of micelles (such as polarity and degree of hydration) are not uniform inside the carrier, a drug, depending on its properties, can be placed either close to the surface or in the inner core [134]. Typically, hydrophobic drugs are loaded and placed in the inner core. In certain cases, a drug may also be covalently linked to a polymer (polymer–drug conjugate).
The hydrophilic part usually consists of PEG, but other polymers can be used: poly(vinylpyrrolidone), poly(acryloylmorpholine), or poly(trimethylene carbonate). The hydrophobic segment may consist of poly (propylene oxide) or polyesters (poly (ε-caprolactone) or polymers and copolymers of glycolic and lactic acids [109].
Most examples of clinically approved polymer micellar drugs or polymer micellar drugs in clinical development are in the field of cancer therapy. There have been reports of preclinical studies using small molecule drugs in polymeric micelles for the treatment of autoimmune, cardiovascular, skin, and eye diseases; dementia; microbial infections; pulmonary arterial hypertension; and spinal cord injury and wound healing. Polymer micelles significantly improve drug solubility, stability, and bioavailability.
5.3 Stability of Polymer Micelles
Drug release from polymeric micelles can occur either by drug diffusion from intact micelles or by disassembly of micelles. Micelles must have a good thermodynamic and kinetic stability to avoid uncontrolled drug release [135,136,137]. Therefore, several physicochemical strategies have been proposed to stabilize the encapsulated drug in the micellar core to avoid the rapid disaggregation of the system [138].
It is known that the CMC can be decreased by increasing the length of the hydrophobic part of the unimer [135, 139]. Block copolymers conjugated with lipid molecules were synthesized. Other strategies include hydrophobic block functionalization, cross-linking of the micelle core, or formation of a conjugate between the polymer and the drug [140].
The structural stability of micelles should be investigated under relevant conditions, because proteins of plasma or intracellular fluids can be absorbed on the surface of micelles, which leads to the formation of the so-called protein corona. Such a corona partially masks the functional groups of the outer shell, modifying the physiological response of the nanocarriers [141, 142].
Serum proteins play a key role in the stability of micelles by promoting their degradation or aggregation [143]. Micellar disaggregation can be observed as a result of interaction with mucus, epithelium, lipids of the stratum corneum, and sebum [144, 145].
The use of hydrophilic blocks with “anti-fouling” properties reduces the binding of serum components (serum proteins and the complement system) and protects the encapsulated drug from loss of cargo in the circulatory system. Polymer micelles should be designed in such a way as to resist their excretion from the body owing to the adsorption of plasma proteins and/or activation by the reticuloendothelial system (RES) [146]. For imparting “anti-fouling” properties to polymer micelles, several hydrophilic blocks were added to the structure of block copolymers, as described in [147]. It has been established that the physicochemical properties of hydrophilic polymers (molecular weight and surface density) are closely related to the stability, system circulation time, and biodistribution of polymer micelles in vivo [148].
The possibility of regulating drug release depending on the pH medium or under the influence of ultrasound, magnetic field, or temperature changes has been shown [149].
The amount of drugs loaded into micelles also can affect the stability, morphology, and size of micelles in an aqueous solution. Hydrophobic interactions between drugs and the hydrophobic block of amphiphilic block copolymers are some of the main factors of drug solubilization in polymer micelles. Additional molecular interactions that exist in the core, such as hydrogen bonds, are no less significant, because they can enhance the molecular interactions between the polymer and the drug in the core.
5.4 Polymeric Micelles and Questions of Kinetics and Biodynamics of Drugs
The purpose of studying micelle pharmacokinetics is to quantitatively characterize micelle absorption, distribution, and elimination (metabolism and excretion). Pharmacokinetic data are needed to establish a relationship that “concentration effect” is less than a “dose effect.” The results of pharmacokinetics help to choose an approximate dosing regimen.
As mentioned above, micelles are nanosized colloidal particles with a hydrophobic interior (core) and a hydrophilic surface (shell). The polymer micelle consists of two separate regions: an inner hydrophobic region of the polymer chain (central region) and an outer region of well-solvated hydrophilic polymer chains (crown or shell region), which imparts colloidal stability to the system [150, 151]. Drugs and contrast agents can either be placed into the micelle lipid core or covalently bond to its surface. Micelles are somewhat smaller (about 50 nm) than liposomes. To ensure the long-term circulation of micelles in the bloodstream, various modifications of their shells have been proposed, making them thermodynamically stable and biocompatible [152]. The kinetics of drug release from polymer micelles is highly dependent on many factors: the size of the micelles, the length, crystallinity, and polarity of the hydrophobic block, as well as the compatibility between the micelle core and the drug molecules. The larger the micelles, the slower is the release of the drug [153]. Long hydrophobic blocks cause a slow drug release rate, and the closer is the temperature to room temperature, the higher is the viscosity of the medium and the slower is the release [154]. A larger core diameter may result in higher core crystallinity, which slows drug release [155]. In [156, 157], it has been established that the greater is the poly(γ-benzyl L-glutamate (PBLG) content in the copolymer, the larger is the micelle particle size. It has been found that the length of the hydrophobic block of a micelle is not significant in measuring the release rate. Also, the higher is the hydrophobicity, the slower is the release rate [158, 159]. The rate of drug release from micelles decreases with increasing drug/polymer ratio at a constant copolymer concentration [160].
Drug release is highly dependent on where the drug molecules are located [161]. If the drug is located predominantly in the crown, then the length of the core of the forming block, the micelle size, and the molecular volume of the drug are less important for determining the release rate [162]. The amount of drug loaded into the micelle core is the determining factor for its release rate. Gref et al. [163] observed a faster release of lidocaine if it was dispersed in the micelle cores. In [156, 157], it was shown that the release of adriamycin and clonazepam from micelles was slower at higher concentrations of the respective drugs in the micelle core. At high loads of lidocaine, crystallization of the drug in micelles is observed [153]. Drug release is possible only after the drug dissolves and diffuses into the external solution, so crystallization slows down drug release [164]. So far, there has been no clear picture of how the drug is released from micelles and freely diffuses from the core of an intact micelle, or release is observed after rupturing of the micelle. Some researchers have reported a biphasic release profile [111, 155]. Studies to analyze the release of drugs in vitro in an environment that mimics physiological conditions have been conducted [153, 154, 156, 165,166,167]. The decisive factors influencing the kinetics of drug release from a polymeric micelle include (a) the stability of micelles; (b) their compatibility with the main drug; (c) the molar volume of drugs; and (d) physiological conditions. Mechanical forces acting on polymer micelles in veins and small capillaries can also have a strong effect on drug release rates [111].
5.5 Preclinical and Clinical Trials Using Polymeric Micelles
Preclinical studies are mandatory for all micellar medicinal products, regardless of whether the original or known pharmacological substance is used to make them [168, 169]. When combining several pharmacological substances in one dosage form, the toxicity of the combination as a whole and of each ingredient separately is studied, if it has not been previously approved for use in medical practice [169, 170]. The strategy for the preclinical testing of micellar medicinal products should be a three-tiered approach, which includes (a) physicochemical characteristics of the micellar drug; (b) interaction of medicinal substances in micellar form at the cellular level in vitro (rate of cellular uptake and intercellular persistence); and (c) safety assessment of a micellar medicinal product in laboratory animals [171].
The manifestation of the toxic properties of polymer micelles largely depends on the route of their entry into the body [172,173,174]. Therefore, the acute toxicity of polymer micelles can be judged only from the results of studies using the route of administration that is supposed to be used in clinical trials. On the first day after polymer micelles with the drug are administered, animals should be under continuous observation. The total duration of acute toxicity observation should be at least 30 days. The cumulative properties of pharmacological substances contained in polymer micelles are evaluated in chronic toxicity studies. Three doses of polymer micelles are used in chronic toxicity experiments. Doses are calculated by the amount of active substance in the composition of the dosage form. The route of administration is similar to the clinical one. If multiple routes of administration are recommended, then all routes used should be evaluated [169, 171, 174,175,176]. Tests to assess the functional state of phagocytes, antioxidant system, expression of inflammatory markers, level of oxidative processes, and related damages are also used [177]. Siegrist et al. [171] recommend the following parameters for assessing the damaging effect of potential drugs contained in polymer micelles: complement activation, platelet aggregation, hemolysis, oxidative stress, cell viability, phagocytosis, inflammation, and DNA damage. Mandatory assessment of the damaging effect should be subject to the structure and function of the nervous system, kidneys, and liver as possible main targets of the toxic action of polymer micelles [178]. For the first phase of clinical trials in humans or target animals, on the basis of preclinical studies, the maximum recommended starting dose is calculated [177, 178].
