
Abstract
RANK ligand (RANKL) is a member of the tumor necrosis factor alpha superfamily of cytokines. It is the only known ligand binding to a membrane receptor named receptor activator of nuclear factor-kappa B (RANK), thereby triggering recruitment of TNF receptor-associated factor (TRAF) adaptor proteins and activation of downstream pathways. RANK/RANKL signaling is controlled by a decoy receptor, osteoprotegerin (OPG), but also has additional more complex levels of regulation. It is crucial for the differentiation of bone-resorbing osteoclasts and is deregulated in disease processes such as osteoporosis and cancer bone metastasis. Cells expressing RANK and RANKL are commonly found in the tumor environment. In many tumor types, the RANK/RANKL pathway is overexpressed, and this is in most cases correlated with poor prognosis. RANK signaling plays an important role in the innate and adaptive immune response, generates regulatory T (Treg) cells, and increases the production of cytokines. It is also involved in chemo resistance in vitro. Recent evidence suggests that RANKL blockade improves the efficacy of anti-CTLA-4 antibodies against solid tumors and experimental metastasis. Therefore, there is increasing interest to use RANKL inhibition as an immunomodulatory strategy in an attempt to make immune-resistant tumor responsive to immune therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- RANK
- RANKL
- Osteoprotegerin
- Microenvironment
- Cancer
- Bone health
- Immunomodulation
- PFD-L1
- CTLA-4
- Immune response
- Inflammation
- Immune tolerance
- Angiogenesis
3.1 Background
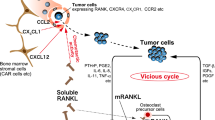
In most cancer types, only a minority of patients have an improved survival after immune therapy. Mutational burden, neoantigen load, quality and clonality of neoantigens, expression of antigen presenting molecules and immune checkpoints, interferon gamma responsiveness, and composition of the microenvironment (hot versus cold tumors), all influence the beneficial effects of immune therapies [1]. Although combinations of immune therapy (e.g., CTLA-4 and PD-L1 blocking) can be synergistic, they do not resolve their diminutive effectiveness in cancer treatment and often induce significant additional toxicity [2,3,2,5]. Hence, there is an increasing interest in combining immune therapy with less toxic immune modulating drugs to sensitize immune unresponsive tumors to immune therapies [6]. Recent data suggest that RANK/RANKL inhibition may be an attractive approach to increase the effectiveness of immunotherapy. Signaling between the receptor activator of nuclear factor-kappa B (RANK) and its ligand (RANKL) is essential for the differentiation of bone-resorbing osteoclasts and is deregulated in pathological processes such as postmenopausal osteoporosis or cancer-induced bone destruction [2]. However, cells expressing RANK and RANKL are also commonly found in the tumor microenvironment. RANK signaling plays an important role in the innate and adaptive immune response as it generates regulatory T (Treg) cells and increases production of cytokines [7, 8]. In this chapter, the effects of RANK/RANKL signaling inhibition on the microenvironment of malignant tumors are reviewed. It is hypothesized that this approach may be used to improve the response to immunotherapy (Fig. 3.1).

Main effects of RANK/RANKL signaling pathway on tumor growth, immune cells, and microenvironment
3.2 The RANK/RANKL Signaling Pathway
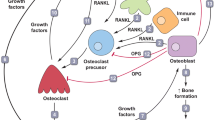
The receptor activator of nuclear factor-kappa B ligand (RANKL) was originally identified in T cells and dendritic cells (DC) [2]. It is a type II homotrimeric transmembrane protein that has three known isoforms. RANKL1 and RANKL2 are expressed as membrane bound proteins. RANKL3 is a soluble secreted protein that is formed by cleavage of the membranous counterparts or by alternative splicing [9]. The RANKL has a large cytoplasmic domain containing four cystein-rich repeat motifs and two N-glycosylation sites. The full length RANKL is called RANKL1, in RANKL2 a part of the intracellular domain is deleted, while in RANKL3 the N-terminal part misses [7, 10]. The RANKL is encoded by the TNFS11 gene in humans and is also named osteoclast differentiation factor (ODF), osteoprotegerin ligand (OPGL), or TNF-related activation induced cytokine (TRANCE) [9, 11]. It is the only known ligand binding to a membrane receptor named receptor activator of nuclear factor-kappa B (RANK), which is a type I transmembrane protein belonging to the TNF receptor superfamily (TNFRSF11A) [11, 12]. Binding between RANKL and RANK induces trimerization of the receptor. This triggers recruitment of TNF receptor-associated factors (TRAF), adaptor proteins, and activation of downstream signaling pathways (such as NF-kB, AKT/PKB, JNK, and the MAP kinase cascade) [7, 13, 14]. A regulatory system is built into the RANK/RANKL signaling pathway by means of a decoy receptor called osteoprotegerin (OPG, TBFRSF11B) interacting with RANKL [2]. OPG is a soluble glycoprotein that can exist either as a 60-kDa monomer or as a 120-kD dimer but lacks transmembrane or cytoplasmatic domains. The dimerization of OPG increases the affinity of OPG to RANKL dramatically and is essential for RANK/RANKL signal inhibition [15]. Several factors can upregulate OPG expression such as estrogen (which is important for bone metabolism), TRAIL, Wnt, and TNFalpha [7]. On the other hand, it can be downregulated by PTH and TGF-beta [9]. The overall inhibitory effect of OPG on RANKL depends on the balance of its binding to these various ligands [11, 12]. The RANK/RANKL signaling network is further complexed by a second, more recently discovered, decoy receptor for RANKL, LGR4 [14]. LGR4 suppresses canonical RANK signaling by competing with RANK to bind RANKL. The binding of RANKL to LGR4 activates the Gαq and GSK3-β signaling pathway. This suppresses the expression and activity of nuclear factor of activated T cells and calcineurin-dependent 1 (NFATC1) during osteoclastogenesis. Furthermore, functional RANK splicing variants have also been identified, implicating several sophisticated levels of the pathway [16].
3.3 The Functional Role of RANK/RANKL Signaling in Humans
RANK and RANKL can be detected in many different tissues, such as the bone, prostate, thymus, mammary glands, and liver, implicating a functional role in these organs [17]. Studies in mice indicate that RANK/RANKL signaling is required for mammary gland development and lymph node formation [7, 17, 18, 19]. The signaling pathway’s crucial role in healthy bone remodeling and bone homeostasis is, however, much better documented [8]. The RANK/RANKL pathway regulates the formation of multinucleated osteoclasts from their monocyte-macrophage precursor cells and subsequently also their activation and survival [8]. By binding RANKL, OPG prevents it to connect to and activate RANK, thereby protecting the skeleton from excessive bone resorption [19, 20]. When deregulated, this pathway may lead to pathological processes such as cancer-induced bone destruction and osteoporosis but also chronic inflammatory processes such as inflammatory bowel diseases and arthritis [14, 20, 21].
Another well-known functional role for RANK/RANKL signaling is that of modulating the immune response. RANK/RANKL and OPG knockout mice showed a disrupted immune phenotype (e.g., impaired T or B cell development) [7, 22]. RANKL can be found in tumor-infiltrating lymphocytes (TILs), immature dendritic cells, B cells, macrophages, and monocytes [2]. RANK activation induces lymphocyte differentiation, T-cell activation, and dendritic cell (DC) survival, triggering intracellular signaling pathways (e.g., MAPK, NFkB, p38, and c-JNK) and even extracellular kinases (ERK) [17, 23, 24, 25]. RANKL can induce the expression of multiple activating cytokines by DCs, including IL-1, IL-6, IL-12, and IL-15, and can enhance DC survival via the induction of the antiapoptotic protein Bcl-xL (B-cell lymphoma-extra large) [2]. Dendritic cells prime and activate T cells during the immune response by processing and presenting antigens to them. The RANKL signal can alter the function of dendritic cells, which may lead to an increase of Foxp3-positive Tregs [7]. Recent evidence suggests that in response to injury, pericytes are also able to modulate local tissue immune responses via several independent pathways including RANKL signaling. In this area, the OPG/RANK/RANKL axis in association with the functions of pericytes may be involved in vasculogenesis, the process of atherosclerosis by altering lipid metabolism, vascular signaling, and angiogenesis [26, 27].
3.4 RANK/RANKL Signaling in Cancer
Several studies documented RANK signaling to be important in a variety of cancers [23,24,25,26,27,28,29,30,31,32,33,34,34,35,36,37,38,39,40,41,42,43]. This was recently nicely reviewed by Renema et al. and de Groot et al. [17]. Tregs are a CD4+ helper T-cell subset that can suppress autoimmune responses in the body and are critical to create an immune suppressive environment in cancers [2]. Together with other partners, such as TAMs, they can create a status of local immunosuppression surrounding the tumor [7]. TAMs express immune checkpoint modulators (such as PD-L1) that directly inhibit activated T cells and produce various chemokines that attract other immunosuppressive cells, such as Tregs and myeloid-derived suppressor cells (MDSCs) [11]. In many situations, the RANK network is an important driver to create an immunosuppressive microenvironment, thereby promoting tumor progression. The central role of RANK/RANKL signaling in bone metastasis has been well studied [9]. The RANK signal network has been shown to drive epithelial to mesenchymal transition (EMT), induce stem cell-like phenotypes, promote osteomimicry, and give cancer cells the ability to home to bone [11, 39]. In a large population of breast cancer patients, strikingly high levels of RANK expression in the primary tumor were predictive for the frequency of the later occurrence of bone metastasis [37]. Recently, it seems that RANK signaling is important in the biology of many tumor types beyond bone metastasis [2, 7, 8, 11, 17, 23,24,25,26,27,28,29,30,31,32,33,34,34,35,36,37,38,39,40,41,42,43]. RANK and RANKL-expressing cells are commonly found in the tumor microenvironment [2]. The RANKL/RANK pathway is often overexpressed in cancers of the prostate, endometrium, stomach, breast, cervix, stomach, bladder, oesophagus, and thyroid, which is correlated with poor prognosis [23,24,25,26,27,28,29,30,31,32,33,34,34,35,36,37,38,39,40,41,42,43]. RANKL has been detected in endothelial cells and implicated in angiogenesis [7].
There is some circumstantial evidence suggesting that paracrine signaling through RANK/RANKL is responsible for the expansion of mammary stem cells observed during pregnancy and luteal cycles [13, 38]. MMTV-RANK transgenic mice are prone to develop mammary tumors, which may be related to activated RANK signaling [35]. Pharmacologic inhibition of RANKL or genetic ablation of RANK reduces (particularly estrogen and progesterone receptor negative) mammary tumor and metastasis development in animal models [32]. Breast cancer cells are able to produce RANKL and stimulate osteoclast differentiation [16, 38, 39]. In humans, high RANK expression is associated with altered mammary differentiation, which suggests that increased RANK signaling may contribute to breast carcinogenesis [13, 40]. High RANK expression was particularly detected in human primary breast adenocarcinomas that lack expression of the hormone receptors, in tumors with high pathologic grade and proliferation index. It is associated with the presence of metastases and poor prognosis [37]. It has been shown in vitro that HIF-1 alpha-induced expression of RANKL initiates increased migration of breast cancer cells via PI3K/AKT signaling, illustrating that the RANK/RANKL pathway also plays an important role in breast cancer progression [41, 42].
Mouse models and randomized studies in humans have shown that combination of antibodies blocking OPG or RANK with chemotherapy, hormone therapy, or targeted drugs resulted in stronger decrease of tumor burden in the bone [8, 17]. However, inhibition of RANK signaling also has a direct effect on tumor cells at other locations [28]. The RANK/RANKL pathway was variably expressed in tumors of the thyroid, and increased serum OPG was also correlated with poor prognosis in gastric, cervical, esophageal, and bladder carcinoma [23,24,25,26,27, 29,30,31,32]. Song et al. found that RANK expression was significantly higher in hepatocellular carcinoma (HCC) than in peritumoral hepatic tissue [33]. HCC cell lines express RANK constitutively, and activation of the RANK-RANKL axis significantly promoted migration and invasion ability of HCC cells in vitro. Recently, it has been demonstrated that RANK/RANKL expression is also significantly elevated in endometrial and prostate cancer tissue, particularly in tumors of higher stage [20, 34, 35, 44]. Therefore, there may be a role for RANKL inhibitors as a therapeutic strategy.
3.5 Effects of the RANK/RANKL Signaling Pathway on the Tumor Microenvironment
RANK and RANKL expressing cells are commonly found in the tumor microenvironment [20]. RANKL modulates the immune response by inducing T-cell proliferation and dendritic cell survival [45]. In human breast carcinomas, RANKL is found in tumor-infiltrating lymphocytes (TILs), and RANK is strongly expressed in tumor-associated macrophages (TAMs) [18]. TAMs accumulate in the microenvironment and, depending on their M2 or M1 phenotype, are involved in tumor growth, angiogenesis, and metastasis. RANKL acts as a chemoattractant for these cells [2]. RANK/RANKL signaling in M2 macrophages modulates production of chemokines, promoting the proliferation of Tregs and thereby creating an immunosuppressive environment. As RANKL is mainly produced by Tregs, a vicious circle is established in conjunction with the TAMs mainly expressing RANK [7, 46]. Tumor-infiltrating Tregs have been shown to stimulate mammary cancer metastasis through RANKL-RANK signaling [47]. RANKL treatment enhances survival of mature dendritic cells (DCs) and triggers generation of proinflammatory cytokines (IL-1, IL-6, and IL-12) that can promote differentiation of CD4+ T cells into Th1 cells, providing a major costimulatory factor for CD4+ T-cell responses [47]. RANK is also expressed on NK cells, playing an important role in immunosurveillance. RANKL/RANK is involved in crosstalk between the bone and the immune system. It stimulates osteoclasts to function as antigen-presenting cells, thereby activating CD4+ and CD8+ T cells. A similar phenomenon might also be present in the microenvironment of solid tumors [7]. The crosstalk of tumor cells with the immune system is not completely understood, but the impact of RANK-RANKL signaling on the tumor immune response is likely to be context specific [4]. Due to sequestering OPG by tumor cells or entrapment of OPG by the proteoglycans and glycosaminoglycans of the extracellular matrix, a microenvironment is created that facilitates the expansion of the tumor cells [48]. In addition, OPG can block TRAIL activity, thereby acting as an antiapoptotic and pro-proliferative stimulus for cancer cells [11, 21]. It has been shown that RANK/RANKL signaling can promote the initial stages of cancer development by inducing stemness and epithelial-mesenchymal transition [19]. RANKL (e.g., produced by osteoblasts or bone marrow stromal cells) attracts RANK-expressing cells and induces their migration by activation of specific signaling pathways, such as the MAP kinase pathway [44]. RANKL was also detected in endothelial cells and has been implicated in angiogenesis through Src and phospholipase C-dependent mechanisms [2, 49].
3.6 RANKL Signaling Inhibition
The only commercially available inhibitor of RANKL is denosumab. This drug is a fully human monoclonal antibody that binds RANKL, thereby blocking its interaction with RANK [2, 8]. Denosumab is approved by the Food and Drug Administration for the treatment of osteoporosis and giant cell tumor of the bone and for the prevention and treatment of skeletal complications caused by bone metastases and lytic bone lesions in multiple myeloma [8]. The drug has a well-known and acceptable toxicity profile [8]. It remains unclear whether RANK/RANKL inhibition with denosumab in patients with cancer has any effect beyond the bone. In a post hoc analysis of patients with non-small-cell carcinoma of the lung (NSCLC) that were included in a phase III randomized trial comparing zoledronic acid versus denosumab, a survival benefit was observed (HR 0.80; 95% CI 0.67–0.95, p = 0.01) for the patients treated in the denosumab arm [50]. There was no difference in the delay of bone events in both groups, and the beneficial effect of denosumab could be observed in patients with visceral metastasis, as well as in patients with bone metastasis only. However, the recent prospective SPLENDOUR trial could not show any improvement in OS or PFS by adding denosumab to standard first-line therapy in patients with metastatic NSCLC [51].
The effect of adjuvant denosumab in women with early breast cancer was recently studied in two large, multicenter, prospective, randomized trials [52, 53]. In the ABCSG-18 study, it was shown that disease-free survival was significantly better in the denosumab group [52]. This study compared placebo or denosumab 60 mg subcutaneously every 6 months for 5 years in 3425 postmenopausal patients with hormone-sensitive early breast cancer treated with an aromatase inhibitor. In the DCARE study, which assessed 4509 high-risk early breast cancer patients treated with standard therapy either with or without denosumab 120 mg SC every month (for 6 months, then 3 monthly up to 5 years), no improvement in bone metastasis-free, disease-free, or overall survival was reported, even though there was an improvement in time to bone metastasis at site of first recurrence in the denosumab group [53]. It is important to mention that most (95.9%) of these patients had received taxane or anthracycline-based chemotherapy. This raises the hypothesis that chemotherapy may reduce some of the tumor suppressive effects of RANK/RANKL inhibition in the cancer microenvironment. Other explanations may be the differences in molecular characteristics of the tumors of these patient populations, or effects of the menopause and endocrine treatment on the tumor behavior. It is clear that more research is necessary to unravel the effect of denosumab on tumor behavior. In the D-BEYOND trial, the biological effects of two neoadjuvant injections of 120 mg denosumab (1 week apart) in 27 patients with premenopausal primary breast cancer were evaluated [54]. The authors concluded that 2 weeks of RANKL inhibition did not have an effect on the tumor proliferation rate, but significantly increased the number of TILS in the tumor environment, making them theoretically more susceptible for immune therapy. Recently, some additional evidence emerged that RANK/RANKL inhibition may have a role as immune modulator. In preclinical studies, RANKL blockade improves the efficacy of anti-CTLA-4-targeted antibodies in solid tumor models of metastasis [53]. Bakhru et al. showed that antibodies blocking RANKL and CTLA-4 cooperate to increase the frequency of tumor-infiltrating CD4+ T cells expressing cytolytic markers, thereby improving antimelanoma immunity [55]. Addition of RANKL blockade to anti-PD-1 and anti CTLA-4 resulted in superior tumor responses and was most effective if RANKL inhibition was given concurrent or following checkpoint blockade [54]. This triple combination therapy improved T-cell effector function in tumor bearing mice by increasing the proportion of tumor-infiltrating CD4+ and CD8+ T cells that can produce both interferon gamma and TNF. In 2014, Smyth et al. described a case of a rapidly advancing metastatic melanoma with aggressive and symptomatic bone metastases requiring treatment with the anti-RANKL antibody denosumab for palliation in a patient who was concomitantly treated with ipilimumab (an anti-CTLA-4 antibody) [56]. She had a spectacular partial response and was alive at 62 weeks. In a melanoma preclinical model, these authors could demonstrate that monoclonal antibodies (mAbs) directed to CTLA-4 or RANKL have modest antimetastatic activities in monotherapy, but when these drugs were combined at the time of intravenous melanoma inoculation, the development of metastases was significantly reduced. Mechanistically, the combined effect of anti-CTLA-4 and anti-RANKL depends on lymphocytes or natural killer cells. In a retrospective study, Afzal and Shirai evaluated the synergistic effect of immune checkpoint inhibitors and denosumab in metastatic melanoma patients [57]. Eleven (29.72%) out of 37 patients were treated with immune checkpoint inhibitors and denosumab, and the others only immune checkpoint inhibitors. The median progression-free and overall survival in the cohort having the combination treatment, respectively, was 11.6 and 57 months compared with 4.15 and 22.8 months in the control group. Although there are potential confounders, this suggests that adding denosumab to immune checkpoint inhibitors may have a beneficial effect on outcome. In a subsequent study, Ahern et al. assessed the efficacy of a combination of RANKL and CTLA-4 blockade by analysis of tumor-infiltrating lymphocytes, tumor growth, and metastasis in a model using a variety of neutralizing antibodies and gene-targeted mice [58]. RANKL blockade improved the efficacy of anti-CTLA-4 mAbs against solid tumors and experimental metastases. Treg-depleting anti-CTLA-4 mAbs of the mouse IgG2a isotype showed the highest combinatorial activity. The optimal combination depended on the presence of activating Fc receptors and lymphocytes (particularly natural killer and CD8+ T cells), whereas anti-RANKL alone did not require Fc receptors. T-cell infiltration into solid tumors post anti-RANKL and anti-CTLA-4 was significantly higher, and this was accompanied by increased T-cell effector function. Several studies are currently ongoing, studying the effect of denosumab monotherapy and the combination of RANKL inhibition and immunotherapy [2, 7].
3.7 RANK/RANKL Signaling and Chemo- or Radiotherapy
The role of the RANK/RANKL signaling in drug resistance remains unclear. There is some in vitro evidence suggesting that RANK/RANKL signaling can induce chemoresistance through the activation of multiple signal transduction pathways [59, 60]. However, in a mouse model, RANKL blockade increases the efficacy of cisplatin chemotherapy [60]. At the moment, there are no objective data that RANK/RANKL signaling inhibition has an influence on the effectivity of chemotherapy or radiotherapy in humans [7].
3.8 Conclusion
The role of RANK/RANKL inhibition as an immunomodulatory strategy in combination with other treatment modalities should be further investigated. As denosumab has clear immune-stimulating effects and an interesting toxicity profile, the drug has an attractive potential to be coadministered with immunotherapies for cancer treatment, thereby reinforcing the antitumor immune response. Optimal dosage and sequencing of treatment with other drug combinations warrants further investigation.
References
Aspelagh S, Morel D, Soria JC, Postel-Vinay S (2018) Epigenetic modifiers as new immunomodulatory therapies in solid tumors. Ann Oncol 29:812–824. https://doi.org/10.1093/annonc/mdy50
van Dam PA, Verhoeven Y, Trinh XB, Wouters A, Lardon F, Prenen H, Smits E, Baldewijns M, Lammens M (2019) RANK/RANKL signaling inhibition may improve the effectiveness of checkpoint blockade in cancer treatment. Crit Rev Oncol Hematol 133:85–91
Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor R, Decker WK, Whelan RL, Shanta Kumara HM, Signori E, Honoki K et al (2015) Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 35:S185–S198
Tilborghs S, Corthouts J, Verhoeven Y, Arias D, Rolfo C, Trinh XB, van Dam PA (2017) The role of nuclear factor-kappa B signaling in human cervical cancer. Crit Rev Oncol Hematol 120:141–150. https://doi.org/10.1016/j.critrevonc.2017.11.001. Epub 2017 Nov 7
van Dam PA, van Dam PH, Rolfo C, Giallombardo M, Van Berckelaer C, Trinh XB, Altintas S, Huizing M, Papadimitriou K, Tjalma WA, Van Laere S (2016) In silico pathway analysis in cervical carcinoma reveals potential new targets for treatment. Oncotarget 7(3):2780–2795. https://doi.org/10.18632/oncotarget.6667
Casey SC, Amedei A, Aquilano K, Azmi AS, Benecia F, Bhakta D, Bilsland AE, Boosani CS, Chen S, Ciriolo MR, Crawford S, Fujii H, Georgakilas AG, Guha G et al (2015) Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin Cancer Biol 35:S199–S223
van Dam P, Verhoeven Y, Jacobs J, Wouters A, Tjalma W, Lardon F, Van den Weyngaert, De Wulf J, Smits E, Colpaert C, Prene H, Peeters M, Lammens M, Trinh XB (2019) RANK_RANKL signaling in cancer of the uterine cervix: a review. Int J Mol Sci 20(9):2183
Castellano D, Sepulveda JM, Garcia-Escobar I, Rodriguez-Antolin A, Sundlov A, Cortes-Funes H (2011) The role of RANK-ligand inhibition in cancer: the story of denosumab. Oncologist 16(2):136–145
Millian MM (2015) The role of estrogen receptor in bone cells. Clin Rev Bone Miner Metabol 13:105–112
Ikeda T, Kasai M, Utsuyama M, Harikowa K (2001) Determination of three isoforms of the receptor activator of nuclear factor-kappa B ligand and their differential expression in bone and thymus. Endocrinology 142:1419–1426
Renema N, Navet B, Heymann MF, Lezot F, Heymann D (2016) RANK-RANKL signaling in cancer. Biosci Rep 36:e00366. https://doi.org/10.1042/BSR20160150
Theolaire S, Wittrant Y, Kwantat S, Fortun Y, Redeni F, Heymann D (2004) The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiologic bone remodeling. Cytokine Growth Factor Rev 15:457–475
Gonzalez-Suarez E, Sanz-Moreno A (2016) RANK as a therapeutic target in cancer. FEBS J 283:2018–2033
Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, Huang J, Dai W, Li C, Zheng C, Xu L, Chen H, Wang J, Li D, Siwko S, Penninger JM, Ning G, Xiao J, Liu M (2016) LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med 22:539–546. https://doi.org/10.1038/nm.4076. Epub 2016 Apr 11
Schneeweis LA, Willard D, Milla ME (2005) Functional dissection of osteoprotegerin and its interactions with receptor activator of NF-kappaB ligand. J Biol Chem 280:41155–41164
Papanastasiou AD, Sirinian C, Kalofonos HP (2012) Identification of novel human receptor activator of NF-kB isoforms generated through alternative spicing: implications in breast cancer cell survival and migration. Breast Cancer Res 1:R112
de Groot AF, Appelman-Dijkstra NM, van der Burg SH, Kroep JR (2018) The anti-tumor effect of RANKL inhibition in malignant solid tumors - A systematic review. Cancer Treat Rev 62:18–28. https://doi.org/10.1016/j.ctrv.2017.10.010. Epub 2017 Nov 2. Review
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R et al (2010) RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468:103–107
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernadez-Ortega S, Urricoechea A, Climent F, Soler MT, Munoz P, Vinals F, Tometsko M, Branstetter D, Dougall WC, Gonzalez-Suarez E (2012) RANK induces epithelial-mesenchymal transition and stem ness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res 72(11):2678–2688
Chu CY, Chung LW (2014) RANK-mediated signaling network and cancer metastasis. Cancer Metastasis Rev 33:497–509
Reid PE, Brown NJ, Holen I (2009) Breast cancer cells stimulate osteoprotegerin production by endothelial cells through direct cell contact. Mol Cancer 8:49. https://doi.org/10.1186/1476-4598-8-9
Sigl V, Jones LP, Penninger JM (2016) RANKL/RANK: from bone loss to the prevention of breast cancer. Open Biol 6:160230. https://doi.org/10.1098/rsob.160230
De Castro J, Garcia R, Garrido P, Isla D, Massuti B, Blanca B, Vazquez J (2015) Therapeutic potential of denosumab in patients with lung cancer: beyond prevention of skeletal complications. Clin Lung Cancer 16:431–446
Heymann MF, Riet A, Le Goff B, Battaglia S, Paineau J, Heymann D (2008) OPG, RANK and RANK ligand expression in thyroid lesions. Regul Pept 148:46–53
Mizutani Y, Matsubara H, Yamamoto K, Nan Li Y, Mikami K, Okihara K, Kawauchi A, Bonavida B, Miki T (2004) Prognostic significance of serum osteoprotegrin levels in patients with bladder carcinoma. Cancer 101:1794–1802
Rochette L, Meloux A, Rigal E, Zeller M, Cottin Y, Vergely C (2019) The role of osteoprotegerin and its ligands in vascular function. Int J Mol Sci 20(3). pii: E705. https://doi.org/10.3390/ijms20030705. Review
Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O (2014) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307(1):C25–C38. https://doi.org/10.1152/ajpcell.00084.2014. Epub 2014 Apr 30
Yamada T, Tsuda M, Takahashi T, Totsuka Y, Shindoh M, Ohba Y (2011) RANKL expression specifically observed in vivo promotes epithelial mesenchymal transition and tumor progression. Am J Pathol 178:2845–2856
Naumnik W, Płońska I, Ossolińska M, Nikliński J, Naumnik B (2018) Prognostic value of osteoprotegerin and sRANKL in Bronchoalveolar Lavage fluid of patients with advanced non-small cell lung cancer. Adv Exp Med Biol 1047:1–6. https://doi.org/10.1007/5584_2017_111
Zhang X, Song Y, Song N, Zhang L, Wang Y, Li D, Wang Z, Qu X, Liu Y (2018) RANKL expression predicts poor prognosis in gastric cancer patients: results from a retrospective and single-center analysis. Braz J Med Biol Res 51(3):e6265. https://doi.org/10.1590/1414-431X20176265
Shang WQ, Li H, Liu LB, Chang KK, Yu JJ, Xie F, Li MQ, Yu JJ (2015) RANKL/RANK interaction promotes the growth of cervical cancer cells by strengthening the dialogue between cervical cancer cells and regulation of IL-8 secretion. Oncol Rep 34(6):3007–3016
Ma D, Chang LY, Zhao S, Zhao JJ, Xiong YJ, Cao FY, Yuan L, Zhang Q, Wang XY, Geng ML, Zheng HY, Li O (2017) KLF5 promotes cervical cancer proliferation, migration and invasion in a manner partly dependent on TNFRSF11a expression. Sci Rep 7(1):15683. https://doi.org/10.1038/s41598-017-15979-1
Song FN, Duan M, Liu LZ, Wang ZC, Shi JY, Yang LX, Zhou J, Fan J, Gao Q, Wang XY (2014) RANKL promotes migration and invasion of hepatocellular carcinoma cells via NFkB mediated epithelial mesenchymal transition. PLoS One 9:e108507. https://doi.org/10.1371/journal.pone
Wang J, Zhang H, Wang Y, Li Y (2015) MPA influences tumor cell proliferation, migration and invasion induced by RANKL through PRB involving the MAPK pathway in endometrial cancer. Oncol Rep 33:799–809
Christoph F, König F, Lebentrau S, Jandrig B, Krause H, Strenziok R, Schostak M (2018) RANKL/RANK/OPG cytokine receptor system: mRNA expression pattern in BPH, primary and metastatic prostate cancer disease. World J Urol 36(2):187–192. https://doi.org/10.1007/s00345-017-2145-y. Epub 2017 Dec 4
Schultz MA, Hagan SS, Datta A, Zhang Y, Freeman ML, Sikka SC, Abdel-Mageed AB, Mondal D (2014) Nrf1 and Nrf2 transcription factors regulate androgen receptor transactivation in prostate cancer cells. PLoS One 9(1):e87204. https://doi.org/10.1371/journal.pone.0087204
Pellegrini P, