
Abstract
One of the most widely studied subjects in nanoscience technology is the creation of supramolecular architectures with well-defined structures and functionalities. These supramolecular structures are generated as a result of self-assembly of amphiphilic block polymers. Self-assembly of block polymers via hydrophobic and hydrophilic effects, electrostatic interactions, hydrogen bonding, and metal complexation has shown tremendous potential for creating such supramolecular structures with a wide array of applications. Polymeric micelles have gathered considerable attention in the field of drug and gene delivery due to their excellent biocompatibility, low toxicity, enhanced blood circulation time, and ability to solubilize a large number of drugs in their micellar core.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Amphiphilic molecules or surfactant monomers that possess a polar head and a lipophilic tail could show changes in their physicochemical properties in solutions. These changes are associated with the orientation and association of amphiphilic molecules in solution resulting in the formation of structures called micelles. The micelles internally have a hydrophobic core and externally a hydrophilic surface. Micelles are generally made up of 50–200 monomers (an average number of monomers forming micelle at any given time is termed as the aggregation number). The radius of a spherical micelle is 1–3 nm, and thus they lie in the colloidal range [1, 2]. The major driving force behind self-association of amphiphilic molecules is the decrease of free energy of the system. The decrease in free energy is a result of removal of hydrophobic fragments from the aqueous surroundings with the formation of a micelle core stabilized with hydrophilic fragments exposed into water. The factors affecting the process of micelle formation are the size of the hydrophobic domain in the amphiphilic molecule, concentration of amphiphiles, temperature, and solvent. The minimum concentration of amphiphilic molecules to form assembly is called critical micelle concentration (CMC). At low concentrations in medium, these amphiphilic molecules exist separately, and are so small that they appear to be subcolloidal. Below the CMC, the amphiphile undergo adsorption at the air–water interface. As the total concentration of the amphiphile is increased up to CMC, the interface as well as the bulk phase is saturated with monomers. Any further amphiphile added in excess of CMC results in the aggregation of monomers in the bulk phase, such that the free energy of the system is reduced. The temperature below which amphiphilic molecules exist as unimers and above which as aggregates is the critical micellization temperature (CMT) [1, 3, 4].
2 Polymeric Micelle
A polymeric micelle is a macromolecular assembly that forms from synthetic amphiphilic block copolymers or graft copolymers. It has a spherical inner core and an outer shell [5]. As shown in Fig.16.1, which features an AB-type block copolymer, a micellar structure forms in an aqueous medium if one segment of the block copolymer can provide interchain cohesive interactions sufficient for the micelle formation.

Design of a polymeric micelle carrier system
Here, Amphiphilic block or graft copolymers behave in the same manner as that of conventional amphiphiles and in aqueous solution, above CMC, these polymers form polymeric micelles. In contrast to the micelles of conventional surfactant monomers, in polymeric micelles there is a covalent linkage in individual surfactant molecules within the hydrophobic core. This linkage prevents dynamic exchange of monomers between free solution and the micellar pseudo-phase which confers rigidity and stability to the polymeric micelles [6]. The aggregation number of polymeric micelles is of the magnitude of several hundreds and the diameter ranges from 10 to 100 nm. Factors controlling the size of the polymeric micelles include molecular weight of the amphiphilic block copolymer, aggregation number of the amphiphiles, relative proportion of hydrophilic and hydrophobic chains, and the preparation process [7]. In aqueous medium amphiphilic block copolymers can principally self-assemble into spherical micelles, worm-like or cylindrical micelles, and polymer vesicles or polymersomes. Main factor governing the morphology of micelles is the hydrophilic–hydrophobic balance of the block copolymer defined by the hydrophilic volume fraction, f. For amphiphilic block copolymers with value of nearly 35 %, polymer vesicles are formed, whereas, for f value more than 45 %, spherical micelles are formed from self-assembly [8, 9]. By using amphiphiles of more complicated molecular design, e.g., miktoarm star copolymers, or by varying the experimental conditions for self-assembly more complex morphologies such as that of crew-cut micelles, multicompartment micelles, toroids, etc. may be obtained [10].
3 Types of Polymeric Micelles
On the basis of the type of intermolecular forces governing the segregation of the core segment from the aqueous environment, polymeric micelles can be classified in three main categories, i.e., micelles formed by hydrophobic interactions, those resulting from electrostatic interactions (polyion complex micelles), and micelles from metal complexation.
3.1 Conventional
These micelles are formed by hydrophobic interactions between the core segment and the corona region of amphiphilic block copolymer in the aqueous environment. One of the simplest example of this type is the micelle formed by hydrophobic interactions in poly(ethylene oxide)-b-poly(propylene oxide)-bpoly(ethylene oxide) [11].
3.2 Polyion Complex Micelles
Polymeric micelles are also formed by electrostatic interactions between two oppositely charged moieties, such as polyelectrolytes and these micelles are termed polyion complex micelles (PICMs). When oppositely charged polymers are added in the solution, they can penetrate in the corona of the micelle and give rise to polyionic micelle. The electrostatic forces and the van der Waals force of interaction control the structure and size of the charged micelle coronas. PICMs have some peculiar features such as simple synthetic route, easy self-assembly in aqueous medium, structural stability, high drug loading capacity, and prolonged circulation in the blood. The preparation of micelles being carried out in aqueous medium without involvement of any organic solvents, they overcome the associated side-effects produced by the residual organic solvents. The core of the PICMs can entrap many therapeutic agents such as hydrophobic compounds, hydrophilic compounds, metal complexes, and charged macromolecules through electrostatic, hydrophobic, hydrogen bonding interactions and release them after receiving a suitable trigger. Because of these reasons, the PICMs have a great potential for drug release, especially for the delivery of charged drugs, antisense oligonucleotides, DNA, and enzymes [12, 13]. Recently, Jung et al. prepared polymeric micelles of methoxypoly(ethylene glycol)-grafted-chitosan encapsulating all trans retinoic acid through the formation of a polyion complex between the amine group of chitosan and the carboxylic acid group of all-trans retinoic acid. The PICMs were designed for drug delivery to the brain tumor. The sizes of PICMs were about 50–200 nm and the loading efficiency of micelle was higher than 80 % (w/w).
3.3 Non-covalently Connected Polymeric Micelles
Polymeric micelles are also prepared by a novel “block-copolymer-free” technique. Here, polymeric micelles are obtained via self-assemblage of homopolymer, random copolymer, graft copolymer or oligomer by inter polymer hydrogen bonding complexation. Core and shell are non-covalently connected at their homopolymer chain end by specific inter molecular interactions such as H-bonding or metal-ligand interactions in the resultant structures and hence these are termed as non-covalently connected micelles. Jiang et al. prepared the intermolecular complexes with poly(4-vinylpyridine) as the backbone and carboxyl terminated poly butadiene as the grafts due to hydrogen bonding using chloroform as a common solvent.
4 Polymers used in Polymeric Micelle
Amphiphilic copolymers, either block copolymers (di, tri, or tetra) or graft copolymers, can be used to form polymeric micelle. A graft copolymer is one which comprises a polymer chain as a backbone and another polymer chain as side “grafted” parts. These copolymers usually demonstrate properties of both the polymeric backbones as well as of the graft [14]. Table 16.1 shows different possible structures of amphiphilic copolymers with representative example of each class.
Amphiphilic diblock AB-type or triblock ABA-type copolymers with the length of a hydrophilic block exceeding to some extent that of a hydrophobic one would self-assemblage to form spherical micelles in aqueous solutions. If the length of a hydrophilic block is too large, copolymers exist in water as individual molecules (unimers), and molecules with lengthy hydrophobic blocks develop various structures. Examples of different amphiphilic copolymers that have been investigated for producing micelles are given in Table 16.2.
5 Preparation of Polymeric Micelle
Polymeric micelles are prepared by either of the two following methods.
-
1.
By direct dissolution: This method is used for block copolymers with low molecular weight and short length of the insoluble block. Here, micelles are prepared by direct dissolution in a selective solvent for one of the blocks. To facilitate dissolution, stirring, thermal, or ultrasound treatments can be used. The micelle formed gets trapped in a solvent that is a strong nonsolvent for the core and thus become stable.
-
2.
By counter solvent method: molecularly dissolved chains of block copolymer in a selective solvent for one of the blocks is added with a non-solvent for the other block to form micelle. Alternatively, temperature or pH variations may be used for micelle formation [27].
6 Preparation of Drug-Loaded Micelles
Drug-loaded polymeric micelles can be prepared mainly by three common approaches: direct dissolution, solvent evaporation, and dialysis.
Direct dissolution method: In this method, the amphiphilic copolymer and drug in water is prepared at or above CMC. The copolymer and the drug self-assemble in water to form drug-loaded micelles. But this method usually is associated with low drug loading which can be enhanced by increase in temperature. Alternately a thin evaporated film of drug can be prepared before the addition of copolymer.
Solvent evaporation or solution-casting technique: In this technique, copolymer and the drug are dissolved in a volatile organic solvent and a thin film is formed by evaporating the solvent. Drug-loaded polymeric micelles are obtained by reconstitution of film with water. When the core forming blocks are long and more hydrophobic, the two above-mentioned techniques are unsuitable.
Dialysis technique: Micelles from copolymers having long and more hydrophobic core forming blocks have the potential to solubilize large amounts of poorly water-soluble drugs. In these cases, the dialysis technique can be used to prepare drug-loaded micelles. Solutions of the drug and the polymer in organic solvent are placed in the dialysis bag, and the solvent is exchanged with water by immersing bag into water, inducing micelle assembly [28, 29]. However, emulsification involving use of chlorinated solvents is not safer and dialysis process often requires more than 36 h for efficient loading.
Lyophilization technique: The above mentioned limitations can be overcome by employing a simple and cost-effective method in which water/tert-butanol mixture is used for dissolving drug as well as polymer and then the solution is lyophilized. Drug-loaded polymeric micelles are then obtained by redispersing the lyophilized product in a suitable vehicle [30, 31].
6.1 Drug Loading Capacity of Polymer Micelles
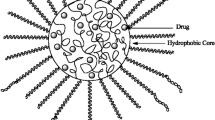
Almost one-third of newly discovered drugs are highly insoluble in water, but there is no standard method to solubilize such drugs [32]. As a result of the capability to load lipophilic molecules into the hydrophobic core, polymer micelles have been widely used to solubilize and deliver poorly water-soluble drugs (Fig. 16.2).

Schematic of a hydrophobically assembled polymer micelle. The hydrophobic core loading lipophilic drugs is protected from the environment by the hydrophilic shell
6.2 Theories of Drug Solubilization by Polymeric Micelles
The first-generation polymer micelle were used to solubilize (or load) highly lipophilic drugs like paclitaxel using PEG-b-poly(d,l-lactic acid) (PDLLA or PLA) as polymer. As reported the loading capacity of paclitaxel is ~ 10–20 % (wt/wt) [17]. This enhancement of drug solubility in water, which is derived from hydrophobic interaction between hydrophobic polymer blocks and drugs [33]. The hydrophobic interaction, more exactly hydrophobic effect, is a phenomenon induced by the London dispersive force that exists between any kinds of molecule. The hydrophobic effect is induced when hydrophobic molecules are mixed with water, because the London dispersive force between lipophilic drugs and hydrophobic blocks is much stronger than that between the lipophilic drug and water.
Although the hydrophobic effect is a major driving force, drug loading capacity and efficiency also depend on the miscibility between polymers and drugs. The mechanism of drug loading into a polymer micelle is explained by the Hildebrand–Scatchard solubility parameter (δ),
where ∆E vap is the energy of vaporization and V is the molar volume of the solvent [34].
The drug loading into a polymer micelle is in a way mixing of the polymer with drugs, the loading capacity can be described by the Flory–Huggins theory, expressed by
where χ drug-polymer is the Flory–Huggins interaction parameter between the drug and the polymer, V drug is the volume of the drug, R is the ideal gas constant, T is the temperature, and δ drug and δ polymer are the Hildebrand–Scatchard solubility parameters of the drug and the polymer, respectively [35, 36].
The above equation describes the miscibility between polymers and drugs. Letchford and colleagues investigated the miscibility of PEG-b-PCL with five different drugs [37] and observed that etoposide, paclitaxel, plumbagin, curcumin, and indomethacin followed the ascending order of χ drug-polymer. As a lower value (<0.5) of the Flory–Huggins parameter means a better solubility, indomethacin is the drug best solubilized in PEG-b-PCL micelle among the five. It should be noted that the hydrophobicity of each drug does not follow the order of the Flory–Huggins parameter, indicating that a hydrophobic effect is not the only mechanism to explain the efficiency of drug loading into polymer micelles. Similarly, the heat of mixing is another parameter to describe the miscibility between polymers and drugs. The drug loading efficiency was found to be highly dependent on the heat of mixing and the order was poly(benzyl-l-aspartate) (PBLA) >PCL > PDLLA> PGA. It is therefore obvious that the drug loading in to a polymer micelle is not only forced by the hydrophobic effect, but also facilitated by other interactions between polymers and drugs to increase the miscibility.
In addition, another important parameter to govern the drug loading capacity of polymer micelles is the hydrophilic–lipophilic balance (HLB) of block copolymers. For example, block copolymers with longer hydrophobic block showed better drug loading property, which was confirmed by determining the partition coefficient of drugs into PEG-b-PCL micelles [37].
The hydrotropy also significantly enhances the aqueous solubility of poorly soluble compounds [38, 39]. The mechanism of hydrotropic solubilization has been explained in several ways, such as hydrophobic effect, hydrogen bonding and stacking interaction [38]. The hydrotropes form non-covalent aggregates only above a certain concentration, which is called the minimal hydrotrope concentration (MHC) [40]. As hydrotropes are small molecules containing both hydrophobic and hydrophilic moieties, the hydrophobic effect can be a driving force to generate aggregates such as surfactants. In addition, polar groups of the hydrophilic moieties can interact with drugs by means of hydrogen bonding [41]. Hypothetically, hydrotropes may break the hydrogen bonding between drugs that is considered as one of the drug crystallization mechanisms.
Most hydrotropes have an aromatic ring substituted by heteroatoms. Depending on the substituted atom species, the benzene rings can interact with each other and be stacked (П– П stacking) [42, 43]. As most poorly soluble drugs consist of one or multiple benzene rings and polar groups, the self-aggregation property of hydrotropes can be expanded to complexation between hydrotropes and lipophilic drugs [44, 45].
The significance of multiple interaction parameters in describing the solubilization phenomenon is indicated in the linear solvent free energy relationship (LSER) equation [43],
where SP is the property of interest for a drug (i.e., partition coefficient), R 2 is the excess molar refraction of the solution derived from the London dispersion force, p 2 is the drug dipolarity/polarizability, Σa 2 is the hydrogen bonding acidity of the drug, Σβ2is the hydrogen bonding basicity of the drug, and Vx is the McGowan’s characteristic volume calculated from molecular structure. The c, r, s, a, b, and v are regression coefficients.
Based on the LSER theory, drug partition in two immiscible phases of water and micelle core-forming polymer is explained by transferring the free energy of drugs in water to that in polymer. This free energy is proportional to the sum of multiple independent interactions. Therefore, the LSER equation suggests that the important parameters to maximize the miscibility are hydrophobicity, electrostatic interaction, dipole–dipole interaction, hydrogen bonding, and size of the drug. When the miscibility between polymers and drugs is optimized, it is expected that much improved drug loading capacity of polymer micelles will be obtained. Introduction of the hydrotropy into polymer micelles is one solution to accomplish the optimized miscibility.
6.3 Stability Issues
It is known that polymeric micelles possess high structural stability provided by the entanglement of polymer chains in the inner core. This stability has two aspects: static and dynamic [46–49]. Static stability is described by a critical micelle concentration (CMC). Generally, polymeric micelles show very low CMC values in a range from 1 mg/mL to 10 mg/mL. These values are much smaller than typical CMC values of micelles forming from low-molecular weight surfactants. The second aspect, dynamic stability, is described by the low dissociation rates of micelles
Intravenous injections of polymeric micelles could get destabilized due to extreme dilutions they undergo in blood. This would lead to leakage and burst release of loaded drugs. Such problems could be overcome by improving the interaction between the drug and polymer in the shell via chemical conjugation or by cross-linking [50]. Other causes for decrease in stability of polymeric micelle are: Dis-balance in the HLB of the system due to over-loading of hydrophobic moiety (drug) into the core region; Hydrolytic cleavage of drugs or copolymers in aqueous systems. However, lyophilized polymeric micelle formulations have shown to possess improved long-term stability for intravenously administered preparations [51]. Other strategy used for stabilization is cross-linking of hydrophilic shell and/or hydrophobic core.
6.3.1 Cross-Linking the Shell
Cross-linking of hydrophilic shell leads to a stabilization of the micelle system and delays the degradation of the micelle. This strategy involves introduction of cross-linkable groups within the hydrophilic portion of the copolymer and then using polymer chemistry to cross-link the hydrophilic shell portion after the micellization of the polymer. Other approaches, such as conjugation of the core with the drug, can also be useful in preparing sustained-release micellar systems. For example, multifunctional, multi-armed PEG can be copolymerized with some degradable hydrophobic polymers to form an amphiphilic block. PEG branching in this polymer can be used to create cross-linkable groups in the system to prepare a shell cross-linked micelle system. Thurmond et al. [52] prepared micelles from polyvinyl pyridine-b-poly styrene (M n = 52,500 Da) block copolymer, which on self-assembly, forms shell cross-linked knedel-like micelles. Li et al. reported similar stabilization of the micelles by cross-linking the shell of the micelles made from a poly(styrene-b-butadiene-b-styrene) polymer [53] by first preparing micelles in aqueous medium and then cross-linking the hydrophobic portion of the micelles using chloromethylation and amination.
6.3.2 Cross-Linking the Core
Shell cross-linking strategies needs preparation at a high dilution to avoid inter micelle cross-link formation which decreases the efficiency of the process [54]. Hence, the strategy of core cross-linking was developed to form a core matrix that traps the drug inside it, thereby controlling the diffusion of the drug from the core. Many approaches have been tried to stabilize the core by cross-linking it with different functional groups. Thiol group can be introduced to cross-link the core with a disulfide group and the cross-linked core polymer be used to prepare polyion complex micelles. In such a polyion complex, the electrostatic interaction between two polymer segments drives association. Kazikawa et al. synthesized micelles using PEG-5,000-b-poly(lysine) diblock copolymer [55]. The cross-linking of the poly(lysine) core was achieved using thiolation chemistry. A completely biodegradable system was prepared by Hu et al. using the polymer PEG-b-PLA with a 5-methyl-5-allyloxycarbonyl-1,3-dioxane-2–1 (M n = 4,500 Da) group as the polymerizable group for cross-linking the core [56]. The cross-linking was achieved post micellization by reaction with 2,2-azoisobutyronitrile. These micelles (130 nm size) were shown to survive water dilution and temperature better than non-cross-linked micelles. Core cross-linked strategy can also be used to prepare drug-loaded micelles that offer a longer sustained release than non-modified regular micelles.
6.3.3 Use of a Low Critical Solution Temperature Hydrogel
Low critical solution temperature (LCST) hydrogel can be used to stabilize the micelles by polymerizing LCST gel along with the core of the micelle to stabilize the core. LCST gels remain in a swollen state at room temperature, allowing drug loading while at physiological temperatures, these gels collapse and lock the hydrophobic portion of the micelle forming a locked core that contains the drug. Such a locked interpenetrating network in the core prevents the breakdown of the core upon dilution and also the drug loaded in the core would remain in the micelles for prolonged release. Such a system with pluronic micelles and an LCST gel was reported by Rapoport [57] where he suggested three ways to stabilize pluronic micelles, namely, core cross-linking, introducing vegetable oil in the hydrophobic portion to stabilize the micelles and polymerizing an LCST gel with the hydrophobic portion of the micelle to stabilize the core. However, one major disadvantage of using an LCST gel in the core of the micelle is that it increases the micellar size by severalfold. Rapoport reported a size increase from 12–15 nm to 30–400 nm [57].
7 Characterization of Polymeric Micelles
7.1 Critical Micelle Concentration (CMC)
Amphiphilic polymers exist in the form of micelles in aqueous media and when the delivery system gets diluted below CMC the micelles may collapse. Hence, CMC is the critical parameter for the formation and the static stability of polymeric micelles. Some of the methods used for determination of CMC in aqueous dispersions of micelles include surface tension measurements, chromatography, light scattering, small angle neutron scattering, small angle X-ray scattering, differential scanning calorimetry, viscometry, and utilization of fluorescent probes. However, the simplest method is by plotting the surface tension as a function of the logarithm of the concentration. The CMC is said to be attained when the surface tension stops decreasing and reaches a plateau value. Use of pyrene as a fluorescent probe for estimating CMC is also much reported [58].
7.2 Size and Shape Determination
Polydispersity index of the prepared micellar solutions are best studies by quasielastic light scattering technique. Monodisperse micelles produce blue color from light scattering while the scattered light is white for aggregates [59]. Size of polymeric micelles usually falls in the colloidal range and so scanning electron microscopy (SEM) and transmission electron microscopy (TEM) techniques have been widely used for the direct visualization, size and shape determination of particularly block copolymer micelles. Recently, cryo-TEM technique has increasingly started gaining importance for characterization of block copolymer micelles in aqueous medium. SEM or atomic force microscopy (AFM) reveals information regarding size distribution when chemically attached micelles to surfaces are presented. Hydrodynamic diameters and poly-dispersity indices of micelles are also obtained using photon correlation spectroscopy. Recently size characterization of drug-loaded polymeric micelles using asymmetrical flow field-flow fractionation and the structure of assemblies by small angle neutron scattering are also reported [60, 61].
7.3 In Vitro Drug Release Behavior
In vitro drug release behavior from micelles is studied by placing the micellar solution in a dialysis tube immersed into a flask containing release medium, kept at a constant temperature. At predetermined time intervals, aliquots of the release medium are taken and replaced by fresh medium. The content of drug released in the medium are measured by spectroscopic or other suitable method [62]. The parameters affecting the drug release are;
7.3.1 Micelle Stability
Dissociation of micelles into single chains will obviously free the entrapped molecules. Similarly, erosion or biodegradation of the carrier could provoke the escape of drug molecules. However, biodegradation of polymeric micelle follow controlled degradation and hence may release the entrapped drug molecules in a sustained manner [63].
In cases of stable polymeric micelle that are slowly biodegradable or non-biodegradable and above the CMC, the drug release will depend on the rate of diffusion from the micelle. The rate of diffusion was shown to be influenced by various factors mentioned below;
-
The core-forming block length—increase in the length of the core forming block will favor slow and continuous release as the drugs located in the core have to diffuse through a longer path [64].
-
The micelle morphology—spherical, cylindrical, bilayer, etc. are associated with different diameters and surface (and interface) areas per micelle, thus affecting the release rate.
-
The physical state of the core—physical state of the micelle core, whether solid-like or liquid-like, was also shown to influence micelle stability and, in parallel, the release of the entrapped drug. For example, diffusion of a drug through a glassy core was slower than through a more mobile core [65] and this tendency could be qualitatively correlated with the respective glass transition temperatures of the core components.
-
The presence of cross-links (within either the core or the corona segments), and the compatibility of the copolymer–drug pair cross-linking of the corona-forming blocks affects the permeability of the corona and the period within which drugs diffuse [66].
-
The polymer/drug compatibility–polymer/drug compatibility can also influence drug release and drug incorporation. The Flory–Huggins interaction parameter can be used to infer the characteristics of drug release of a system. Generally speaking, the stronger the interaction between the drug and the core-forming block, the slower its release from the micelle.
-
Drug localization—the localization of the drug within the micellar assembly is expected to influence the release, with molecules located at the core/corona interface or within the corona diffusing faster than those located in the core [65].
7.3.2 Drug Properties
Properties inherent to the drug molecule such as its molecular volume, physical state in the micelle core, relationship between the molecular volume of the drug and diffusion constant are other parameters to influence the drug release. The physical state of a drug in the micelle core can alter the drug release profile. Jeong et al. [64] showed that an increase in the amount of clonazepam loaded into PEG-b-PBLG micelles (12.1–32.8 % (w/w)) resulted in as lower drug release. Differential scanning calorimetry thermograms revealed that crystallization of the drug occurred at the higher loading.
Drug release profiles determined in vitro are useful to compare drug formulations but they rarely correlate with the in vivo behavior. Most of the time, the release gets accelerated in vivo. For example, the in vitro release of hydroxylcamptothecin loaded in PEG-b-PCL micelles occurred over several days while the drug was cleared from plasma within few hours following i.v. administration of the micelles [67]. Similar results of premature micelle disassembly were reported [68, 69] where the loss of integrity of polymeric micelle within an hour after intramuscular or subcutaneous injection was observed. It was recently demonstrated that the destabilization of the polymeric micelle and the release of the loaded drug in vivo is possibly due to interactions with plasma proteins [70]. In addition, factors of in vivo micelle destabilization such as interaction with other blood components, the translocation of hydrophobic drugs to the lipid components in the blood and the degradation of the copolymers, can also contribute to the disassembly of the micelles and the fast release of the drug.
7.3.3 Drug Release from Specialized Micellar Systems
In the case of polyion complex micelles (PICM), drug release is not diffusion-based but rather occurs through dissociation of electrostatic interactive forces between oppositely charged ions, of the assemblies. The dissociation in vivo, mostly takes place by exchange events with charged ions (i.e., salts, heparin). Similarly, the dissociation of the polymer–metal complex micelles occurs through the substitution of the metal from the coordinating groups of the copolymer by ions in the medium, thus resulting in the micellar dissociation and subsequent release of the drug.
In addition to the above mentioned mechanisms, the drug release from the micellar carriers are also triggered by a change in pH, temperature or in the redox state of the surrounding medium. Ultrasounds have also been utilized to trigger drug release from micellar systems in vitro and in vivo [71, 72].
7.3.3.1 pH-Triggered Drug Release
pH variations occur at different pathological/physiological sites permitting drug release with change in the environmental pH. For example, the microenvironment pH in tumors is generally more acidic than in normal tissues [73]. Changes in pH are also encountered upon cellular internalization of the drug-loaded carriers via clathrin-mediated endocytosis, resulting in an increase of acidity inside the endosome. Orally administered formulations experience a pH gradient as they transit from the stomach to the jejunum. Several strategies have been exploited to achieve pH-sensitivity, most of which are based on changes in the polymer properties following the protonation/deprotonation of acidic and basic groups present along the polymer chain or on the hydrolytic cleavage of hydrophobic functionalities or cross-links.
For example, polymeric micelle with a corona composed of a PNIPAM copolymer bearing carboxylic acid functionalities show the pH-sensitivity. At neutral pH, the carboxylic acid groups make the PNIPAM segment soluble but a sharp decrease in the solubility of the corona is observed as the pH is lowered. As a result, mixing of the PNIPAM chains and core region takes place, increasing the polarity of the core and promoting the release of the entrapped drug [74]. Another approach is to impart amphiphilicity to a copolymer by the conjugation of hydrophobic moieties to one of the polymer block (the core block) via pH-sensitive links. Decrease in pH result in the hydrolysis of the link and exposes polar groups on the core forming block, resulting in the destabilization of the micelles and drug release. A system like this was reported by Gillies et al. [75, 76] who developed block copolymers of PEG and either PLL or polyester dendrons and used highly acid-sensitive cyclic acetals to attached hydrophobic groups to the dendrimer periphery. These polymers self-assembled into micelles that were stable in neutral aqueous solution but disintegrated into unimers at mildly acidic pH [65] following loss of the hydrophobic groups upon acetal hydrolysis [76]. The pH-sensitive micelles were produced by directly conjugating a hydrophobic anticancer agent (DOX) to copolymers to release the drug at acidic pH found in tumor tissues [75].
7.3.3.2 Temperature Sensitivity
Local hyperthermia is observed in some disease states and local increases in body temperature can also be induced by exterior means, making temperature-directed drug release as another viable strategy for localized drug release. To achieve this, polymers presenting a lower critical solution temperature (LCST) transition like PNIPAM can be incorporated in the composition of micelles. The LCST of PNIPAM can be adjusted within a desired range by copolymerizing it with hydrophilic or hydrophobic monomers which strengthen or weaken the interactions between polymer chains and water, resulting in an increase or decrease in water solubility, respectively [77–79]. Below the LCST (at normal temperature), the non-polar core is segregated from the hydrated PNIPAM corona while, at higher temperature (above the LCST), corona collapses and results in the increased mixing of the NIPAM corona units and hydrophobic core units. This increases the core polarity triggers release of the drug incorporated in the micelle [74, 77, 80, 81].
7.3.3.3 Redox Sensitivity
Another stimulus to trigger the release of drugs from the polymeric carriers is redox sensitivity due to the presence of oxygen-reactive species released by activated macrophages in the inflamed tissues and certain tumors [82]. The hydrophobic poly(propylene sulfide) (PPS) in PEG-b-PPS block copolymer respond to such oxidative condition and readily gets converted to hydrophilic poly(sulfoxide) or poly(sulfone) by mild oxidizing agents. Micelles obtained from this polymer was demonstrated to release the incorporated hydrophobic drugs during the solubilization or swelling of the polysulfide upon oxidation [83, 84]. Another mechanism could be to take advantage of the reductive conditions met in the cytosol which could cleave disulphide bond generally used to link drugs or siRNA to polymer like PEG. This cleavage eventually causes release of the drug in cytosol [85, 86].
8 Micelle–Cell Interaction and In Vivo Fate of Polymeric Micelle
A polymer micelle hardly interacts with cell membrane if its hydrophilic corona is biologically inert but the hydrophilic shell of polymer micelles is not totally inert. It is well known that polymer micelles enter cells by means of endocytosis by specific or nonspecific interaction [87]. The mechanism explaining endocytosis of polymer micelles has not been fully clarified. One possibility based on the role of the labeled dye is charge on the micelle. It is known that positively charged macromolecules can be effectively internalized by electrostatic interaction with heparan sulfate on cell surface [88].On the other hand, polymer micelles are shown to increase the drug accumulation inside cells without endocytosis also. For example, polymer micelle made of PluronicP-85 or P-105 enabled effective accumulation of a hydrophobic dye inside cells by inhibiting P-glycoprotein (P-gp) [89, 90]. A polymer micelle consisting of PEG-b-PCL also showed a similar effect on blocking the P-gp function [91].
Expression of targeting moieties onto micelle surface provides a major strategy to enhance the therapeutic effect of micellar drug carriers. Micelles conjugated with different targeting moieties such as biotin, folate, antibodies, growth factors, or homing peptide shave been developed especially for intracellular delivery of anticancer drugs [92]. However, most of the micelles are based on the physical assembly of block copolymers so their stability in blood is not guaranteed. Therefore, improving the micelle stability in blood should be considered in order to optimize the active targeting strategy using targeting moieties.
In vivo pharmacokinetics and pharmacodynamics of drugs formulated using polymer micelles have been widely studied, using radioisotope as a tool to monitor the biodistribution. Polymeric micelles are mostly located in liver, kidney, spleen, and blood indicating their capability for prolonging circulation time in blood, which is an important rationale to develop micellar formulation of lipophilic drugs. Table 16.3 demonstrates that micelles (or unimers) are highly distributed to organs that have excretion and metabolism functions.
9 Applications of Polymeric Micelle
Most drug carrier applications have been studied with AB- or ABA-type block copolymers because the close relationship between micelles’ properties and the structure of polymers can be evaluated more easily with AB- or ABA-type block copolymers than with the other types of copolymers.
Advantages of polymeric micelle as a drug carrier
Advantages of polymeric micelles as drug carriers are:
-
1.
Very small size (diameter ¼ 10e100 nm)
-
2.
High structural stability
-
3.
Large amount of drug loading
-
4.
High water solubility
-
5.
Low toxicity
-
6.
Incorporation of various chemical species
-
(a)
Polymeric micelles are formed typically in a diameter range from 10 nm to 100 nm with a substantial narrow distribution. This size range is considered ideal for the attainment of stable, long-term circulation of the carrier system in the bloodstream. The small size of polymeric micelles is also a big benefit in the sterilization processes in pharmaceutical productions.
-
(b)
Polymeric micelles possess high structural stability provided by the entanglement of polymer chains in the inner core. This stability has two aspects: static and dynamic [46–49]. Static stability is by a critical micelle concentration (CMC). Generally, polymeric micelles show very low CMC values in a range from 1 mg/mL to 10 mg/mL. These values are much smaller than typical CMC values of micelles forming from low-molecular weight surfactants. The second aspect, dynamic stability, is described by the low dissociation rates of micelles, and this aspect may be more important than the static one for in vivo drug delivery in physiological environments that are in non-equilibrium conditions.
-
(c)
Polymeric micelle carrier system as a drug carrier has advantage of high water solubility even when hydrophobic drugs are incorporated [109]. Generally, in conventional synthetic polymer–drug conjugate systems and antibody–drug conjugate systems, a loss of the carrier’s water solubility resulting from the conjugation of a hydrophobic drug creates a serious problem. Several research groups reported this problem of the polymer–drug conjugates in syntheses [110–112] and in their intravenous injections [113].
-
(d)
Polymeric micelles can incorporate a large number of hydrophobic drug molecules in the micelles’ inner core, and simultaneously, the micelles can maintain their water solubility by inhibiting intermicellar aggregation of the hydrophobic cores with a hydrophilic outer shell layer that works as a barrier against intermicellar aggregation. This is a great advantage because many potent drugs that have been developed in recent years are very hydrophobic and are, therefore, water insoluble.
-
(e)
Polymeric surfactants are known to be less toxic than low-molecular-weight surfactants, such as sodium dodecyl sulfate. Furthermore, in theory, polymeric micelles are considered very safe in relation to chronic toxicity. Possessing a much larger size than that for critical filtration in the kidney, polymeric micelles can evade renal filtration, even if the molecular weight of the constituting block copolymer is lower than the critical molecular weight for renal filtration. On the other hand, all polymer chains can be dissociated (as single polymer chains) from the micelles over a long time period. This phenomenon results in the complete excretion of the block copolymers from the renal route if the polymer chains are designed with a lower molecular weight than the critical value for renal filtration.
-
(f)
The sixth advantage is the fact that various chemical species can be incorporated into polymeric micelles. As explained previously, the most commonly examined chemical species are hydrophobic low-molecular-weight organic compound drugs. These drugs can be incorporated into the micelle inner core either by chemical conjugation to the inner-core-forming polymer block or by physical entrapment owing to hydrophobic interactions between the entrapped drug molecules and the hydrophobic inner-core forming polymer block. Hydrophobic interactions also work as a driving force for micelle formation. On the other hand, polymeric micelles are formed through ionic interactions between charged polymer chains. For example, polymeric micelles form from poly(ethylene glycol) (PEG)-b-poly(lysine) block copolymers and poly(aspartic acid) (ASP) homopolymers where the poly(lysine) chain is positively charged and the poly(ASP) chain is negatively charged. If negatively charged polypeptides [114] or nucleic acid [115] are used in place of poly(ASP), these pharmacologically active macromolecules are incorporated into polymeric micelles for protein, gene, and small interfering RNA delivery purposes. Furthermore, metal ions or metal ions’ chelates can be incorporated into polymeric micelles through coordination bonds or ionic interactions. A platinum chelate cisplatin, which is a widely used anticancer drug, was successfully incorporated into polymeric micelles forming from PEG-b-poly(ASP) through a ligand exchange reaction between a carboxylic acid residue of the poly(ASP) chain and a chloride ion of cisplatin [116, 117]. Alternatively, gadolinium (Gd) ions, which can work as a magnetic resonance imaging (MRI) contrast agent, were incorporated into polymeric micelles by the use of a chelatemoiety-conjugated block copolymer [118, 119]. As stated above, various pharmaceutical drugs, genes, and contrast agents can be incorporated into polymeric micelles with appropriate choices of block copolymer structures.
-
(a)
Disadvantages of polymeric micelle as a drug carrier
Disadvantages of polymeric micelle drug carriers are:
-
1.
Specific disadvantages of polymeric micelle carriers
-
(a)
Difficult polymer synthesis
-
(b)
Immature drug-incorporation technology
-
(a)
-
2.
Common disadvantages of polymeric carriers
-
(a)
Slow extravasation
-
(b)
Possible chronic liver toxicity due to slow metabolic process
-
(a)
The first disadvantage is a fact that relatively high levels of polymer chemistry are needed in the polymeric micelle studies. AB type of block copolymer is one of the most favorable structures for polymeric micelle carriers. The architecture of the AB block copolymer is very simple. However, its synthesis is more difficult than that of random polymers particularly in large industrial scale.
The second disadvantage of the polymeric micelle systems is the immature technology for drug incorporation in a physical manner. Yokoyama et al. reported that physical incorporation efficiencies is dependent on various factors in drug-incorporation processes [116]. Presently, there seem to be no universal incorporation method applicable to any polymer. Furthermore, in some methods the drug incorporation may be difficult on a large industrial scale.
The third disadvantage is much slower extravasation of polymeric carrier systems than that of low-molecular weight drugs. The polymeric systems translocate from the bloodstream to the interstitial space of organs and tissues through intra-cellular channels and inter-cellular junctions, whereas the drugs permeate directly through lipid bilayer cell membranes. Therefore, a long circulation character of the polymeric systems is an essential requirement for delivery of a therapeutic amount owing to compensation of the slow extravasation with a large area under the curve (AUC) value that results from the long circulation.
The forth disadvantage is a risk of chronic liver toxicity. Drugs conjugated or incorporated in the polymeric carrier systems are metabolized in liver in a slower manner than free drug, since access of metabolic enzymes to drugs is inhibited because of the conjugation and incorporation. Therefore, toxic side effects of the conjugated and incorporated drug may be exhibited for a longer period than a case of free drug whose toxic effects can be lowered through metabolism in a short period.
9.1 Drug Solubilization
The micellar core is a compatible microenvironment for incorporating water-insoluble guest molecules as the hydrophobic molecules can be covalently coupled to the block copolymers or physically incorporated into the hydrophobic core of micelles. The solubilization process leads to enhancement of their water solubility and thereby bioavailability [57]. The extent of solubilization depends upon the process of micelle preparation, the compatibility between the drug and the core forming block, chain length of the hydrophobic block, concentration of polymer, and temperature [120]. Above CMC of the polymer, there is a sharp increase in the solubility of drug as it gets more space to occupy in the aggregates of the hydrophobic part of the micelle. However, the core region has limited capacity for accommodation, for instance, Pluronic P85 has a core region which is 13 % of the whole micelle weight [121]. The influence of hydrophobic block length on solubilization of griseofulvin in polyoxy ethylene and polyoxy butylene copolymer micelles investigated showed that the solubilization capacity was dependent on the hydrophobic block length up to a certain extent (15 units of hydrophobic block), after which the solubilization capacity became independent of the same [122]. Dong and coworkers also concluded that solubilization capacity of polyurethane surfactants increased with an increase in the hydrophobic segment of the diblock and triblock polyurethane surfactants [123].
9.2 Sustained Release Using Polymeric Micelle
Current approaches to achieve sustained drug release from the micelles include use of prodrug synthesis, novel polymers, layer by layer assembly of micelles on a solid support, reverse micelles, drug–polymer conjugate micelles, and polymer films that form micelles in vivo. However, the sustained release achieved by these strategies lasts only up to a maximum of few weeks.
9.2.1 Prodrug/Drug Polymer Conjugate Micelle
In this approach a prodrug that is most compatible with the micelle-forming amphiphilic molecule is desirable. The two limiting processes controlling drug release are prodrug release from the micelles and prodrug conversion to drug. One such example is paclitaxel palmitate, a paclitaxel prodrug, which was encapsulated in PEG-b-polycaprolactone (PEG-b-PCL) (Mw of PEG: 5,000, Mw of PCL: 10,500) micelles [124]. Other examples of this approach are summarized in Table 16.4.
9.2.2 Novel Polymers
This is the most common approach used to prepare sustained release micelles. Polymers with very low CMC (<0.1 μg/ml) can be used for prolonging the circulation time before the micelle degrades. The micelles undergo dilution in the body after intravenous injection leading to drop in the concentration of the polymer or surfactant below the CMC if the CMC value is higher. Therefore, a higher concentration of the polymer or surfactant has to be used to prepare the micelles so that they can withstand the dilution in the blood. But, in most cases, the use of high concentrations is not feasible due to toxicity related dose limitations. If the polymer or surfactant has a CMC lower than 0.1 μg/ml, concentrations as low as 5 mg/ml may be used to prepare a micelle formulation in order to counter the dilution effects in the blood. A variety of polymers including diblock copolymers, triblock copolymers and graft copolymers have been investigated for this purpose. Some polymers investigated for sustained release micelle are listed below.
9.2.2.1 Block Copolymers with Lipids
Block copolymers between a polymer and a lipid are one useful approach in preparing micelles. Lipids are more hydrophobic than most polymers and hence, a micelle made with a lipid as its hydrophobic part might lower the CMC. Hence, using fatty acyl chains as hydrophobic segments in an amphiphilic copolymer might be a useful approach. Further, it was observed that increasing the length of the hydrophobic portion of a micelle will lead to a decrease in its CMC [125]. Among diblock copolymer, distearoyl phosphatidyl ethanolamine (DSPE) has been used as the hydrophobic block with hydrophilic polyethylene oxide (PEO) to form 22 nm micelles. These micelles sustained release of lipophilic beclomethasone dipropionate for up to 6 days [126]. Lavasanifar et al. prepared micelles of polyethylene oxide-poly[N-(6-hexyl stearate-l-aspartamide)] (PEO-PHSA) to encapsulate amphotericin B [127]. The release of encapsulated drug was sustained (20 % released in 1 h) while the plain drug was released within 10 min. The release depended inversely on the degree of fatty acid substitution in the core.
9.2.2.2 Block Copolymers with Cyclodextrins
Another approach for drug delivery is supramolecular polymeric micelles which involves non-covalent interactions between a macromolecular polymer, and a small polymer molecule (guest molecule). One such attempt was made using α-cyclodextrins (α-CDs) as the hydrophilic macromolecular host and PCL (Mn = 37,000) as the hydrophobic guest molecule [128]. The formed supramolecular polymeric micelles having a mean diameter of 30 nm resulted in sustained release of an anti-inflammatory drug up to 700 h.
9.2.2.3 Diblock Copolymer Micelles
Diblock polymer that can physically interact with the drug can result in drug retention and sustained release from such polymer micelles. If the drug can form hydrogen bonds with the core of the micelle, then the release obtained from the micelle will be much more sustained. Micelles prepared from PEG-b-poly-l-lactic acid (PEG-b-PLLA; Mw: 8,500 Da) and PEG-b-PCL (Mw: 10,050 Da) block copolymers showed sustained release of the loaded drug, quercetin for approximately 160 h [129].
9.2.2.4 Triblock Copolymer Micelles
A triblock copolymer with small hydrophobic ends and a long hydrophilic midsection can assemble to form flower-like micellar structure in aqueous environment. These flower-like micelles can dissolve the drug in the hydrophobic core and sustain drug release for long periods of time. Zero-order release of sulindac and tetracaine has been reported using PLA–PEO–PLA, triblock flower-like micelles (7–13 nm) for 20 days and 10 days respectively [80].
9.3 Drug Targeting Potentials of Polymeric Micelle
Drug targeting is defined as selective drug delivery to specific physiological sites like organs, tissues, or cells, where the drug’s pharmacological activities are required. Drug-targeting strategies are classified as active targeting and passive targeting [130, 131]. Active targeting aims at an increase in the delivery of drugs to the target by using biologically specific interactions, such as antigen-antibody binding or by utilizing locally applied signals, such as heating and sonication. On the other hand, passive targeting is defined as a method whereby the physical and chemical properties of carrier systems increase the target/non-target ratio of the administered drug.
In most of the active targeting processes particularly for tumors, passive transfer phenomena precede biologically specific interactions except in cases of intravascular targets, such as vascular endothelial targeting. Most tumor targets are located in extra-vascular space. To reach these targets through the bloodstream, translocation through the vascular endothelium is a necessary step, followed by diffusion in the interstitial space.
9.3.1 Passive Drug Targeting to Solid Tumors
The passive targeting of polymeric micelles on solid tumors can be achieved owing to the enhanced permeability and retention effect (EPR effect). Vascular permeability of tumor tissues is enhanced due to malformation of tumor vasculature during rapid angiogenesis and by the actions of secreted factors, such as kinin and vascular permeability factor. As a result of this increased vascular permeability, macromolecules selectively increase their transport from blood vessels to tumor tissues. Furthermore, the lymphatic drainage system does not operate effectively in tumor tissues. Therefore, macromolecules are selectively retained for a prolonged time in the tumor interstitium. However, the carrier systems must fulfill the following two requirements to avoid nonspecific capture at non-tumor sites:
-
1.
The drug carrier systems must possess an appropriate size or molecular weight. The diameter of carriers must be smaller than approximately 200 nm if the reticuloendothelial system’s uptake is to be evaded [132]. Additionally, molecular weights greater than a critical value (approximately 40,000) are favorable for evading renal filtration.
-
2.
The drug carrier systems must not exhibit strong interactions or uptake with or by normal organs (especially the reticuloendothelial systems). These strong interactions and uptakes are typically seen for cationic [133] and hydrophobic carriers [134]. Therefore, the carrier systems should preferably possess hydrophilic surfaces, and their surface charge must be neutral or weakly negative. However, hydrophobic carriers can be used but hydrophilic coating using polymers like PEG is needed to avoid this interaction. Furthermore, the carrier systems must possess no other chemical structures that would be biologically recognizable to normal tissues.
Considering the two afore mentioned requirements, polymeric micelles are very much suited for passive targeting to tumors because they are formed in a diameter ranging from 10 nm to 100 nm. The second requirement can be easily fulfilled through a choice of hydrophilic and neutrally or weakly negatively charged polymers for the outer shell-forming block. With this choice, polymeric micelles can circulate in the bloodstream for a long time period by evading nonspecific capture, resulting in successful attainment of the EPR effect.
The first successful example of tumor targeting with a polymeric micelle carrier was reported by Yokoyama et al, where, doxorubicin (DOX) was chemically conjugated to ASP residues of PEG-poly(ASP) block copolymers (PEG-poly(Asp)) by amide bond formation and was presented as a polymeric micelle system [59, 135–137]. The PEG segment was hydrophilic, whereas the DOX conjugated poly(ASP) chain was hydrophobic. Therefore, the obtained drug-block copolymer conjugate (PEG-poly(Asp)-DOX) formed micellar structures owing to its amphiphilic character. In addition, DOX was also incorporated into the inner core by physical entrapment using hydrophobic interactions with the chemically conjugated DOX molecules. As a result, polymeric micelles containing both the chemically conjugated and the physically entrapped DOX in the inner core were obtained with the PEG outer shell. The DOX entrapped polymeric micelle circulated in the bloodstream for a long time and was delivered to the solid tumor site at much higher concentrations than that of free DOX [137].
Shiraishi et al prepared a polymeric micelle containing an MRI contrast agent using a poly(ethylene glycol)-b-poly(l-lysine) block copolymer derivative which was found to enhance MRI contrasts by shortening the T1 relaxation times of protons of water. This polymeric micelle was found to be targeted to a murine tumor C26, and the tumorwas successfully visualized with the targeted MRI contrast agent [119, 138].
9.3.2 Targeted Micelles
Selective biodistribution of micelles for improved efficiency could theoretically be achieved by using systems which respond to external stimuli like pH and temperature variations or by attaching specific ligands to the exposed hydrophilic ends of the carriers. These targeting mechanisms are referred to as active targeting.
9.3.2.1 pH-Responsive Micelles
Active targeting of drugs from polymeric micelle (PM) can be triggered by pH changes if the change in pH is associated to a pathological process like solid tumors presenting acidosis [139, 140]. Hence, micellar devices have been designed to trigger and/or enhance drug release in response to pH. An extended application of the pH-sensitive micelles has recently been introduced for the formulation of multiple anticancer agents in the context of combinatorial therapy for better patient compliance and higher efficacy through a synergistic mechanism, thereby reducing the therapeutic dose and toxicity of the drugs. In this approach, pH-sensitive micelles are prepared using polymer conjugated with drugs at a precise ratio. For instance, Bae, Y. et al. [141] conjugated DOX and wortmannin to PEG-poly(aspartate hydrazide) through an acid-sensitive hydrazone bond. The polymer–drug conjugates then assembled into micelles in which the drug mixing ratio between DOX and wortmannin was critical. These mixed PM could reduce the DOX dose required for cytotoxicity through a synergistic drug action.
9.3.2.2 Temperature-Sensitive Micelles
The efficiency of the micellar carriers can also be improved by combining the EPR effect with temperature sensitivity. Thermo-responsive PMs were shown to release the loaded drug when the temperature increases beyond the lowest critical solution temperature (LCST), thereby increasing their therapeutic efficacy. For example, PM of (PNIPAM-co-N,N dimethyl-acrylamide)-b-poly(benzyl methacrylate) [80] and PNIPAM-b-poly(butyl methacrylate) (PNIPAM-b-PBMA) [142] loaded with DOX not only showed a thermo-responsive drug release behavior but also showed increased cytotoxicity towards bovine aortic endothelial cells in vitro above their LCST compared to the free drug.
9.3.2.3 Functionalized Micelles
The shell-components of PM, primarily selected to hinder nonspecific interactions and increase blood circulation times, may prevent internalization of the carriers by target cells [143]. Hence, systems presenting ligands at their water-exposed surface have been designed to enhance their selective binding to specific receptors on the cells and to promote the uptake of the drug loaded micelles by receptor-mediated endocytosis and enhance efficacy. For efficient targeting, the receptors must be overexpressed by target cells (e.g., tumor cells) compared to normal tissues [144]. A variety of molecules including antibodies [145, 146] peptides [147, 148], aptamers [149, 150], vitamins and sugar moieties [151] have been used to achieve targeting for anticancer drugs (Table 16.5) and genetic materials (Table 16.6).
The use of antibodies as the targeting moiety presents the advantage of selectivity, high affinity, and minimal competition for the receptor, contrary to what is observed with endogenous molecules such as folic acid (FOL) or transferrin [172, 173]. Antibodies, however, might induce immunogenicity, can be difficult to produce/handle and present a large size, potentially putting strain on micelle self-assembly. The use of small molecules such as sugars and vitamins can then become advantageous. As reported in many studies, it is important that the functional groups be readily available on the surface for efficient attachment to the receptors. The ligands can be attached either before or after the assembly of the particulate carrier.
9.3.2.3.1 Monoclonal Antibodies and Antigen Binding Fragments
The whole antibody or its fragments such as the fragment antigen binding (Fab′) and F(ab′)2 or scFV (single-chain variable) can be used for targeting [174]. Using an antigen binding fragment (e.g., Fab′) instead of the whole monoclonal antibody provides an advantage of avoiding steric hindrance during complex formation due to its lower molecular weight (Fig. 16.3).

Targeting can be achieved with the whole antibody (a) or its fragments such as F(ab′)2 (b), (Fab′) (c)
In addition, it can induce less immunogenicity when in vivo applications are sought. Micelles functionalized with either monoclonal antibody or Fab′ have both shown higher cellular uptake compared to non-targeted micelles. For instance, Merdan et al. [146] observed more than sixfold binding of the targeted micelles to the ovarian carcinoma cells (OVCAR-3 cells) using Fab′-PEG-PEI/pDNA PICM compared to the unmodified system. Increased cellular uptake and fourfold higher tumor accumulation of the drug (paclitaxel) in vivo was reported by Torchilin and coworkers [175] for whole antibody-PM targeting lung cancer cells.
9.3.2.3.2 Aptamers
Nucleic acid ligands (aptamers) have been used recently for the targeting of drug encapsulated PMs. Aptamers selected by screening a random library of nucleic acids to specific molecular targets can fold by intramolecular interactions into unique three-dimensional conformations capable of binding to target antigens with high affinity and specificity [176, 177]. An RNA aptamer targeting the prostate specific membrane antigen, overexpressed on prostate acinar epithelial cells was used to decorate PEG-b- PLA or PEG-b-PLGA micelles loaded with docetaxel [149, 150]. This targeted delivery system showed a marked increase in the cellular uptake and increased cytotoxicity in vitro and in increased antitumor efficacy in vivo over the non-targeted PM. However, the instability of DNA or RNA molecules in the blood may limit the use of these ligands in the clinic.
9.3.2.3.3 Non-immune Peptides and Proteins
Proteins like transferrin, an iron transporter, are used to target rapidly dividing cells such as tumor cells. Hu- Lieskovan et al. [187] developed a cyclodextrin-containing polycation that self-assembled with a siRNA inhibiting EWS-FLI1 (a gene that is found in 85 % of patients with Ewing’s tumor) for multicomponent delivery for metastatic tumor treatment. The surface of the complexes was then decorated with PEG and targeted with transferrin which downregulated onco-proteins and suppressed the spread of metastatic tumors up on systemic administration of the system. Transferrin, being a big molecule (80 kDa), can put strain on micelles and so a smaller peptide might be advantageous. One such example is the cyclic RGD (cRGD) peptide, a cellular transmembrane protein that has a marked role in tumor growth and metastasis and targets the αvβ3 integrin [190, 191]. The cRGD peptide was conjugated to maleimide-terminated PEG-b-PCL PM encapsulating DOX. The delivery system showed a threefold increase in cellular uptake when the surface density was adjusted to 5 % cRGD while, a more pronounced 30-fold increase was observed with 76 % cRGD attachment [158]. Similar studies with a PEGylated branched PEI modified with an RGD peptide at the distal end of the PEG further demonstrated the possibility of in vivo targeting with this peptide [188].
9.3.2.3.4 Vitamins
The expression levels of folic acid (FOL) receptors in tumors have been reported to be 100–300 times higher than those observed in normal tissues [144]. Functionalized PEG-b-PLGA chains with FOL covalently derivatized via its gamma-carboxyl group showed high affinity for the FOL receptor (especially for FOL receptors alpha) and retains its receptor binding affinity [157, 163, 192]. This polymeric system physically mixed with PEG-b-PLGA-DOX and free-DOX to produce targeted micelles decreased the tumor growth rate compared to control non-targeted micelles and enhanced the antitumor efficacy when administered at the same dose level.
9.3.2.3.5 Sugars
One of the most common cancers affecting human is the hepatocarcinoma and so the development of liver-targeted drug carriers is therefore highly desirable. Advances in this area rely on the fact that hepatocytes express carbohydrate receptors, i.e., asialoglycoprotein receptors (ASGPR), that recognize different sugar moieties such as lactose, galactose or mannose, allowing for liver-specific delivery [193, 194]. Jeong et al. [181] have prepared paclitaxel-loaded galactose-PEG-b-PBLG micelles, and showed a greater in vitro uptake and cytotoxicity of the micelles in an ASGPR-expressing cancer cell line compared to an analogous non-ASGPR expressing cell line. Alternatively, delivery system having lactose attached to the surface of PICM for siRNA delivery exhibited gene silencing of firefly luciferase expression in HuH-7 cells expressing ASGPR that was comparable to cationic liposomes (oligofectamine) [189].
10 Conclusions
Micelles are a promising drug delivery vehicle for drugs and genetic materials. These core-shell self-assemblies can also be tailored to increase the solubility of poorly water-soluble drugs just as protect labile hydrophilic drugs from premature degradation. Because of their nanometer size and hydrated outer layer, micelles can prolong the circulation time of an encapsulated drug and passively accumulate at tumor sites, thereby reducing its systemic toxicity and enhancing its efficacy. Micelles that actively target tissues can also be prepared by utilizing stimuli-responsive components or by attaching recognition groups at their surface. It is also important that the polymeric carriers need to be stable and retain the encapsulated drug long enough for any of these applications to be achievable. To meet this purpose, both the thermodynamic stability and kinetic stability of the micelles can be improved by varying the nature of the hydrophobic block, increasing the hydrophobic/hydrophilic balance, increasing the hydrophobic block length or by accommodating hydrophobic molecules in the core. In addition, micelle stabilization can also be achieved by cross-linking either the core or the corona of preformed micelles, by preparing crystalline micelles or by designing intrinsically stable unimolecular polymeric micelles (UPM). The parameters affecting micelle stability, however, need to be carefully optimized with respect to their influence on the extent of drug solubilization and drug release kinetics. Much progress has been achieved to modulate the stability and stimuli responsiveness of PM in vitro, while, these strategies still remain to be tested in vivo to demonstrate real control over the pharmacological properties of the encapsulated drugs. With the structural requirements for micelle stability and drug release are still under conflict, future work should focus on the development and clinical application of multifunctional micelles capable of delivering drugs at target sites in a controlled/triggered fashion.
Abbreviations
- AFM:
-
Atomic force microscopy
- ATRA:
-
All-trans retinoic acid
- AUC:
-
Area under the curve
- CMC:
-
Critical micellar concentration
- CMT:
-
Critical micellization temperature
- CPT:
-
Camptothecin
- CsA:
-
Cyclosporine A
- DNA:
-
Deoxy ribonucleic acid
- DOX:
-
Doxorubicin
- DSPE:
-
Distearoyl phosphatidyl ethanolamine
- EPR:
-
Enhanced permeability and retention
- F-5-CADA:
-
Fluorescein-5-carbonyl azide diacetate
- FA:
-
Folic acid
- Gd:
-
Gadolinium
- HEMAm:
-
N-(2-hydroxyethyl) methacrylamide
- HLB:
-
Hydrophilic–lipophilic balance
- LCST:
-
Low critical solution temperature
- MHC:
-
Minimal hydrotrope concentration
- MRI:
-
Magnetic resonance imaging
- PAsp:
-
Poly(aspartic acid)
- PBLA:
-
Poly(benzyl-l-aspartate)
- PCL:
-
Poly(e-caprolactone)
- PDLLA:
-
PEG-b-poly(d,l-lactic acid)
- PEG:
-
Poly(ethylene glycol)
- PEO:
-
Polyethylene oxide
- PET:
-
Positron emission tomography
- P-gp:
-
P-glycoprotein
- PICMs:
-
Polyion complex micelles
- PM:
-
Polymeric micelle
- PMMA:
-
Poly(methacrylate)
- PPO:
-
Poly(propylene oxide)
- PTX:
-
Paclitaxel
- PVA:
-
Poly(vinyl alcohol)
- RNA:
-
Ribonucleic acid
- RT:
-
Room temperature
- SEM:
-
Scanning electron microscopy
- TEM:
-
Transmission electron microscopy
References
Zana R (2005) Dynamics of surfactant self-assemblies: micelles, microemulsions, vesicles and lyotropic phases. CRC press, Florida
Sinko PJ, Allen LV Jr, Popovich NG, Ansel HC (2006) Martin’s physical pharmacy and pharmaceutical sciences, 5th edn. Lippincott Williams & Wilkins, Philadelphia
Adams ML, Lavasanifar A, Kwon GS (2003) Amphiphilic block copolymers for drug delivery. J Pharm Sci 92:1343–1355. doi:10.1002/jps.10397
Moroi Y (1992) Micelles: theoretical and applied aspects. Springer, New York
Tuzar Z, Kratochvil P (1976) Block and graft copolymer micelles in solution. Adv Colloid Interface Sci 6:201–232. doi:10.1016/0001-8686(76)80009-7
Hickok RS, Wedge SA, Hansen AL, Morris KF, Billiot FH, Warner IM (2002) Pulsed field gradient NMR investigation of solubilization equilibria in amino acid and dipeptide terminated micellar and polymeric surfactant solutions. Magn Reson Chem 40:755–761. doi:10.1002/mrc.1099
Jones M, Leroux J (1999) Polymeric micelles—a new generation of colloidal drug carriers. Eur J Pharm Biopharm 48:101–111. doi:10.1016/S0939-6411(99)00039-9
Erhardt R, Böker A, Zettl H, Kaya H, Pyckhout-Hintzen W, Krausch G, Abetz V, Müller AH (2001) Janus micelles. Macromolecules 34:1069–1075. doi:10.1021/ma000670p
Riess G, Hurtrez G, Bohadur P (1985) Block copolymers. Wiley-Interscience, Encyclopedia of Polymer Science and Engineering 2: 324–434
Xu J, Ge Z, Zhu Z, Luo S, Liu H, Liu S (2006) Synthesis and micellization properties of double hydrophilic A2BA2 and A4BA4 non-linear block copolymers. Macromolecules 39: 8178–8185. doi:10.1021/ma061934w
Bouchemal K, Agnely F, Koffi A, Ponchel G (2009) A concise analysis of the effect of temperature and propanediol-1, 2 on Pluronic F127 micellization using isothermal titration microcalorimetry. J Colloid Interface Sci 338:169–176. doi:10.1016/j.jcis.2009.05.075
Ranger M, Jones MC, Yessine MA, Leroux JC (2001) From well‐defined diblock copolymers prepared by a versatile atom transfer radical polymerization method to supramolecular assemblies. J Polym Sci A Polym Chem 39:3861–3874. doi:10.1002/pola.10029
Zhang J, Ma PX (2009) Host-guest interaction mediated polymeric core-shell assemblies: versatile nanocarriers for drug delivery. Angew Chem Int Ed Engl 48:964. doi:10.1002/anie.200804135
Liu YL, Lin GC, Wu CS (2008) Preparation of polysulfone‐g‐poly (N‐isopropylacrylamide) graft copolymers through atom transfer radical polymerization and formation of temperature‐responsive nanoparticles. J Polym Sci A Polym Chem 46:4756–4765. doi:10.1002/pola.22809
Peng X, Zhang L (2007) Formation and morphologies of novel self-assembled micelles from chitosan derivatives. Langmuir 23:10493–10498
Kriz J, Pleštil J, Tuzar Z, Pospíšil H, Brus J, Jakeš J, Masar B, Vlcek P, Doskocilova D (1999) Interface affected polymer dynamics: NMR, SANS, and DLS study of the influence of shell-core interactions on the core chain mobility of poly (2-ethylhexyl acrylate)-block-poly (acrylic acid) micelles in water. Macromolecules 32:397–410. doi:10.1021/ma9809334
Procházka K, Martin TJ, Munk P, Webber SE (1996) Polyelectrolyte poly (tert-butyl acrylate)-block-poly (2-vinylpyridine) micelles in aqueous media. Macromolecules 29:6518–6525. doi:10.1021/ma960630e
Geng Y, Discher DE (2005) Hydrolytic degradation of poly (ethylene oxide)-block-polycaprolactone worm micelles. J Am Chem Soc 127:12780–12781
Hu Y, Zhang L, Cao Y, Ge H, Jiang X, Yang C (2004) Degradation behavior of poly (ε-caprolactone)-b-poly (ethylene glycol)-b-poly (ε-caprolactone) micelles in aqueous solution. Biomacromolecules 5:1756–1762
Xu B, Yuan J, Ding T, Gao Q (2010) Amphiphilic biodegradable poly (ε-caprolactone)-poly (ethylene glycol)-poly (ε-caprolactone) triblock copolymers: synthesis, characterization and their use as drug carriers for folic acid. Polym Bull 64:537–551. doi:10.1007/s00289-009-0157-5
Lee SC, Kim KJ, Jeong Y-K, Chang JH, Choi J (2005) pH-Induced reversible complexation of poly (ethylene glycol) and poly (ε-caprolactone)-b-poly (methacrylic acid) copolymer micelles. Macromolecules 38:9291–9297. doi:10.1021/ma051380h
Danhier F, Magotteaux N, Ucakar B, Lecouturier N, Brewster M, Préat V (2009) Novel self-assembling PEG-p-(CL-< i> co-TMC) polymeric micelles as safe and effective delivery system for Paclitaxel. Eur J Pharm Biopharm 73:230–238
Arimura H, Ohya Y, Ouchi T (2004) The formation of biodegradable polymeric micelles from newly synthesized poly (aspartic acid)‐block‐polylactide AB‐type diblock copolymers. Macromol Rapid Commun 25:743–747
Alani AW, Bae Y, Rao DA, Kwon GS (2010) Polymeric micelles for the pH-dependent controlled, continuous low dose release of paclitaxel. Biomaterials 31:1765–1772
Huang CK, Lo CL, Chen HH, Hsiue GH (2007) Multifunctional micelles for cancer cell targeting, distribution imaging, and anticancer drug delivery. Adv Funct Mater 17:2291–2297. doi:10.1002/adfm.200600818
Hu F-Q, Liu L-N, Du Y-Z, Yuan H (2009) Synthesis and antitumor activity of doxorubicin conjugated stearic acid-< i> g-chitosan oligosaccharide polymeric micelles. Biomaterials 30:6955–6963
Webber S (1998) Polymer micelles: an example of self-assembling polymers. J Phys Chem B 102:2618–2626. doi:10.1021/jp980386o
Gohy JF (2005) Block copolymer micelles. Adv Polym Sci 190:65–136. doi:10.1007/12_048
Jette KK, Law D, Schmitt EA, Kwon GS (2004) Preparation and drug loading of poly (ethylene glycol)-block-poly (ε-caprolactone) micelles through the evaporation of a cosolvent azeotrope. Pharm Res 21:1184–1191. doi:10.1023/B:PHAM.0000033005.25698.9c
Fournier E, Dufresne MH, Smith DC, Ranger M, Leroux JC (2004) A novel one-step drug-loading procedure for water-soluble amphiphilic nanocarriers. Pharm Res 21:962–968. doi:10.1023/B:PHAM.0000029284.40637.69
La SB, Okano T, Kataoka K (1996) Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly(ethylene oxide)-poly(beta-benzyl L-aspartate) block copolymer micelles. J Pharm Sci 85:85–90. doi:10.1021/js950204r
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249. doi:10.1016/S1056-8719(00)00107-6
Attwood D, Booth C, Yeates SG, Chaibundit C, Ricardo NM (2007) Block copolymers for drug solubilisation: relative hydrophobicities of polyether and polyester micelle-core-forming blocks. Int J Pharm 345:35–41. doi:10.1016/j.ijpharm.2007.07.039
Hildebrand JH (1949) A critique of the theory of solubility of non-electrolytes. Chem Rev 44:37–45. doi:10.1021/cr60137a003
Flory PJ (1953) Principles of polymer chemistry. Cornell University Press, New York
Liu RR, Forrest ML, Kwon GS (2008) 13 Micellization and drug solubility enhancement part II: polymeric micelles. Water-Insoluble Drug Formulation, 307
Letchford K, Liggins R, Burt H (2008) Solubilization of hydrophobic drugs by methoxy poly(ethylene glycol)-block-polycaprolactone diblock copolymer micelles: theoretical and experimental data and correlations. J Pharm Sci 97:1179–1190. doi:10.1002/jps.21037
Ooya T, Sang CL, Kang MH, Park K (2006) Hydrotropic nanocarriers for poorly soluble drugs. In: Mozafari MR (ed) Nanocarrier technologies. Springer, Netherlands
Coffman RE, Kildsig DO (1996) Hydrotropic solubilization—mechanistic studies. Pharm Res 13:1460–1463. doi:10.1023/A:1016011125302
Bauduin P, Renoncourt A, Kopf A, Touraud D, Kunz W (2005) Unified concept of solubilization in water by hydrotropes and cosolvents. Langmuir 21:6769–6775. doi:10.1021/la050554l
Charman W, Lai C, Craik D, Finnin B, Reed B (1993) Self-association of nicotinamide in aqueous-solution: NMR studies of nicotinamide and the mono-and di-methyl-substituted amide analogs. Aust J Chem 46:377–385. doi:10.1071/CH9930377
Landauer J, McConnell H (1952) A study of molecular complexes formed by aniline and aromatic nitrohydrocarbons1, 2. J Am Chem Soc 74:1221–1224. doi:10.1021/ja01125a025
Quina FH, Alonso EO, Farah JP (1995) Incorporation of nonionic solutes into aqueous micelles: a linear solvation free energy relationship analysis. J Phys Chem 99:11708–11714. doi:10.1021/j100030a014
Rasool AA, Hussain AA, Dittert LW (1991) Solubility enhancement of some water‐insoluble drugs in the presence of nicotinamide and related compounds. J Pharm Sci 80:387–393. doi:10.1002/jps.2600800422
Sanghvi R, Evans D, Yalkowsky SH (2007) Stacking complexation by nicotinamide: a useful way of enhancing drug solubility. Int J Pharm 336:35–41. doi:10.1016/j.ijpharm.2006.11.025
Calderara F, Hruska Z, Hurtrez G, Lerch J, Nugay T, Riess G (1994) Investigation of polystyrene-poly (ethylene oxide) block copolymer micelle formation in organic and aqueous solutions by nonradiative energy transfer experiments. Macromolecules 27:1210–1215. doi:10.1021/ma00083a020
Wang Y, Kausch CM, Chun M, Quirk RP, Mattice WL (1995) Exchange of chains between micelles of labeled polystyrene-block-poly (oxyethylene) as monitored by nonradiative singlet energy transfer. Macromolecules 28:904–911. doi:10.1021/ma00108a016
Wilhelm M, Zhao CL, Wang Y, Xu R, Winnik MA, Mura JL, Riess G, Croucher MD (1991) Poly (styrene-ethylene oxide) block copolymer micelle formation in water: a fluorescence probe study. Macromolecules 24:1033–1040. doi:10.1021/ma00005a010
Desjardins A, Eisenberg A (1991) Colloidal properties of block ionomers. 1. Characterization of reverse micelles of styrene-b-metal methacrylate diblocks by size-exclusion chromatography. Macromolecules 24:5779–5790
Trivedi R, Kompella UB (2010) Nanomicellar formulations for sustained drug delivery: strategies and underlying principles. Nanomedicine (Lond) 5:485–505. doi:10.2217/nnm.10.10
Nishiyama N, Kataoka K (2006) Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther 112:630–648. doi:10.1016/j.pharmthera.2006.05.006
Thurmond KB, Kowalewski T, Wooley KL (1996) Water-soluble knedel-like structures: the preparation of shell-cross-linked small particles. J Am Chem Soc 118:7239–7240. doi:10.1021/ja961299h
Li T, Ning F, Xie J, Chen D, Jiang M (2002) Preparation and morphologies of shell cross-linked micelles based on commercial poly (styrene-block-ethylene-co-butene-block-styrene). Polym J 34:529–533. doi:10.1295/polymj.34.529
Zhang JX, Yan MQ, Li XH, Qiu LY, Li XD, Li XJ, Jin Y, Zhu KJ (2007) Local delivery of indomethacin to arthritis-bearing rats through polymeric micelles based on amphiphilic polyphosphazenes. Pharm Res 24:1944–1953. doi:10.1007/s11095-007-9322-4
Kakizawa Y, Harada A, Kataoka K (1999) Environment-sensitive stabilization of core-shell structured polyion complex micelle by reversible cross-linking of the core through disulfide bond. J Am Chem Soc 121:11247–11248. doi:10.1021/ja993057y
Hu X, Chen X, Wei J, Liu S, Jing X (2009) Core crosslinking of biodegradable block copolymer micelles based on poly(ester carbonate). Macromol Biosci 9:456–463. doi:10.1002/mabi.200800158
Rapoport N (1999) Stabilization and activation of Pluronic micelles for tumor-targeted drug delivery. Colloids Surf B: Biointerfaces 16:93–111. doi:10.1016/S0927-7765(99)00063-6
Bogdanov AA Jr, Mt L, Weissleder R (1999) Approaches and agents for imaging the vascular system. Adv Drug Deliv Rev 37:279–293. doi:10.1016/S0169-409X(98)00098-2
Yokoyama M, Okano T, Sakurai Y, Ekimoto H, Shibazaki C, Kataoka K (1991) Toxicity and antitumor activity against solid tumors of micelle-forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res 51:3229–3236
Lebduskova P, Kotek J, Hermann P, Vander Elst L, Muller RN, Lukes I, Peters JA (2004) A gadolinium(III) complex of a carboxylic-phosphorus acid derivative of diethylenetriamine covalently bound to inulin, a potential macromolecular MRI contrast agent. Bioconjug Chem 15:881–889. doi:10.1021/bc049966g
Caravan P, Ellison JJ, McMurry TJ, Lauffer RB (1999) Gadolinium (III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem Rev 99:2293–2352. doi:10.1021/cr980440x
Pathak AP, Gimi B, Glunde K, Ackerstaff E, Artemov D, Bhujwalla ZM (2004) Molecular and functional imaging of cancer: advances in MRI and MRS. Methods Enzymol 386:3–60
Jeong JH, Park TG (2001) Novel polymer-DNA hybrid polymeric micelles composed of hydrophobic poly (D, L-lactic-co-glycolic acid) and hydrophilic oligonucleotides. Bioconjug Chem 12:917–923
Jeong YI, Cheon JB, Kim SH, Nah JW, Lee YM, Sung YK, Akaike T, Cho CS (1998) Clonazepam release from core-shell type nanoparticles in vitro. J Control Release 51:169–178. doi:10.1016/S0168-3659(97)00163-6
Teng Y, Morrison M, Munk P, Webber S, Procházka K (1998) Release kinetics studies of aromatic molecules into water from block polymer micelles. Macromolecules 31:3578–3587. doi:10.1021/ma971721u
Rösler A, Vandermeulen GW, Klok H-A (2012) Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv Drug Deliv Rev 64:270–279. doi:10.1016/j.addr.2012.09.026
Shi B, Fang C, You MX, Zhang Y, Fu S, Pei Y (2005) Stealth MePEG-PCL micelles: effects of polymer composition on micelle physicochemical characteristics, in vitro drug release, in vivo pharmacokinetics in rats and biodistribution in S180 tumor bearing mice. Colloid Polym Sci 283:954–967. doi:10.1007/s00396-004-1243-8
Savic R, Azzam T, Eisenberg A, Maysinger D (2006) Assessment of the integrity of poly (caprolactone)-b-poly(ethylene oxide) micelles under biological conditions: a fluorogenic-based approach. Langmuir 22:3570–3578. doi:10.1021/la0531998
Burt HM, Zhang X, Toleikis P, Embree L, Hunter WL (1999) Development of copolymers of poly (D, L-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel. Colloids Surf B: Biointerfaces 16:161–171. doi:10.1016/S0927-7765(99)00067-3
Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng JX (2008) Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivo forster resonance energy transfer imaging. Langmuir 24:5213–5217. doi:10.1021/la703570m
Gao Z, Fain HD, Rapoport N (2004) Ultrasound-enhanced tumor targeting of polymeric micellar drug carriers. Mol Pharm 1:317–330. doi:10.1021/mp049958h
Gao ZG, Fain HD, Rapoport N (2005) Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. J Control Release 102:203–222. doi:10.1016/j.jconrel.2004.09.021
Tannock IF, Rotin D (1989) Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 49:4373–4384
Schild HG, Tirrell DA (1991) Microheterogeneous solutions of amphiphilic copolymers of N-isopropylacrylamide. An investigation via fluorescence methods. Langmuir 7:1319–1324
Gillies ER, Frechet JM (2005) pH-Responsive copolymer assemblies for controlled release of doxorubicin. Bioconjug Chem 16:361–368. doi:10.1021/bc049851c
Gillies ER, Jonsson TB, Frechet JM (2004) Stimuli-responsive supramolecular assemblies of linear-dendritic copolymers. J Am Chem Soc 126:11936–11943. doi:10.1021/ja0463738
Chung J, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T (1999) Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly (N-isopropylacrylamide) and poly (butylmethacrylate). J Control Release 62:115–127
Chung J, Yokoyama M, Aoyagi T, Sakurai Y, Okano T (1998) Effect of molecular architecture of hydrophobically modified poly (N-isopropylacrylamide) on the formation of thermoresponsive core-shell micellar drug carriers. J Control Release 53:119–130. doi:10.1016/S0168-3659(97)00244-7
Chen G, Hoffman AS (1995) Graft copolymers that exhibit temperature-induced phase transitions over a wide range of pH. Nature 373:49–52
Nakayama M, Okano T (2005) Polymer terminal group effects on properties of thermoresponsive polymeric micelles with controlled outer-shell chain lengths. Biomacromolecules 6:2320–2327
Taillefer J, Jones MC, Brasseur N, van Lier JE, Leroux JC (2000) Preparation and characterization of pH-responsive polymeric micelles for the delivery of photosensitizing anticancer drugs. J Pharm Sci 89:52–62. doi:10.1002/(SICI)1520-6017(200001)89:1<52:: AID-JPS6>3.0.CO;2-D
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867. doi:10.1038/nature01322
Rehor A, Schmoekel H, Tirelli N, Hubbell JA (2008) Functionalization of polysulfide nanoparticles and their performance as circulating carriers. Biomaterials 29:1958–1966. doi:10.1016/j.biomaterials.2007.12.035
Rehor A, Hubbell JA, Tirelli N (2005) Oxidation-sensitive polymeric nanoparticles. Langmuir 21:411–417. doi:10.1021/la0478043
Kim SH, Jeong JH, Lee SH, Kim SW, Park TG (2006) PEG conjugated VEGF siRNA for anti-angiogenic gene therapy. J Control Release 116:123–129
Lee SH, Kim SH, Park TG (2007) Intracellular siRNA delivery system using polyelectrolyte complex micelles prepared from VEGF siRNA-PEG conjugate and cationic fusogenic peptide. Biochem Biophys Res Commun 357:511–516. doi:10.1016/j.bbrc.2007.03.185
Allen C, Yu Y, Eisenberg A, Maysinger D (1999) Cellular internalization of PCL20-b-PEO44 block copolymer micelles. BBA Biomembranes 1421:32–38. doi:10.1016/S0005-2736(99)00108-X
Poon GM, Gariepy J (2007) Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem Soc Trans 35:788–793. doi:10.1042/BST0350788
Miller DW, Batrakova EV, Waltner TO, Alakhov V, Kabanov AV (1997) Interactions of pluronic block copolymers with brain microvessel endothelial cells: evidence of two potential pathways for drug absorption. Bioconjug Chem 8:649–657. doi:10.1021/bc970118d
Rapoport N, Marin A, Luo Y, Prestwich GD, Muniruzzaman MD (2002) Intracellular uptake and trafficking of Pluronic micelles in drug-sensitive and MDR cells: effect on the intracellular drug localization. J Pharm Sci 91:157–170. doi:10.1002/jps.10006
Zastre JA, Jackson JK, Wong W, Burt HM (2008) P-glycoprotein efflux inhibition by amphiphilic diblock copolymers: relationship between copolymer concentration and substrate hydrophobicity. Mol Pharm 5:643–653. doi:10.1021/mp7001347
Mikhail AS, Allen C (2009) Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels. J Control Release 138:214–223. doi:10.1016/j.jconrel.2009.04.010
Park YJ, Lee JY, Chang YS, Jeong JM, Chung JK, Lee MC, Park KB, Lee SJ (2002) Radioisotope carrying polyethylene oxide–polycaprolactone copolymer micelles for targetable bone imaging. Biomaterials 23:873–879. doi:10.1016/s0142-9612(01)00196-x
Liu J, Zeng F, Allen C (2007) In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur J Pharm Biopharm 65:309–319. doi:10.1016/j.ejpb.2006.11.010
Lin WJ, Chen YC, Lin CC, Chen CF, Chen JW (2006) Characterization of pegylated copolymeric micelles and in vivo pharmacokinetics and biodistribution studies. J Biomed Mater Res B Appl Biomater 77:188–194. doi:10.1002/jbm.b.30418
Aliabadi HM, Brocks DR, Lavasanifar A (2005) Polymeric micelles for the solubilization and delivery of cyclosporine A: pharmacokinetics and biodistribution. Biomaterials 26:7251–7259. doi:10.1016/j.biomaterials.2005.05.042
Novakova K, Laznicek M, Rypacek F, Machova L (2003) Pharmacokinetics and distribution of 125I-PLA-b-PEO block copolymers in rats. Pharm Dev Technol 8:153–161. doi:10.1081/PDT-120018484
Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Wan Kim S, Seo MH (2001) In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release 72:191–202. doi:10.1016/S0168-3659(01)00275-9
Yamamoto Y, Nagasaki Y, Kato Y, Sugiyama Y, Kataoka K (2001) Long-circulating poly(ethylene glycol)–poly(d, l-lactide) block copolymer micelles with modulated surface charge. J Control Release 77:27–38. doi:10.1016/s0168-3659(01)00451-5
Hu Z, Luo F, Pan Y, Hou C, Ren L, Chen J, Wang J, Zhang Y (2008) Arg-Gly-Asp (RGD) peptide conjugated poly(lactic acid)-poly(ethylene oxide) micelle for targeted drug delivery. J Biomed Mater Res A 85:797–807. doi:10.1002/jbm.a.31615
Kawano K, Watanabe M, Yamamoto T, Yokoyama M, Opanasopit P, Okano T, Maitani Y (2006) Enhanced antitumor effect of camptothecin loaded in long-circulating polymeric micelles. J Control Release 112:329–332. doi:10.1016/j.jconrel.2006.03.012
Kawakami S, Opanasopit P, Yokoyama M, Chansri N, Yamamoto T, Okano T, Yamashita F, Hashida M (2005) Biodistribution characteristics of all-trans retinoic acid incorporated in liposomes and polymeric micelles following intravenous administration. J Pharm Sci 94: 2606–2615. doi:10.1002/jps.20487
Batrakova EV, Li S, Li Y, Alakhov VY, Elmquist WF, Kabanov AV (2004) Distribution kinetics of a micelle-forming block copolymer Pluronic P85. J Control Release 100:389–397. doi:10.1016/j.jconrel.2004.09.002
Wang Y, Li Y, Zhang L, Fang X (2008) Pharmacokinetics and biodistribution of paclitaxel-loaded pluronic P105 polymeric micelles. Arch Pharm Res 31:530–538. doi:10.1007/s12272-001-1189-2
Le Garrec D, Gori S, Luo L, Lessard D, Smith D, Yessine M-A, Ranger M, Leroux J-C (2004) Poly (N-vinylpyrrolidone)-block-poly (d, l-lactide) as a new polymeric solubilizer for hydrophobic anticancer drugs: in vitro and in vivo evaluation. J Control Release 99:83–101. doi:10.1016/j.jconrel.2004.06.018
Bae Y, Nishiyama N, Kataoka K (2007) In vivo antitumor activity of the folate-conjugated pH-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjug Chem 18:1131–1139. doi:10.1021/bc060401p
Tian HY, Deng C, Lin H, Sun J, Deng M, Chen X, Jing X (2005) Biodegradable cationic PEG-PEI-PBLG hyperbranched block copolymer: synthesis and micelle characterization. Biomaterials 26:4209–4217. doi:10.1016/j.biomaterials.2004.11.002
Rijcken CJ, Snel CJ, Schiffelers RM, van Nostrum CF, Hennink WE (2007) Hydrolysable core-crosslinked thermosensitive polymeric micelles: Synthesis, characterisation and in vivo studies. Biomaterials 28:5581–5593. doi:10.1016/j.biomaterials.2007.08.047
Yokoyama M, Kwon GS, Okano T, Sakurai Y, Seto T, Kataoka K (1992) Preparation of micelle-forming polymer-drug conjugates. Bioconjug Chem 3:295–301. doi:10.1021/bc00016a007
Hoes C, Potman W, Van Heeswijk W, Mud J, De Grooth B, Greve J, Feijen J (1985) Optimization of macromolecular prodrugs of the antitumor antibiotic adriamycin. J Control Release 2:205–213. doi:10.1016/0168-3659(85)90046-X
Duncan R, Kopeckova-Rejmanova P, Strohalm J, Hume I, Cable HC, Pohl J, Lloyd JB, Kopecek J (1987) Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of daunomycin and puromycin conjugates in vitro. Br J Cancer 55:165–174
Endo N, Umemoto N, Kato Y, Takeda Y, Hara T (1987) A novel covalent modification of antibodies at their amino groups with retention of antigen-binding activity. J Immunol Methods 104:253–258. doi:10.1016/0022-1759(87)90512-6
Zunino F, Pratesi G, Micheloni A (1989) Poly (carboxylic acid) polymers as carriers for anthracyclines. J Control Release 10:65–73. doi:10.1016/0168-3659(89)90018-7
Harada A, Kataoka K (1998) Novel polyion complex micelles entrapping enzyme molecules in the core: preparation of narrowly-distributed micelles from lysozyme and poly (ethylene glycol)-poly (aspartic acid) block copolymer in aqueous medium. Macromolecules 31:288–294. doi:10.1021/ma971277v
Kataoka K, Togawa H, Harada A, Yasugi K, Matsumoto T, Katayose S (1996) Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline. Macromolecules 29:8556–8557. doi:10.1021/ma961217+
Yokoyama M, Okano T, Sakurai Y, Suwa S, Kataoka K (1996) Introduction of cisplatin into polymeric micelle. J Control Release 39:351–356. doi:10.1016/0168-3659(95)00165-4
Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K (2003) Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res 63:8977–8983
Nakamura E, Makino K, Okano T, Yamamoto T, Yokoyama M (2006) A polymeric micelle MRI contrast agent with changeable relaxivity. J Control Release 114:325–333. doi:10.1016/j.jconrel.2006.05.030
Shiraishi K, Kawano K, Maitani Y, Yokoyama M (2010) Polyion complex micelle MRI contrast agents from poly(ethylene glycol)-b-poly(l-lysine) block copolymers having Gd-DOTA; preparations and their control of T1-relaxivities and blood circulation characteristics. J Control Release 148:160–167. doi:10.1016/j.jconrel.2010.08.018
Clark SL, Crowley AJ, Schmidt PG, Donoghue AR, Piché CA (2004) Long-term delivery of ivermectin by use of poly (D, L-lactic-co-glycolic) acid microparticles in dogs. Am J Vet Res 65:752–757. doi:10.2460/ajvr.2004.65.752
Sartor O (2003) Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology 61:25–31. doi:10.1016/S0090-4295(02)02396-8
Singh SR, Grossniklaus HE, Kang SJ, Edelhauser HF, Ambati BK, Kompella UB (2009) Intravenous transferrin, RGD peptide and dual-targeted nanoparticles enhance anti-VEGF intraceptor gene delivery to laser-induced CNV. Gene Ther 16:645–659. doi:10.1038/gt.2008.185
Raghuvanshi RS, Singh O, Panda AK (2001) Formulation and characterization of immunoreactive tetanus toxoid biodegradable polymer particles. Drug Deliv 8:99–106. doi:10.1080/107175401750177089
Forrest ML, Yanez JA, Remsberg CM, Ohgami Y, Kwon GS, Davies NM (2008) Paclitaxel prodrugs with sustained release and high solubility in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelle nanocarriers: pharmacokinetic disposition, tolerability, and cytotoxicity. Pharm Res 25:194–206. doi:10.1007/s11095-007-9451-9
Zhang N, Guo SR, Li HQ, Liu L, Li ZH, Gu JR (2006) Synthesis of Three Types of Amphiphilic Poly (ethylene glycol)‐block‐Poly (sebacic anhydride) Copolymers and Studies of their Micellar Solutions. Macromol Chem Phys 207:1359–1367. doi:10.1002/macp.200600100
Gaber NN, Darwis Y, Peh KK, Tan YT (2006) Characterization of polymeric micelles for pulmonary delivery of beclomethasone dipropionate. J Nanosci Nanotechnol 6:3095–3101. doi:10.1166/jnn.2006.426
Lavasanifar A, Samuel J, Kwon GS (2002) The effect of fatty acid substitution on the in vitro release of amphotericin B from micelles composed of poly(ethylene oxide)-block-poly(N-hexyl stearate-l-aspartamide). J Control Release 79:165–172. doi:10.1016/S0168-3659(01)00537-5
Dong H, Li Y, Cai S, Zhuo R, Zhang X, Liu L (2008) A facile one-pot construction of supramolecular polymer micelles from alpha-cyclodextrin and poly(epsilon-caprolactone). Angew Chem Int Ed Engl 47:5573–5576. doi:10.1002/anie.200800952
Yang X, Zhu B, Dong T, Pan P, Shuai X, Inoue Y (2008) Interactions between an anticancer drug and polymeric micelles based on biodegradable polyesters. Macromol Biosci 8:1116–1125. doi:10.1002/mabi.200800085
Yokoyama M, Okano T (1996) Targetable drug carriers: present status and a future perspective. Adv Drug Deliv Rev 21:77–80. doi:10.1016/S0169-409X(96)00439-5
Sugiyama Y (1996) Importance of pharmacokinetic considerations in the development of drug delivery systems. Adv Drug Deliv Rev 19:333–334. doi:10.1016/0169-409X(96)00007-5
Litzinger DC, Buiting AM, van Rooijen N, Huang L (1994) Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim Biophys Acta 1190:99–107. doi:10.1016/0005-2736(94)90038-8
Takakura Y, Hashida M (1996) Macromolecular carrier systems for targeted drug delivery: pharmacokinetic considerations on biodistribution. Pharm Res 13:820–831. doi:10.1023/A:1016084508097
Illum L, Davis S, Müller R, Mak E, West P (1987) The organ distribution and circulation time of intravenously injected colloidal carriers sterically stabilized with a blockcopolymer-poloxamine 908. Life Sci 40:367–374. doi:10.1016/0024-3205(87)90138-X
Yokoyama M, Inoue S, Kataoka K, Yui N, Sakurai Y (1987) Preparation of adriamycin‐conjugated poly (ethylene glycol)‐poly (aspartic acid) block copolymer. A new type of polymeric anticancer agent. Die Makromolekulare Chemie, Rapid Commun 8:431–435
Yokoyama M, Miyauchi M, Yamada N, Okano T, Sakurai Y, Kataoka K, Inoue S (1990) Characterization and anticancer activity of the micelle-forming polymeric anticancer drug adriamycin-conjugated poly(ethylene glycol)-poly(aspartic acid) block copolymer. Cancer Res 50:1693–1700
Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kataoka K (1999) Selective delivery of adiramycin to a solid tumor using a polymeric micelle carrier system. J Drug Target 7:171–186. doi:10.3109/10611869909085500
Shiraishi K, Kawano K, Minowa T, Maitani Y, Yokoyama M (2009) Preparation and in vivo imaging of PEG-poly(L-lysine)-based polymeric micelle MRI contrast agents. J Control Release 136:14–20. doi:10.1016/j.jconrel.2009.01.010
Engin K, Leeper DB, Cater JR, Thistlethwaite AJ, Tupchong L, McFarlane JD (1995) Extracellular pH distribution in human tumours. Int J Hyperthermia 11:211–216. doi:10.3109/02656739509022457
Helmlinger G, Yuan F, Dellian M, Jain RK (1997) Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med 3:177–182. doi:10.1038/nm0297-177
Bae Y, Diezi TA, Zhao A, Kwon GS (2007) Mixed polymeric micelles for combination cancer chemotherapy through the concurrent delivery of multiple chemotherapeutic agents. J Control Release 122:324–330. doi:10.1016/j.jconrel.2007.05.038
Chung J, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T (1999) Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly (N-isopropylacrylamide) and poly (butylmethacrylate). J Control Release 62:115–127. doi:10.1016/S0168-3659(99)00029-2
Jaeghere F, Allemann E, Feijen J, Kissel T, Doelker E, Gurny R (2000) Cellular uptake of PEO surface-modified nanoparticles: evaluation of nanoparticles made of PLA: PEO diblock and triblock copolymers. J Drug Target 8:143–153. doi:10.3109/10611860008996860
Ross JF, Chaudhuri PK, Ratnam M (1994) Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer 73:2432–2443
Ulbrich K, Etrych T, Chytil P, Jelinkova M, Rihova B (2003) HPMA copolymers with pH-controlled release of doxorubicin: in vitro cytotoxicity and in vivo antitumor activity. J Control Release 87:33–47. doi:10.1016/S0168-3659(02)00348-6
Merdan T, Callahan J, Petersen H, Kunath K, Bakowsky U, Kopečková P, Kissel T, Kopecek J (2003) Pegylated polyethylenimine-Fab′ antibody fragment conjugates for targeted gene delivery to human ovarian carcinoma cells. Bioconjug Chem 14:989–996, 10.1021/bc0340767
Vinogradov S, Batrakova E, Li S, Kabanov A (1999) Polyion complex micelles with protein-modified corona for receptor-mediated delivery of oligonucleotides into cells. Bioconjug Chem 10:851–860. doi:10.1021/bc990037c
Nah J-W, Yu L, S-o H, Ahn C-H, Kim SW (2002) Artery wall binding peptide-poly(ethylene glycol)-grafted-poly(l-lysine)-based gene delivery to artery wall cells. J Control Release 78:273–284. doi:10.1016/S0168-3659(01)00499-0
Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R (2004) Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res 64:7668–7672. doi:10.1158/0008-5472.CAN-04-2550
Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R (2006) Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A 103:6315–6320. doi:10.1073/pnas.0601755103
Lim DW, Yeom YI, Park TG (2000) Poly (DMAEMA-NVP)-b-PEG-galactose as gene delivery vector for hepatocytes. Bioconjug Chem 11:688–695. doi:10.1021/bc000014u
Yoo HS, Lee EA, Park TG (2002) Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J Control Release 82:17–27. doi:10.1016/S0168-3659(02)00088-3
Danson S, Ferry D, Alakhov V, Margison J, Kerr D, Jowle D, Brampton M, Halbert G, Ranson M (2004) Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br J Cancer 90:2085–2091. doi:10.1038/sj.bjc.6601856
Alakhov V, Klinski E, Li S, Pietrzynski G, Venne A, Batrakova E, Bronitch T, Kabanov A (1999) Block copolymer-based formulation of doxorubicin. From cell screen to clinical trials. Colloids Surf B: Biointerfaces 16:113–134
Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Watanabe N (2004) Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br J Cancer 91:1775–1781. doi:10.1038/sj.bjc.6602204
Venne A, Li S, Mandeville R, Kabanov A, Alakhov V (1996) Hypersensitizing effect of pluronic L61 on cytotoxic activity, transport, and subcellular distribution of doxorubicin in multiple drug-resistant cells. Cancer Res 56:3626–3629
Yoo HS, Park TG (2004) Folate receptor targeted biodegradable polymeric doxorubicin micelles. J Control Release 96:273–283. doi:10.1016/j.jconrel.2004.02.003
Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J (2004) cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem Int Ed Engl 43:6323–6327. doi:10.1002/anie.200460800
Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K (2005) Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem 16:122–130. doi:10.1021/bc0498166
Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ (2004) Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res 10:3708–3716. doi:10.1158/1078-0432.CCR-03-0655
Jeong YI, Seo SJ, Park IK, Lee HC, Kang IC, Akaike T, Cho CS (2005) Cellular recognition of paclitaxel-loaded polymeric nanoparticles composed of poly(gamma-benzyl L-glutamate) and poly(ethylene glycol) diblock copolymer endcapped with galactose moiety. Int J Pharm 296:151–161. doi:10.1016/j.ijpharm.2005.02.027
Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B (2003) Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci U S A 100: 6039–6044. doi:10.1073/pnas.0931428100
Elsabahy M, Perron ME, Bertrand N, Yu GE, Leroux JC (2007) Solubilization of docetaxel in poly(ethylene oxide)-block-poly(butylene/styrene oxide) micelles. Biomacromolecules 8:2250–2257. doi:10.1021/bm070226v
Taillefer J, Brasseur N, van Lier JE, Lenaerts V, Le Garrec D, Leroux JC (2001) In-vitro and in-vivo evaluation of pH-responsive polymeric micelles in a photodynamic cancer therapy model. J Pharm Pharmacol 53:155–166. doi:10.1211/0022357011775352
Le Garrec D, Taillefer J, Van Lier JE, Lenaerts V, Leroux JC (2002) Optimizing pH-responsive polymeric micelles for drug delivery in a cancer photodynamic therapy model. J Drug Target 10:429–437. doi:10.1080/1061186021000001887
Mizumura Y, Matsumura Y, Hamaguchi T, Nishiyama N, Kataoka K, Kawaguchi T, Hrushesky WJ, Moriyasu F, Kakizoe T (2001) Cisplatin‐incorporated polymeric micelles eliminate nephrotoxicity, while maintaining antitumor activity. Cancer Sci 92:328–336. doi:10.1111/j.1349-7006.2001.tb01099.x
Nishiyama N, Kato Y, Sugiyama Y, Kataoka K (2001) Cisplatin-loaded polymer-metal complex micelle with time-modulated decaying property as a novel drug delivery system. Pharm Res 18:1035–1041. doi:10.1023/A:1010908916184
Nishiyama N, Yokoyama M, Aoyagi T, Okano T, Sakurai Y, Kataoka K (1999) Preparation and characterization of self-assembled polymer-metal complex micelle from cis-dichlorodiammineplatinum (II) and poly (ethylene glycol)-poly (α, β-aspartic acid) block copolymer in an aqueous medium. Langmuir 15:377–383. doi:10.1021/la980572l
Torchilin VP (2001) Structure and design of polymeric surfactant-based drug delivery systems. J Control Release 73:137–172. doi:10.1016/S0168-3659(01)00299-1
Adams ML, Andes DR, Kwon GS (2003) Amphotericin B encapsulated in micelles based on poly(ethylene oxide)-block-poly(L-amino acid) derivatives exerts reduced in vitro hemolysis but maintains potent in vivo antifungal activity. Biomacromolecules 4:750–757. doi:10.1021/bm0257614
Benahmed A, Ranger M, Leroux JC (2001) Novel polymeric micelles based on the amphiphilic diblock copolymer poly(N-vinyl-2-pyrrolidone)-block-poly(D, L-lactide). Pharm Res 18:323–328. doi:10.1023/A:1011054930439
Singh M (1999) Transferrin as a targeting ligand for liposomes and anticancer drugs. Curr Pharm Des 5:443–452
Qian ZM, Li H, Sun H, Ho K (2002) Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev 54:561–587
Allen TM (2002) Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer 2:750–763. doi:10.1038/nrc903
Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B (2003) Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci 100: 6039–6044
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–822. doi:10.1038/346818a0
Lupold SE, Hicke BJ, Lin Y, Coffey DS (2002) Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 62:4029–4033
Elsabahy M, Zhang M, Gan S-M, Waldron KC, Leroux J-C (2008) Synthesis and enzymatic stability of PEGylated oligonucleotide duplexes and their self-assemblies with polyamidoamine dendrimers. Soft Matter 4:294–302. doi:10.1039/B714221H
Yessine MA, Dufresne MH, Meier C, Petereit HU, Leroux JC (2007) Proton-actuated membrane-destabilizing polyion complex micelles. Bioconjug Chem 18:1010–1014. doi:10.1021/bc060159m
Jeong JH, Kim SW, Park TG (2003) A new antisense oligonucleotide delivery system based on self-assembled ODN-PEG hybrid conjugate micelles. J Control Release 93:183–191. doi:10.1016/j.jconrel.2003.07.002
Jeong JH, Kim SH, Kim SW, Park TG (2005) Polyelectrolyte complex micelles composed of c-raf antisense oligodeoxynucleotide-poly(ethylene glycol) conjugate and poly(ethylenimine): effect of systemic administration on tumor growth. Bioconjug Chem 16:1034–1037. doi:10.1021/bc0497315
Jeong JH, Kim SW, Park TG (2003) Novel intracellular delivery system of antisense oligonucleotide by self-assembled hybrid micelles composed of DNA/PEG conjugate and cationic fusogenic peptide. Bioconjug Chem 14:473–479. doi:10.1021/bc025632k
Kim SH, Jeong JH, Mok H, Lee SH, Kim SW, Park TG (2007) Folate receptor targeted delivery of polyelectrolyte complex micelles prepared from ODN-PEG-folate conjugate and cationic lipids. Biotechnol Prog 23:232–237. doi:10.1021/bp060243g
Harada A, Togawa H, Kataoka K (2001) Physicochemical properties and nuclease resistance of antisense-oligodeoxynucleotides entrapped in the core of polyion complex micelles composed of poly(ethylene glycol)-poly(L-lysine) block copolymers. Eur J Pharm Sci 13:35–42. doi:10.1016/S0928-0987(00)00205-0
Kakizawa Y, Harada A, Kataoka K (2001) Glutathione-sensitive stabilization of block copolymer micelles composed of antisense DNA and thiolated poly (ethylene glycol)-b lock-poly (l-lysine): a potential carrier for systemic delivery of antisense DNA. Biomacromolecules 2:491–497. doi:10.1021/bm000142l
Itaka K, Kanayama N, Nishiyama N, Jang WD, Yamasaki Y, Nakamura K, Kawaguchi H, Kataoka K (2004) Supramolecular nanocarrier of siRNA from PEG-based block catiomer carrying diamine side chain with distinctive pKa directed to enhance intracellular gene silencing. J Am Chem Soc 126:13612–13613. doi:10.1021/ja047174r
Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ (2005) Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res 65:8984–8992. doi:10.1158/0008-5472.CAN-05-0565
Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, Molema G, Lu PY, Scaria PV, Woodle MC (2004) Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res 32:e149. doi:10.1093/nar/gnh140
Oishi M, Nagasaki Y, Itaka K, Nishiyama N, Kataoka K (2005) Lactosylated poly (ethylene glycol)-siRNA conjugate through acid-labile β-thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J Am Chem Soc 127:1624–1625. doi:10.1021/ja044941d
Woodle MC, Scaria P, Ganesh S, Subramanian K, Titmas R, Cheng C, Yang J, Pan Y, Weng K, Gu C, Torkelson S (2001) Sterically stabilized polyplex: ligand-mediated activity. J Control Release 74:309–311. doi:10.1016/S0168-3659(01)00339-X
Mizejewski GJ (1999) Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med 222:124–138
Kamen BA, Smith AK (2004) A review of folate receptor alpha cycling and 5-methyltetrahydrofolate accumulation with an emphasis on cell models in vitro. Adv Drug Deliv Rev 56:1085–1097. doi:10.1016/j.addr.2004.01.002
Jule E, Nagasaki Y, Kataoka K (2003) Lactose-installed poly(ethylene glycol)-poly(d, l-lactide) block copolymer micelles exhibit fast-rate binding and high affinity toward a protein bed simulating a cell surface. A surface plasmon resonance study. Bioconjug Chem 14:177–186
Sugahara K, Togashi H, Takahashi K, Onodera Y, Sanjo M, Misawa K, Suzuki A, Adachi T, Ito J, Okumoto K (2003) Separate analysis of asialoglycoprotein receptors in the right and left hepatic lobes using 99mTc‐GSA SPECT. Hepatology 38:1401–1409. doi:10.1016/j.hep.2003.09.031
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Controlled Release Society
About this chapter
Cite this chapter
Murthy, R.S.R. (2015). Polymeric Micelles in Targeted Drug Delivery. In: Devarajan, P., Jain, S. (eds) Targeted Drug Delivery : Concepts and Design. Advances in Delivery Science and Technology. Springer, Cham. https://doi.org/10.1007/978-3-319-11355-5_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-11355-5_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11354-8
Online ISBN: 978-3-319-11355-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)