5.6 Prospects for the Use of Micelles in Diseases, Including Cancer
Polymeric micelles are of interest as carriers of hydrophobic drugs. In particular, micelles can be used for the parenteral administration of drugs such as amphotericin B, propofol, and paclitaxel [179]. Like liposomes, micelles can be used for targeted drug delivery to target cells. This is achieved by attaching pH-sensitive elements to the surface of micelles.
Compared to other nanocarriers, polymer micelles are smaller, have a simple sterilization preparation process, and have a good solubilization property. The latter, unfortunately, is associated with lower stability in biological fluids. Especially difficult is the study of micelle interaction with the biological environment, which is necessary to predict the drug behavior after administration in vivo.
The use of nanoplatforms can lead to both therapy and diagnostics, hence the term “theranostics” [180]. The polymer micellar base was first used for anti-cancer drugs by Professor Kataoka in the late 1980s or early 1990s to increase the accumulation of drugs in tumor tissues. The size of micelles can be adjusted in the diameter range of 20–100 nm, ensuring that they do not pass through the walls of the vessels, so a reduction in the incidence of side effects can be expected [181].
The successful development of theranostic nanoplatforms requires the concomitant development of quantitative imaging techniques that will enable early disease detection and measure therapeutic response. Depending on the ratio of phospholipids and hydrophobic compounds, the micelle core can be adapted to accommodate several single hydrophobic molecules/nanoparticles. In [180], the encapsulated drug showed sustained release from the micellar core for 7 days. The biocompatibility of the micellar system was confirmed by cell viability analysis. The great potential of these theranostic micelles has been established for the imaging and therapy of various diseases, including cancer. Three widely studied classes of block copolymers are characterized by the presence of hydrophobic blocks and are poly(propylene oxide), poly(L-amino acids), and polyesters. These classes of block copolymers have been applied to some complex molecules in the pharmaceutical industry. Polymeric micelles can reduce toxicity, improve delivery to desired biological sites, and improve the therapeutic efficacy of active pharmaceutical ingredients [182].
Numerous types of biodegradable and synthetic block copolymers with different architectures (diblock, triblock, and graft copolymers) and physical properties (charged and neutral) are used to obtain various nanostructures such as vesicles [183,184,185] and spherical and rod-shaped micelles for targeted drug delivery [111].
Most polymeric micelles are designed to deliver hydrophobic anticancer drugs, which often have to be infused with surfactants and organic solvents. When administered systemically, such low-molecular-weight antitumor agents are distributed throughout the body, reducing the effective dose in target tissues and causing intoxication. In addition, the rapid clearance of anticancer drugs from the body leads to repeated administration of an effective drug concentration, which increases chronic toxicity and leads to acquired drug resistance. Thus, polymer micelles are much more beneficial for stabilizing drugs under aqueous conditions, protecting these agents inside their core from the external environment, circulating stably in the bloodstream, and selectively accumulating in tumors, where they can release drugs in a programmed manner [186].
Polymeric micelles can be designed to respond to specific stimuli to achieve spatiotemporal control over their functions, such as reporting on the conditions of their environment, releasing their cargo, and exerting therapeutic effects. Such signals may be endogenously present in the body and enhanced in affected tissues. For example, compared to healthy tissues, the tumor microenvironment provides unique stimuli for selective micelle activation, including acidic pH between 6.5 and 7.2 through aerobic glycolysis and lactate production and an altered redox potential. Moreover, endosomal/lysosomal pH (pH 6.5–4.5), enzymes, adenosine triphosphate (ATP), intracellular ROS, and redox potential can additionally be used to control the action of micelles inside cells [186].
The promising therapeutic potential of micelles loaded with oligonucleotides, especially for cancer therapy (particularly for RNAi-based cancer therapy), has been described in [187]. Shen et al. [188] developed an oligonucleotide delivery system using pH-sensitive polymeric micelle-like nanoparticles and showed that this system effectively delivers oligonucleotides of various lengths (20,100 bp) into cells and has significant potential for cancer treatment.
Micellar forms have been proposed to create new forms of drugs:
-
1.
Anticancer drug
Doxorubicin [89]
Paclitaxel [189]
Camptothecin [68]
Carboplatin [68]
Docetaxel [190]
Cisplatin [191]
Methotrexate [192]
Ethaselen [193]
5-fluorouracil [194]
Indisulam [195]
Disulfiram [196]
Amifostine [197]
Cyclosporine [198]
Gemcitabine [199]
-
2.
Hormones
Estradiol [190]
Dexamethasone [200]
-
3.
Anti-infective (antiviral, antibacterial, antifungal, antiparasitic) drugs
Adamantane [201]
Ciprofloxacin [202]
Amphotericin B [203]
Ivermectin [204]
Also, small organic molecules, siRNA, aptamers, peptides, carbohydrates, and antibodies have been proposed [64].
5.7 Current Status and Prediction
Among the nanoparticles used in pharmaceuticals, polymer micelles have clear advantages, because they contain amphiphilic polymers that self-assemble in an aqueous medium. These amphiphilic polymers are built with different polymer blocks. These blocks can be selected depending on the stability of the hydrophobic/lipophilic balance, size, ability to use the drug, the ability of micellization, and stability in the systemic circulation. The nano-size of micelles allows more efficient exit through the vasculature, as compared with other drug delivery systems. Hydrophilic polymer coating will remain unrecognized by the reticuloendothelial system during circulation [205]. Therefore, micelle-based nanoparticles can be considered a system with unique characteristics, as compared with other nanocarriers. These characteristics include their smaller size, which allows passive targeting of target organs (even poorly permeable ones) [206] and more efficient cell internalization [141]; good solubilizing properties of the hydrophobic compounds assimilated into the lipophilic core; and increased circulation time in blood [207, 208]. Micelles are characterized by simple preparation and high scalability, as compared with polymeric nanoparticles and liposomes requiring complex, time-consuming, and costly manufacturing procedures [141, 208]. Despite the disagreement among scientists about the degree of accumulation of nanodrugs in tumors [209], it is important to point out that several clinically successful drugs, such as doxil and abraxane, are used to provide therapeutic assistance and effectively deliver a sufficient amount of active drugs to target tissues [210].
In the past few decades, polymer micelles have emerged as one of the most promising nanodelivery systems for cancer treatment. Micelles on PEG–PLA are well studied owing to their excellent biodegradability and biocompatibility. Genexol-PM is already approved for the treatment of breast cancer in South Korea [211]. With the progressive development of cancer therapies, the PEG–PLA micelles are increasingly used in combination with photodynamic, photothermal, gene, and immune therapy. The controlled delivery of therapeutic agents is an actively developing field and depends on the uniqueness of the tumor, the microenvironment around the tumor, and the combination with different therapeutic loads.
Nevertheless, the safety of new concepts being developed should not be forgotten, because there are data that are related not only to the toxicity of micelles [212] but also to their ability to cause neuroendocrine disorders [213]. Thus, polymeric micelles still need to be carefully studied in animal models before they can be recommended for human treatment.
6 Conclusion
More than 100 years have passed since the date of the emergence of the idea of targeted drug transport. Today, in pharmacy chains in different countries, we can buy drugs with targeted delivery ability. Such drugs are still few, but there is no doubt that their number will constantly grow. Most of the drugs that have reached the pharmaceutical market are made by using liposomal technology or CDs. An even greater number of drugs are at the stages of preclinical and clinical trials. The main trends in the development of research in the field of targeted drug transport are as follows:
-
(a)
Development of effective vectors.
-
(b)
Development of non-liposomal carriers with low toxicity. For example, the use of blood cells is attractive but requires the establishment of special units in medical institutions, because each patient needs an individual remedy.
-
(c)
Determination of the optimum between the loaded amount of drug carrier and the necessary therapeutic minimum.
-
(d)
Optimization of drug release methods.
-
(e)
A significant increase in the stability of drugs and an increase in their shelf life.
-
(f)
Cheaper drug production.
-
(g)
Development of legislative norms for the regulation, certification, and production of directed transport systems.
As already mentioned, there is considerable interest in the development of polymeric micelles capable of acting as true delivery vehicles for various potent drugs that are not found in therapeutic formulations owing to their water-insoluble, hydrophobic nature. Micelles have advantages as nanocarriers for drug delivery and treatments owing to their excellent physicochemical properties, drug loading and release capacities, facile preparation methods, biocompatibility, and tumor targetability.
Abbreviations
- ATP:
-
Adenosine triphosphate
- AuNPs:
-
Gold nanoparticles
- CDs:
-
Cyclodextrins
- CMC:
-
Critical micelle concentration
- DDS:
-
Drug delivery system
- DOX:
-
Doxorubicin
- PBLG:
-
Poly(γ-benzyl L-glutamate)
- PEG:
-
Polyethylene glycol
- PEG–PLA:
-
Polyethylene glycol–polylactide
- RES:
-
Reticuloendothelial system
References
Maehle AH. “Receptive substances”: John Newport Langley (1852–1925) and his path to a receptor theory of drug action. Med Hist. 2004;48(2):153–74. https://doi.org/10.1017/s0025727300000090.
Bosch F, Rosich L. The contributions of Paul Ehrlich to pharmacology: a tribute on the occasion of the centenary of his Nobel Prize. Pharmacology. 2008;82(3):171–9. https://doi.org/10.1159/000149583.
Tiwari G, Tiwari R, Sriwastawa B, Bhati L, Pandey S, Pandey P, Bannerjee SK. Drug delivery systems: an updated review. Int J Pharm Investig. 2012;2(1):2–11. https://doi.org/10.4103/2230-973X.96920.
Elvira C, Gallardo A, Roman JS, Cifuentes A. Covalent polymer-drug conjugates. Molecules (Basel, Switzerland). 2005;10(1):114–25. https://doi.org/10.3390/10010114.
Ferraris C, Rimicci C, Garelli S, Ugazio E, Battaglia L. Nanosystems in cosmetic products: a brief overview of functional, market. Regul Saf Concerns Pharm. 2021;13:1408. https://doi.org/10.3390/pharmaceutics13091408.
Zhu M, Whittaker AK, Han FY, Smith MT. Journey to the market: the evolution of biodegradable drug delivery systems. Appl Sci. 2022;12:935. https://doi.org/10.3390/app12020935.
Walsh EE, Frenck RW Jr, Falsey AR, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N Engl J Med. 2020;383:2439–50. https://doi.org/10.1056/NEJMoa2027906.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403–16. https://doi.org/10.1056/NEJMoa2035389.
Leung SSF, Mijalkovic J, Borrelli K, Jacobson MP. Testing physical models of passive membrane permeation. J Chem Inf Model. 2012;52(6):1621–36. https://doi.org/10.1021/ci200583t.
Leung SSF, Sindhikara D, Jacobson MP. Simple predictive models of passive membrane permeability incorporating size-dependent membrane-water partition. J Chem Inf Model. 2016;56(5):924–9. https://doi.org/10.1021/acs.jcim.6b00005.
Sharifian GM. Recent experimental developments in studying passive membrane transportof drug molecules. Mol Pharm. 2021;18(6):2122–41. https://doi.org/10.1021/acs.molpharmaceut.1c0.
Marsh D. Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys J. 2007;93(11):3884–99. https://doi.org/10.1529/biophysj.107.107938.
Gullingsrud J, Schulten K. Lipid bilayer pressure profiles and mechanosensitive channel gating. Biophys J. 2004;86(6):3496–509.
Lindahl E, Edholm O. Spatial and energetic-entropic decomposition of surface tension in lipid bilayers from molecular dynamics simulations. J Chem Phys. 2000;113:3882–93.
Yu W, Mookherjee S, Chaitankar V, Hiriyanna S, Kim JW, Brooks M, Ataeijannati Y, Sun X, Dong L, Li T, et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun. 2017;8:14716. https://doi.org/10.1038/ncomms14716.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. https://doi.org/10.1126/science.1247005.
Humphreys IR, Sebastian S. Novel viral vectors in infectious diseases. Immunology. 2018;153:1–9. https://doi.org/10.1111/imm.12829.
Lehrman S. Virus treatment questioned after gene therapy death. Nature. 1999;401:517–8. https://doi.org/10.1038/43977.
Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–58. https://doi.org/10.1038/nrg1066.
Ahi YS, Bangari DS, Mittal SK. Adenoviral vector immunity: its implications and circumvention strategies. Curr Gene Ther. 2011;11(4):307–20. https://doi.org/10.2174/156652311796150372.
Li L, Hu S, Chen X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities. Biomaterials. 2018;171:207–18. https://doi.org/10.1016/j.biomaterials.2018.04.031.
Zhang Y, Ren T, Gou J, Zhang L, Tao X, Tian B, Tian P, Yu D, Song J, Liu X, et al. Strategies for improving the payload of small molecular drugs in polymeric micelles. J Control Release. 2017;261:352–66. https://doi.org/10.1016/j.jconrel.2017.01.047.
Sung Y, Kim S. Recent advances in the development of gene delivery systems. Biomater Res. 2019;23:8. https://doi.org/10.1186/s40824-019-0156-z.
Li L, He ZY, Wei XW, Gao GP, Wei YQ. Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther. 2015;26(7):452–62. https://doi.org/10.1089/hum.2015.069.
Wang M, Cheng Y. The effect of fluorination on the transfection efficacy of surface-engineered dendrimers. Biomaterials. 2014;35(24):6603–13. https://doi.org/10.1016/j.biomaterials.2014.04.065.
Zinchenko A. DNA conformational behavior and compaction in biomimetic systems: toward better understanding of DNA packaging in cell. Adv Colloid Interf Sci. 2016;232:70–9. https://doi.org/10.1016/j.cis.2016.02.005.
Jeong GW, Nah JW. Evaluation of disulfide bond-conjugated LMWSC-g-bPEI as non-viral vector for low cytotoxicity and efficient gene delivery. Carbohydr Polym. 2017;178:322–30. https://doi.org/10.1016/j.carbpol.2017.09.048.
Vijayanathan V, Agostinelli E, Thomas T, Thomas TJ. Innovative approaches to the use of polyamines for DNA nanoparticle preparation for gene therapy. Amino Acids. 2014;46(3):499–509. https://doi.org/10.1007/s00726-013-1549-2.
Takahashi Y, Chen Q, Rajala RVS, Ma JX. MicroRNA-184 modulates canonical Wnt signaling through the regulation of frizzled-7 expression in the retina with ischemia-induced neovascularization. FEBS Lett. 2015;589(10):1143–9. https://doi.org/10.1016/j.febslet.2015.03.010.
Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, Buganim Y, Schroeder A, Langer R, Anderson DG. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31(7):653–8. https://doi.org/10.1038/nbt.2614.
Zhi D, Bai Y, Yang J, Cui S, Zhao Y, Chen H, Zhang S. A review on cationic lipids with different linkers for gene delivery. Adv Colloid Interf Sci. 2018;253:117–40. https://doi.org/10.1016/j.cis.2017.12.006.
Hong SJ, Ahn MH, Sangshetti J, Choung PH, Arote RB. Sugar-based gene delivery systems: current knowledge and new perspectives. Carbohydr Polym. 2018;181:1180–93. https://doi.org/10.1016/j.carbpol.2017.11.105.
Wang P, Lin L, Guo Z, Chen J, Tian H, Chen X, Yang H. Highly fluorescent gene carrier based on Ag-Au alloy nanoclusters. Macromol Biosci. 2016;16:160–7. https://doi.org/10.1002/mabi.201500235.
Ren S, Wang M, Wang C, Wang Y, Sun C, Zeng Z, Cui H, Zhao X. Application of non-viral vectors in drug delivery and gene therapy. Polymers. 2021;13:3307. https://doi.org/10.3390/polym13193307.
Ashfaq UA, Riaz M, Yasmeen E, Yousaf MZ. Recent advances in nanoparticle-based targeted drug-delivery systems against cancer and role of tumor microenvironment. Crit Rev Ther Drug Carrier Syst. 2017;34:317–53. https://doi.org/10.1615/CritRevTherDrugCarrierSyst.2017017845.
Liyanage PY, Hettiarachchi SD, Zhou Y, Ouhtit A, Seven ES, Oztan CY, Celik E, Leblanc RM. Nanoparticle-mediated targeted drug delivery for breast cancer treatment. Biochim Biophys Acta Rev Cancer. 2019;1871:419–33. https://doi.org/10.1016/j.bbcan.2019.04.006.
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3:133–49. https://doi.org/10.2147/ijn.s596.
Fu J, Wang D, Mei D, Zhang H, Wang Z, He B, Dai W, Zhang H, Wang X, Zhang Q. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J Control Release. 2015;204:11–9. https://doi.org/10.1016/j.jconrel.2015.01.039.
Evangelopoulos M, Yazdi IK, Acciardo S, Palomba R, Giordano F, Pasto A, Sushnitha M, Martinez JO, Basu N, Torres A, Hmaidan S, Parodi A, Tasciotti E. Biomimetic cellular vectors for enhancing drug delivery to the lungs. Sci Rep. 2020;10(1):172. https://doi.org/10.1038/s41598-019-55909-x.
Kaneda Y. New vector innovation for drug delivery: development of fusigenic non-viral particles. Curr Drug Targets. 2003;4(8):599–602. https://doi.org/10.2174/1389450033490740.
Shirinsky VP. Liposome. In: Glossary of nanotechnology and related terms. http://thesaurus.rusnano.com/wiki/article1075.
Norvaisas P, Kojic M, Milosevic M, Ziemys A. Prediction and analysis of drug delivery systems: from drug-vector compatibility to release kinetics. Scientifically speaking September 2013. Conference: CRS Newsletter for 41st annual meeting & exposition of the Controlled Release Society. At: 14–15, vol. 30(5).
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–22. https://doi.org/10.1126/science.1095833.
Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8(6):2101–41. https://doi.org/10.1021/mp200394t.
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Del Rev. 2011;63(3):136–51. https://doi.org/10.1016/j.addr.2010.04.009.
Jubeli E, Moine L, Vergnaud-Gauduchon J, Barratt G. E-selectin as a target for drug delivery and molecular imaging. J Control Release. 2012;158(2):194–206. https://doi.org/10.1016/j.jconrel.2011.09.084.
Maruyama K, Takizawa T, Yuda T, Kennel SJ, Huang L, Iwatsuru M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim Biophys Acta. 1995;1234(1):74–80. https://doi.org/10.1016/0005-2736(94)00263-o.
Mu LM, Ju RJ, Liu R, Bu YZ, Zhang JY, Li XQ, Zeng F, Lu WL. Dual-functional drug liposomes in treatment of resistant cancers. Adv Drug Deliv Rev. 2017;115:46–56. https://doi.org/10.1016/j.addr.2017.04.006.
Chen М, Ma Q-R, Lu W-L. Coupling methods of antibodies and ligands for liposomes. Biomaterial engineering. In: Liposome-based drug delivery systems. Berlin, Heidelberg: Springer; 2021. p. 143–66.
Yu Y, Wang ZH, Zhang L, Yao HJ, Zhang Y, Li RJ, Ju RJ, Wang XX, Zhou J, Li N, Lu WL. Mitochondrial targeting topotecan-loaded liposomes for treating drug-resistant breast cancer and inhibiting invasive metastases of melanoma. Biomaterials. 2012;33(6):1808–20. https://doi.org/10.1016/j.biomaterials.2011.10.085.
Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. https://doi.org/10.1021/nn900002m.
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. https://doi.org/10.1016/j.jconrel.2011.06.001.
Grobmyer SR, Moudgil BM, Grobmyer SR, Moudgil BM. Cancer nanotechnology: methods and protocols. Methods in molecular biology, vol. 624. Totowa: Humana Press; 2010.
Prokop A. Intracellular delivery: fundamentals and applications: fundamental biomedical technologies, vol. 5. Dordrecht: Springer Netherlands; 2011.
De Villiers MM, Aramwit P, Kwon GS. Nanotechnology in drug delivery. New York: Springer New York; 2009.
Torchilin VP, de Villiers MM, Aramwit P, Kwon GS. Nanotechnology for intracellular delivery and targeting. Nanotechnology in drug delivery. Springer New York: New York; 2009.
Rukkumani R, Jatinder V, Yakhmi. Chapter 8: Nanotechnological approaches toward cancer chemotherapy. In: Ficai A, Grumezescu AM, editors. Micro and nano technologies, nanostructures for cancer therapy. Elsevier; 2017. p. 211–40.
Beloqui A, Coco R, Memvanga PB, Ucakar B, des Rieux A, Préat V. pH-sensitive nanoparticles for colonic delivery of curcumin in inflammatory bowel disease. Int J Pharm. 2014;473(1–2):203–12. https://doi.org/10.1016/j.ijpharm.2014.07.009. (a)
Beloqui A, Solinís MÁ, des Rieux A, Préat V, Rodríguez-Gascón A. Dextran–protamine coated nanostructured lipid carriers as mucus-penetrating nanoparticles for lipophilic drugs. Int J Pharm. 2014;468(1–2):105–11. https://doi.org/10.1016/j.ijpharm.2014.04.027. (b)
Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: promising drug delivery systems for future clinics. Nanomedicine. 2016;12(1):143–61. https://doi.org/10.1016/j.nano.2015.09.004.
Duan C, Gao J, Zhang D, Jia L, Liu Y, Zheng D, Liu G, Tian X, Wang F, Zhang Q. Galactose-decorated pH-responsive nanogels for hepatoma-targeted delivery of oridonin. Biomacromolecules. 2011;12(12):4335–43. https://doi.org/10.1021/bm201270m.
Jin C, Bai L, Wu H, Song W, Guo G, Dou K. Cytotoxicity of paclitaxel incorporated in PLGA nanoparticles on hypoxic human tumor cells. Pharm Res. 2009;26(7):1776–84. https://doi.org/10.1007/s11095-009-9889-z.
Schleich N, Po S, Jacobs D, Ucakar B, Gallez B, Danhier F, Préat V. Comparison of active, passive and magnetic targeting to tumors of multifunctional paclitaxel/SPIO-loaded nanoparticles for tumor imaging and therapy. J Control Release. 2014;194:82–91. https://doi.org/10.1016/j.jconrel.2014.07.059.
Gao Y, Xie J, Chen H, Gu S, Zhao R, Shao J, Jia L. Nanotechnology-based intelligent drug design for cancer metastasis treatment. Biotechnol Adv. 2014;32:761–77. https://doi.org/10.1016/j.biotechadv.2013.10.013.
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–5. https://doi.org/10.1016/j.addr.2010.03.011.
Gadekar V, Borade Y, Kannaujia S, Rajpoot K, Anup N, Tambe V, Kalia K, Tekade RK. Nanomedicines accessible in the market for clinical interventions. J Control Release. 2021;330:372–97. https://doi.org/10.1016/j.jconrel.2020.12.034.
Crommelin DJA, van Hoogevest P, Storm G. The role of liposomes in clinical nanomedicine development. What now? Now what? J Control Release. 2020;318:256–63. https://doi.org/10.1016/j.jconrel.2019.12.023.
Webster DM, Sundaram P, Byrne ME. Injectable nanomaterials for drug delivery: carriers, targeting moieties, and therapeutics. Eur J Pharm Biopharm. 2013;84:1–20. https://doi.org/10.1016/j.ejpb.2012.12.009.
Chen Z. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol Med. 2010;16:594–602. https://doi.org/10.1016/j.molmed.2010.08.001.
Salehi B, Calina D, Docea AO, Koirala N, Aryal S, Lombardo D, Pasqua L, Taheri Y, Castillo CMS, Martorell M, Martins N, Iriti M, Suleria HAR, Sharifi-Rad J. Curcumin’s nanomedicine formulations for therapeutic application in neurological diseases. J Clin Med. 2020;9(2):430. https://doi.org/10.3390/jcm9020430.
Caddeo C, Manconi M, Fadda AM, Lai F, Lampis S, Diez-Sales O, Sinico C. Nanocarriers for antioxidant resveratrol: formulation approach, vesicle self- assembly and stability evaluation. Colloids Surf B: Biointerfaces. 2013;111:327–32. https://doi.org/10.1016/j.colsurfb.2013.06.016.
Kumar A, Ahuja A, Ali J, Baboota S. Curcumin-loaded lipid nanocarrier for improving bioavailability, stability and cytotoxicity against malignant glioma cells. Drug Deliv. 2016;23(1):214–29. https://doi.org/10.3109/10717544.2014.909906.
Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20:321–34. https://doi.org/10.1038/s41577-019-0269-6.
Patra JK, Das G, Fraceto LF, Campos EVR, Rodriguez-Torres MDP, Acosta-Torres LS, Diaz-Torres LA, Grillo R, Swamy MK, Sharma S, Habtemariam S, Shin H-S. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnol. 2018;16:71. https://doi.org/10.1186/s12951-018-0392-8.
Makarov VG, Makarova MN, Trofimets EI, Makarova MN, Katelnikova AE, Kryshen KL. Endotracheal administration to laboratory animals. Lab Anim Sci. 2020;2:76–81. https://doi.org/10.29296/2618723X-2020-02-09.
Trineeva OV, Halahakoon AJ, Slivkin AI. Cell carriers as systems of delivery of antitumor drugs. Drug Dev Reg. 2019;8(1):43–57. https://doi.org/10.33380/2305-2066-2019-8-1-43-57.
Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, Amaya A, Tang S, Driscoll K, Kimbung R, Kambhampati SR, Gueorguieva I, Hong DS. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79(4):673–80. https://doi.org/10.1007/s00280-017-3245-5.
Shimizu T, Seto T, Hirai F, Takenoyama M, Nosaki K, Tsurutani J, Kaneda H, Iwasa T, Kawakami H, Noguchi K, Shimamoto T, Nakagawa K. Phase 1 study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in Japanese patients with advanced solid tumors. Investig New Drugs. 2016;34(3):347–54. https://doi.org/10.1007/s10637-016-0347-6.
Terheyden P, Becker JC. New developments in the biology and the treatment of metastatic Merkel cell carcinoma. Curr Opin Oncol. 2017;29:221–6. https://doi.org/10.1097/CCO.0000000000000363.
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–60. https://doi.org/10.1038/nrd1632.
Camacho KM, Menegatti S, Mitragotri S. Low-molecular-weight polymer–drug conjugates for synergistic anticancer activity of camptothecin and doxorubicin combinations. Nanomedicine. 2016;11(9):1139–51. https://doi.org/10.2217/nnm.16.33.
He R, Yin C. Trimethyl chitosan based conjugates for oral and intravenous delivery of paclitaxel. Acta Biomater. 2017:355–6. https://doi.org/10.1016/j.actbio.2017.02.012.
Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8(4):1172–81.
Yang T, Choi MK, Cui FD, Lee SJ, Chung SJ, Shim CK, Kim DD. Antitumor effect of paclitaxel-loaded PEGylated immunoliposomes against human breast cancer cells. Pharm Res. 2007;24(12):2402–11. https://doi.org/10.1007/s11095-007-9425-y.
Onishi H, Suyama K, Yamasaki A, Oyama Y, Fujimura A, Kawamoto M, Imaizumi A. CD24 modulates chemosensitivity of MCF-7 breast cancer cells. Anticancer Res. 2017;37(2):561–5. https://doi.org/10.21873/anticanres.11349.
Weissferdt A, Fujimoto J, Kalhor N, Rodriguez J, Bassett R, Wistuba II, Moran C. Expression of PD-1 and PD-L1 in thymic epithelial neoplasms. Mod Pathol. 2017;30:826–33. https://doi.org/10.1038/modpathol.2017.6.
Gardeck AM, Sheehan J, Low WC. Immune and viral therapies for malignant primary brain tumors. Expert Opin Biol Ther. 2017;17(4):457–74. https://doi.org/10.1080/14712598.2017.1296132.
Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic virotherapy for the treatment of malignant glioma. Neurotherapeutics. 2017;14(2):333–44. https://doi.org/10.1007/s13311-017-0516-0.
Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release. 2008;126:187–204. https://doi.org/10.1016/j.jconrel.2007.12.017.
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–15. https://doi.org/10.1021/mp800051m.
Riggio C, Pagni E, Raffa V, Cuschieri A. Nano-oncology: clinical application for cancer therapy and future perspectives. J Nanomater. 2011;2011:1–10. https://doi.org/10.1155/2011/164506.
Sarfraz M, et al. Development of dual drug loaded nanosized liposomal formulation by a reengineered ethanolic injection method and its pre- clinical pharmacokinetic studies. Pharmaceutics. 2018;10:1–22. https://doi.org/10.3390/pharmaceutics10030151.
Alyautdin R, Khalin I, Nafeeza MI, Haron MH, Kuznetsov D. Nanoscale drug delivery systems and the blood–brain barrier. Int J Nanomedicine. 2014;9:795–811. https://doi.org/10.2147/IJN.S52236.
Sedighi M, Sieber S, Rahimi F, Shahbazi MA, Rezayan AH, Huwyler J, Witzigmann D. Rapid optimization of liposome characteristics using a combined microfluidics and design-of-experiment approach. Drug Deliv Transl Res. 2019;9(1):404–13. https://doi.org/10.1007/s13346-018-0587-4.
Yang W, Liang H, Ma S, Wang D, Huang J. Gold nanoparticle based photothermal therapy: development and application for effective cancer treatment. Sustain Mater Technol. 2019;22:e00109. https://doi.org/10.3389/fchem.2019.00167.
Wang J, Potocny AM, Rosenthal J, Day ES. Gold nanoshell-linear tetrapyrrole conjugates for near infrared-activated dual photodynamic and photothermal therapies. ACS Omega. 2020;5:926–40. https://doi.org/10.1021/acsomega.9b04150.
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle- based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33:2373–87. https://doi.org/10.1007/s11095-016-1958-5.
Huang KW, Hsu FF, Qiu JT, Chern GJ, Lee YA, Chang CC, Huang YT, Sung YC, Chiang CC, Huang RL, Lin CC, Dinh TK, Huang HC, Shih YC, Alson D, Lin CY, Lin YC, Chang PC, Lin SY, Chen Y. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci Adv. 2020;6(3):eaax5032. https://doi.org/10.1126/sciadv.aax5032.
Xu C, Nam J, Hong H, Xu Y, Moon JJ. Positron emission tomography - guided photodynamic therapy with biodegradable mesoporous silica nanoparticles for personalized cancer immunotherapy. ACS Nano. 2019;13:12148–61. https://doi.org/10.1021/acsnano.9b06691.
Arias LS, Pessan JP, Vieira APM, Lima TMT, Delbem ACB, Monteiro DR. Iron oxide nanoparticles for biomedical applications: a perspective on synthesis, drugs, antimicrobial activity, and toxicity. Antibiotics (Basel). 2018;7(2):46. https://doi.org/10.3390/antibiotics7020046.
Morrison PW, Connon CJ, Khutoryanskiy VV. Cyclodextrin-mediated enhancement of riboflavin solubility and corneal permeability. Mol Pharm. 2013;10(2):756–62. https://doi.org/10.1021/mp3005963.
Wimmer T. Cyclodextrins. In: Ullmann’s encyclopedia of industrial chemistry. Wiley-VCH; 2012.
Oconnor MS, Clemens D, Anderson A, Dinh D, Sadrerafi K. Cyclodextrin dimers: a pharmaceutical engineering approach to the therapeutic extraction of toxic oxysterols. Atherosclerosis. 2021;331:e130–1. https://doi.org/10.1016/j.atherosclerosis.2021.06.390.
Valcourt DM, Dang MN, Scully MA, Day ES. Nanoparticle-mediated co-delivery of Notch-1antibodies and ABT-737 as a potent treatment strategy for triple-negative breast cancer. ACS Nano. 2020;14:3378–88. https://doi.org/10.1021/acsnano.9b09263.
Zimmer S, Grebe A, Bakke SS, Latz E. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra50. https://doi.org/10.1126/scitranslmed.aad6100.
Mahjoubin-Tehran M, Kovanen PT, Xu S, Jamialahmadi T, Sahebkar A. Cyclodextrins: potential therapeutics against atherosclerosis. Pharm Ther. 2020:107620. https://doi.org/10.1016/j.pharmthera.2020.107620.
Gaspar J, Mathieu J, Alvarez P. 2-Hydroxypropyl-beta-cyclodextrin (HPβCD) reduces age-related lipofuscin accumulation through a cholesterol-associated pathway. Sci Rep. 2017;7(1):2197. https://doi.org/10.1038/s41598-017-02387-8.
Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–24. https://doi.org/10.1038/s41573-020-0090-8.
Yadav HKS, Almokdad AA, Shaluf SIM, Debe MS. Chapter 17: Polymer-based nanomaterials for drug-delivery carriers. In: Mohapatra SS, Ranjan S, Dasgupta N, Mishra RK, Thomas S, editors. Nanocarriers for drug delivery. Elsevier; 2019.
Fluksman A, Ofra B. A robust method for critical micelle concentration determination using coumarin-6 as a fluorescent probe. Anal Methods. 2019;11:3810–8. https://doi.org/10.1039/C9AY00577C.
Hussein YHA, Youssry M. Polymeric micelles of biodegradable diblock copolymers: enhanced encapsulation of hydrophobic drugs. Materials (Basel). 2018;11(5):688. https://doi.org/10.3390/ma11050688.
Atanase LI, Riess G. Self-assembly of block and graft copolymers in organic solvents: an overview of recent advances. Polymers. 2018;10:62. https://doi.org/10.3390/polym10010062.
Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. https://doi.org/10.1016/j.ijpharm.2005.10.010.
Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE. Polymer vesicles in vivo: correlations with PEG molecular weight. J Control Release. 2003;90:323–34. https://doi.org/10.1016/s0168-3659(03)00201-3.
Osborne DW, Ward AJ, O'Neill KJ. Microemulsions as topical drug delivery vehicles: in-vitro transdermal studies of a model hydrophilic drug. J Pharm Pharmacol. 1991;43(6):450–4.
Nath N, Hyun J, Ma H, Chilkoti A. Surface engineering strategies for control of protein and cell interactions. Surf Sci. 2004;4570:98–110. https://doi.org/10.1016/j.susc.2004.06.182.
Wang S, Lu L, Gruetzmacher JA, Currier BL, Yaszemski MJ. Synthesis and characterizations of biodegradable and crosslink able poly(ε-caprolactone fumarate), poly(ethylene glycol fumarate), and their amphiphilic copolymer. Biomaterials. 2006;27:832–41. https://doi.org/10.1016/j.biomaterials.2005.07.013.
Kim J-Y, Shim S-B, Shim J-K. Comparison of amphiphilic polyurethane nanoparticles to nonionic surfactants for flushing phenanthrene from soil. J Hazard Mater. 2004;116:205–12. https://doi.org/10.1016/j.jhazmat.2004.08.031.
Bader H, Ringsdorf H, Schmidt B. Water soluble polymers in medicine. Angew Macromol Chem. 1984;123:457–85. https://doi.org/10.1002/apmc.1984.051230121.
Simon JA. Estradiol in micellar nanoparticles: the efficacy and safety of a novel transdermal drug-delivery technology in the management of moderate to severe vasomotor symptoms. Menopause. 2006;13:222–31. https://doi.org/10.1097/01.gme.0000174096.56652.4f.
Lee AL, Wang Y, Pervaiz S, Fan W, Yang YY. Synergistic anticancer effects achieved by co-delivery of TRAIL and paclitaxel using cationic polymeric micelles. Macromol Biosci. 2011;11:296–307. https://doi.org/10.1002/mabi.201000332.
Scott-Moncrieff JC, Shao Z, Mitra AK. Enhancement of intestinal insulin absorption by bile salt-fatty acid mixed micelles in dogs. J Pharm Sci. 1994;83:1465–9. https://doi.org/10.1002/jps.2600831020.
Wang B, Ma R, Liu G, Li Y, Liu X, An Y, Shi L. Glucose-responsive micelles from self-assembly of poly(ethylene glycol)-b-poly(acrylic acid-co-acrylamidophenylboronic acid) and the controlled release of insulin. Langmuir. 2009;25:12522–8.
Staroverov SA, Pristensky DV, Yermilov DN, Gabalov KP, Zhemerichkin DA, Sidorkin VA, Shcherbakov AA, Shchyogolev SY, Dykman LA. The effectivity analysis of accumulation of liposomal, micellar, and water-soluble forms of diminazene in cells and in organs. Drug Deliv. 2006;13:351–5. https://doi.org/10.1080/10717540500459084.
Staroverov SA, Sidorkin VA, Fomin AS, Shchyogolev SY, Dykman LA. Biodynamic parameters of micellar diminazene in sheep erythrocytes and blood plasma. J Vet Sci. 2011;12:303–7. https://doi.org/10.4142/jvs.2011.12.4.303.
Vail DM, Von Euler H, Rusk AW, Barber L, Clifford C, Elmslie R, Fulton L, Hirschberger J, Klein M, London C, et al. A randomized trial investigating the efficacy and safety of water soluble micellar paclitaxel (Paccal Vet) for treatment of nonresectable grade 2 or 3 mast cell tumors in dogs. J Vet Intern Med. 2012;26:598–607. https://doi.org/10.1111/j.1939-1676.2012.00897.x.
Rey AI, Segura J, Arandilla E, López-Bote CJ. Short- and long-term effect of oral administration of micellized natural vitamin E (D-α-tocopherol) on oxidative status in race horses under intense training. J Anim Sci. 2013;91:1277–84. https://doi.org/10.2527/jas.2012-5125.
Rey A, Amazan D, Cordero G, Olivares A, López-Bote CJ. Lower oral doses of micellized α-tocopherol compared to α-tocopheryl acetate in feed modify fatty acid profiles and improve oxidative status in pigs. Int J Vitam Nutr Res. 2014;84:229–43. https://doi.org/10.1024/0300-9831/a000209.
Francis MF, Cristea M, Winnik FM. Exploiting the vitamin B12 pathway to enhance oral drug delivery via polymeric micelles. Biomacromolecules. 2005;6:2462–7. https://doi.org/10.1021/bm0503165.
Al-Qushawi A, Rassouli A, Atyabi F, Peighambari SM, Esfandyari-Manesh M, Shams GR, Yazdani A. Preparation and characterization of three tilmicosin-loaded lipid nanoparticles: physicochemical properties and in-vitro antibacterial activities. Iran J Pharm Res. 2016;15:663–76.
Troncarelli MZ, Brandão HM, Gern JC, Guimarães AS, Langoni H. Nanotechnology and antimicrobials in veterinary medicine. Badajoz: Formatex; 2013.
Thipparaboina R, Chavan RB, Kumar D, Modugula S, Shastri NR. Micellar carriers for the delivery of multiple therapeutic agents. Colloids Surf B Biointerfaces. 2015;135:291–308. https://doi.org/10.1016/j.colsurfb.2015.07.046.
Makhmalzade BS, Chavoshy F. Polymeric micelles as cutaneous drug delivery system in normal skin and dermatological disorders. J Adv Pharm Technol Res. 2018;9(1):2–8. https://doi.org/10.4103/japtr.JAPTR_314_17.
Shaw DJ. Introduction to colloid and surface chemistry. Elsevier; 2013.
Trivedi R, Kompella UB. Nanomicellar formulations for sustained drug delivery: strategies and underlying principles. Nanomedicine. 2010;5:485–505. https://doi.org/10.2217/nnm.10.10.
Imran M, Shah MR, Shafiullah. Chapter 10: Amphiphilic block copolymers–based micelles for drug delivery. In: Grumezescu AM, editor. Design and development of new nanocarriers. William Andrew Publishing; 2018. p. 365–400.
Ahmad Z, Shah A, Siddiq M, Kraatz H-B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014;4:17028–38. https://doi.org/10.1039/C3RA47370H.
Shi Y, Lammers T, Storm G, Hennink WE. Physico-chemical strategies to enhance stability and drug retention of polymeric micelles for tumor-targeted drug delivery. Macromol Biosci. 2017;17:1600160. https://doi.org/10.1002/mabi.201600160.
Owen SC, Chan DPY, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7:53–65. https://doi.org/10.1016/j.nantod.2012.01.002.
Lee J, Cho EC, Cho K. Incorporation and release behavior of hydrophobic drug in functionalized poly(d,l-lactide)-block–poly(ethylene oxide) micelles. J Control Release. 2004;94:323–35. https://doi.org/10.1016/j.jconrel.2003.10.012.
Lu Y, Zhang E, Yang J, Cao Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018;11:4985–98. https://doi.org/10.1007/s12274-018-2152-3.
Zeng L, Gao J, Liu Y, Gao J, Yao L, Yang X, Liu X, He B, Hu L, Shi J, Song M, Qu G, Jiang G. Role of protein corona in the biological effect of nanomaterials: investigating methods. TrAC Trends Anal Chem. 2019;118:303–14. https://doi.org/10.1016/j.trac.2019.05.039.
Zhu Y, Meng T, Tan Y, Yang X, Liu Y, Liu X, Yu F, Wen L, Dai S, Yuan H, Hu F. Negative surface shielded polymeric micelles with colloidal stability for intracellular endosomal/lysosomal escape. Mol Pharm. 2018;15:5374–86. https://doi.org/10.1021/acs.molpharmaceut.8b00842.
Pepić I, Lovrić J, Filipović-Grčić J. How do polymeric micelles cross epithelial barriers? Eur J Pharm Sci. 2013;50:42–55. https://doi.org/10.1016/j.ejps.2013.04.012.
Grimaudo MA, Pescina S, Padula C, Santi P, Concheiro A, Alvarez-Lorenzo C, Nicoli S. Topical application of polymeric nanomicelles in ophthalmology: a review on research efforts for the noninvasive delivery of ocular therapeutics. Expert Opin Drug Deliv. 2019;16:397–413. https://doi.org/10.1080/17425247.2019.1597848.
Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32:3435–46. https://doi.org/10.1016/j.biomaterials.2011.01.021.
Logie J, Owen SC, McLaughlin CK, Shoichet MS. PEG-graft density controls polymeric nanoparticle micelle stability. Chem Mater. 2014;26:2847–55. https://doi.org/10.1021/cm500448x.
Shiraishi K, Sanada Y, Mochizuki S, Kawano K, Maitani Y, Sakurai K, Yokoyama M. Determination of polymeric micelles’ structural characteristics, and effect of the characteristics on pharmacokinetic behaviours. J Control Release. 2015;203:77–84. https://doi.org/10.1016/j.jconrel.2015.02.017.
Zhou Q, Zhang L, Yang T, Wu H. Stimuli-responsive polymeric micelles for drug delivery and cancer therapy. Int J Nanomedicine. 2018;13:2921–42. https://doi.org/10.2147/IJN.S158696.
Moffitt M, Khougaz K, Eisenberg A. Micellization of ionic block copolymers. Acc Chem Res. 1996;29:95–102. https://doi.org/10.1021/ar940080+.
Zhang L, Eisenberg A. Formation of crew-cut aggregates of various morphologies from amphiphilic block copolymers in solution. Polym Adv Technol. 1998;9:677–99.
Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109(1–3):169–88. https://doi.org/10.1016/j.jconrel.2005.09.034.
Görner T, Gref R, Michenot D, Sommer F, Tran MN, Dellacherie E. Lidocaine-loaded biodegradable nanospheres. I. Optimization of the drug incorporation into the polymer matrix. J Control Release. 1999;57:259–68. https://doi.org/10.1016/s0168-3659(98)00121-7.
Lee H, Zeng F, Dunne M, Allen C. Methoxy poly(ethylene glycol)-block-poly(δ-valerolactone) copolymer micelles for formulation of hydrophobic drugs. Biomacromolecules. 2005;6:3119–28. https://doi.org/10.1021/bm050451h.
Soo PL, Lovric J, Davidson P, Maysinger D, Eisenberg A. Polycaprolactone-block-poly(ethylene oxide) micelles: A nanodelivery system for 17β-estradiol. Mol Pharm. 2005;2:519–27. https://doi.org/10.1021/mp050049h.
Jeong Y-I, Cheon J-B, Kim S-H, Nah J-W, Lee Y-M, Sung Y-K, Akaike T, Cho C-S. Clonazepam release from core-shell type nanoparticles in vitro. J Control Release. 1998;51:169–78. https://doi.org/10.1016/s0168-3659(97)00163-6.
Jeong Y-I, Nah J-W, Lee H-C, Kim S-H, Cho C-S. Adriamycin release from flower-type polymeric micelle based on star-block copolymer composed of poly(γ-benzyl l-glutamate) as the hydrophobic part and poly(ethylene oxide) as the hydrophilic part. Int J Pharm. 1999;188:49–58. https://doi.org/10.1016/s0378-5173(99)00202-1.
Huh KM, Lee SC, Cho YW, Lee J, Jeong JH, Park K. Hydrotropic polymer micelle system for delivery of paclitaxel. J Control Release. 2005;101:59–68. https://doi.org/10.1016/j.jconrel.2004.07.003.
Huh KM, Min HS, Lee SC, Lee HJ, Kim S, Park K. A new hydrotropic block copolymer micelle system for aqueous solubilization of paclitaxel. J Control Release. 2008;126:122–9. https://doi.org/10.1016/j.jconrel.2007.11.008.
Allen C, Eisenberg A, Mrsic J, Maysinger D. PCL-b-PEO micelles as a delivery vehicle for FK506: assessment of a functional recovery of crushed peripheral nerve. Drug Deliv. 2000;7:139–45. https://doi.org/10.1080/10717540050120179.
De Jaeghere F, Allémann E, Leroux JC, Stevels W, Feijen J, Doelker E, Gurny R. Formulation and lyoprotection of poly(lactic acid-co-ethylene oxide) nanoparticles: influence on physical stability and in vitro cell uptake. Pharm Res. 1999;16(6):859–66. https://doi.org/10.1023/a:1018826103261.
Gorshkova MY, Stotskaya LL. Micelle-like macromolecular systems for controlled release of daunomycin. Polym Adv Technol. 1998;9:362–7. https://doi.org/10.1002/(SICI)1099-1581(199806)9:6<362::AID-PAT793>3.0.CO;2-I.
Gref R, Minamitake Y, Peracchia M, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–3. https://doi.org/10.1126/science.8128245.
Soo PL, Luo L, Maysinger D, Eisenberg A. Incorporation and release of hydrophobic probes in biocompatible polycaprolactone-block-poly(ethylene oxide) micelles: implications for drug delivery. Langmuir. 2002;18:9996–10004. https://doi.org/10.1021/la026339b.
La SB, Okano T, Kataoka K. Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly(ethylene oxide)–poly(β-benzyl L-aspartate) block copolymer micelles. J Pharm Sci. 1996;85:85–90. https://doi.org/10.1021/js950204r.
Satoh T, Higuchi Y, Kawakami S, Hashida M, Kagechika H, Shudo K, Yokoyama M. Encapsulation of the synthetic retinoids Am80 and LE540 into polymeric micelles and the retinoids’ release control. J Control Release. 2009;136:187–95. https://doi.org/10.1016/j.jconrel.2009.02.024.
Shuai X, Merdan T, Schaper AK, Xi F, Kissel T. Core-cross-linked polymeric micelles as paclitaxel carriers. Bioconjug Chem. 2004;15(3):441–8. https://doi.org/10.1021/bc034113u.
Arora S, Rajwade JM, Paknikar KM. Nanotoxicology and in vitro studies: the need of the hour. Toxicol Appl Pharmacol. 2012;258(2):151–65. https://doi.org/10.1016/j.taap.2011.11.010.
Patel P, Shah J. Safety and toxicological considerations of nanomedicines: the future directions. Curr Clin Pharmacol. 2017;12(2):73–82. https://doi.org/10.2174/1574884712666170509161252.
Hock SC, Ying YM, Wah CL. A review of the current scientific and regulatory status of nanomedicines and the challenges ahead. PDA J Pharm Sci Technol. 2011;65(2):177–95.
Siegrist S, Cörek E, Detampel P, Sandström J, Wick P, Huwyler J. Preclinical hazard evaluation strategy for nanomedicines. Nanotoxicology. 2019;13(1):73–99. https://doi.org/10.1080/17435390.2018.1505000.
Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50(4):219–44.
Mandal A, Bisht R, Rupenthal ID, Mitra AK. Polymeric micelles for ocular drug delivery: from structural frameworks to recent preclinical studies. J Control Release. 2017;248:96–116. https://doi.org/10.1016/j.jconrel.2017.01.012.
Shiraishi K, Yokoyama M. Toxicity and immunogenicity concerns related to PEGylated-micelle carrier systems: a review. Sci Technol Adv Mater. 2019;20(1):324–36. https://doi.org/10.1080/14686996.2019.1590126.
Lekshmi UM, Reddy PN. Preliminary toxicological report of metformin hydrochloride loaded polymeric nanoparticles. Toxicol Int. 2012;19(3):267–72. https://doi.org/10.4103/0971-6580.103667.
Talkar S, Dhoble S, Majumdar A, Patravale V. Transmucosal nanoparticles: toxicological overview. Adv Exp Med Biol. 2018;1048:37–57. https://doi.org/10.1007/978-3-319-72041-8_3.
Đorđević S, Gonzalez MM, Conejos-Sánchez I, Carreira B, Pozzi S, Acúrcio RC, Satchi-Fainaro R, Florindo HF, Vicent MJ. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Deliv Transl Res. 2022;12(3):500–25. https://doi.org/10.1007/s13346-021-01024-2.
Yokoyama M. Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Deliv. 2010;7(2):145–58. https://doi.org/10.1517/17425240903436479.
Kwon GS. Polymeric micelles for delivery of poorly water-soluble compounds. Crit Rev Ther Drug Carrier Syst. 2003;20(5):357–403. https://doi.org/10.1615/critrevtherdrugcarriersyst.v20.i5.20.
Kumar R, Kulkarni A, Nagesha DK, Sridhar S. In vitro evaluation of theranostic polymeric micelles for imaging and drug delivery in cancer. Theranostics. 2012;2(7):714–22. https://doi.org/10.7150/thno.3927.
Matsumura Y. Polymeric micellar delivery systems in oncology. Jpn J Clin Oncol. 2008;38(12):793–802. https://doi.org/10.1093/jjco/hyn116.
Croy SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12(36):4669–84. https://doi.org/10.2174/138161206779026245.
Soo PL, Eisenberg A. Preparation of block copolymer vesicles in solution. J Polym Sci B. 2004;42:923–38.
Lecommandoux S, Sandre O, Chécot F, Rodriguez-Hernandez J, Perzynski R. Magnetic nanocomposite, micelles and vesicles. Adv Mater. 2005;17:712–9. https://doi.org/10.1002/adma.200400599.
Zhu Y, Yang B, Chen S, Du J. Polymer vesicles: mechanism, preparation, application, and responsive behavior. Prog Polym Sci. 2017;64:1–22. https://doi.org/10.1016/j.progpolymsci.2015.05.001.
Cabral H, Miyata K, Osada K, Kataoka K. Block copolymer micelles in nanomedicine applications. Chem Rev. 2018;118:6844–92. https://doi.org/10.1021/acs.chemrev.8b00199.
Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, Paz-Ares L, Cho DC, Infante JR, Alsina M, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients. Cancer Discov. 2013;3:406–17. https://doi.org/10.1158/2159-8290.CD-12-0429.
Shen Y, Zhang J, Hao W, Wang T, Liu J, Xie Y, Xu S, Liu H. Copolymer micelles function as pH-responsive nanocarriers to enhance the cytotoxicity of a HER2 aptamer in HER2-positive breast cancer cells. Int J Nanomedicine. 2018;2018(13):537–53. https://doi.org/10.2147/IJN.S149942.
Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 2007;24:1029–46. https://doi.org/10.1007/s11095-006-9223-y.
Castillo PM, Jimenez-Ruiz A, Carnerero JM, Prado-Gotor R. Exploring factors for the design of nanoparticles as drug delivery vectors. ChemPhysChem. 2018;19:2810–28. https://doi.org/10.1002/cphc.201800388.
Xu P, Van Kirk EA, Li S, Murdoch WJ, Ren J, Hussain MD, Radosza M, Shen Y. Highly stable core-surface-crosslinked nanoparticles as cisplatin carriers for cancer chemotherapy. Colloids Surf B Biointerfaces. 2006;48:50–7. https://doi.org/10.1016/j.colsurfb.2006.01.004.
Ren J, Fang Z, Yao L, Dahmani FZ, Yin L, Zhou J, Yao J. A micelle–like structure of poloxamer–methotrexate conjugates as nanocarrier for methotrexate delivery. Int J Pharm. 2015;487:177–86. https://doi.org/10.1016/j.ijpharm.2015.04.014.
Li X, Yang Z, Yang K, Zhou Y, Chen X, Zhang Y, Wang F, Liu Y, Ren L. Self-assembled polymeric micellar nanoparticles as nanocarriers for poorly soluble anticancer drug ethaselen. Nanoscale Res Lett. 2009;4:1502–15. https://doi.org/10.1007/s11671-009-9427-2.
Bhadra D, Bhadra S, Jain S, Jain NK. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int J Pharm. 2003;257:111–24. https://doi.org/10.1016/s0378-5173(03)00132-7.
Cesur H, Rubinstein I, Pai A, Onyuksel H. Self-associated indisulam in phospholipid-based nanomicelles: a potential nanomedicine for cancer. Nanomedicine. 2009;5:178–83. https://doi.org/10.1016/j.nano.2008.09.001.
Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S, Li Y. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7:5858–69. https://doi.org/10.1021/nn4010796.
Tagami T, Ozeki T. Recent trends in clinical trials related to carrier-based drugs. J Pharm Sci. 2017;106:2219–26. https://doi.org/10.1016/j.xphs.2017.02.026.
Bu HZ, Gukasyan HJ, Goulet L, Lou XJ, Xiang C, Koudriakova T. Ocular disposition, pharmacokinetics, efficacy, and safety of nanoparticle-formulated ophthalmic drugs. Curr Drug Metab. 2007;8:91–107. https://doi.org/10.2174/138920007779815977.
Norouzi P, Amini M, Dinarvand R, Arefian E, Seyedjafari E, Atyabi F. Co-delivery of gemcitabine prodrug along with anti NF-κB siRNA by tri-layer micelles can increase cytotoxicity, uptake and accumulation of the system in the cancers. Mater Sci Eng C. 2020;116:111161. https://doi.org/10.1016/j.msec.2020.111161.
Chopra P, Hao J, Li SK. Sustained release micellar carrier systems for iontophoretic transport of dexamethasone across human sclera. J Control Release. 2012;160:96–104. https://doi.org/10.1016/j.jconrel.2012.01.032.
Ren S, Chen D, Jiang M. Noncovalently connected micelles based on a β-cyclodextrin containing polymer and adamantane end-capped poly(ε-caprolactone) via host–guest interactions. J Polym Sci A. 2009;47:4267–78. https://doi.org/10.1002/pola.23479.
Liu LH, Venkatraman SS, Yang YY, Guo K, Lu J, He BP, Moochhala S, Kan LJ. Polymeric micelles anchored with TAT for delivery of antibiotics across the blood-brain barrier. Biopolymers. 2008;90:617–23. https://doi.org/10.1002/bip.20998.
Weissig V, Pettinger TK, Murdock N. Nanopharmaceuticals (part 1): products on the market. Int J Nanomedicine. 2014;9:4357–73. https://doi.org/10.2147/IJN.S46900.
Pristensky DV, Staroverov SA, Ermilov DN, Shchyogolev SY, Dykman LA. Analysis of effectiveness of intracellular penetration of ivermectin immobilized onto corpuscular carriers. Biochem Moscow Suppl Ser B. 2007;1:249–53.
Sawant RR, Torchilin VP. Multifunctionality of lipid-core micelles for drug delivery and tumour targeting. Mol Membr Biol. 2010;27:232–46. https://doi.org/10.3109/09687688.2010.516276.
Zhang Y, Huang Y, Li S. Polymeric micelles: nanocarriers for cancer-targeted drug delivery. AAPS PharmSciTech. 2014;15:862–71. https://doi.org/10.1208/s12249-014-0113-z.
Yousefpour MM, Yari KA. Polymeric micelles as mighty nanocarriers for cancer gene therapy: a review. Cancer Chemother Pharmacol. 2017;79(4):637–49. https://doi.org/10.1007/s00280-017-3273-1.
Paliwal R, Babu RJ, Palakurthi S. Nanomedicine scale-up technologies: feasibilities and challenges. AAPS PharmSciTech. 2014;15:1527–34. https://doi.org/10.1208/s12249-014-0177-9.
Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014. https://doi.org/10.1038/natrevmats.2016.14.
van der Meel R, Lammers T, Hennink WE. Cancer nanomedicines: oversold or underappreciated? Expert Opin Drug Deliv. 2017;14(1):1–5. https://doi.org/10.1080/17425247.2017.1262346.
Wang J, Li S, Han Y, Guan J, Chung S, Wang C, Li D. Poly(ethylene glycol)–polylactide micelles for cancer therapy. Front Pharmacol. 2018;9:202. https://doi.org/10.3389/fphar.2018.00202.
Rollerova E, Jurcovicova J, Mlynarcikova A, Sadlonova I, Bilanicova D, Wsolova L, et al. Delayed adverse effects of neonatal exposure to polymeric nanoparticle poly (ethylene glycol)-block-polylactide methyl ether on hypothalamic–pituitary–ovarian axis development and function in Wistar rats. Reprod Toxicol. 2015;57:165–75. https://doi.org/10.1016/j.reprotox.2015.07.072.
Scsukova S, Mlynarcikova A, Kiss A, Rollerova E. Effect of polymeric nanoparticle poly (ethylene glycol)-block-poly (lactic acid) (PEG-bPLA) on in vitro luteinizing hormone release from anterior pituitary cells of infantile and adult female rats. Neuro Endocrinol Lett. 2015;36:88–94.
Acknowledgments
This work was supported by the Russian Science Foundation [Projects Nos. 22-24-00417 (O. I. Guliy), and 19-14-00077-II (S. A. Staroverov, A. S. Fomin, L. A. Dykman)].
Conflict of Interest
There are no conflicts of interests in this chapter.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Guliy, O.I., Fomin, A.S., Zhnichkova, E.G., Kozlov, S.V., Staroverov, S.A., Dykman, L.A. (2022). Polymeric Micelles for Targeted Drug Delivery Systems. In: Barabadi, H., Mostafavi, E., Saravanan, M. (eds) Pharmaceutical Nanobiotechnology for Targeted Therapy. Nanotechnology in the Life Sciences. Springer, Cham. https://doi.org/10.1007/978-3-031-12658-1_18
Download citation
DOI: https://doi.org/10.1007/978-3-031-12658-1_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12657-4
Online ISBN: 978-3-031-12658-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)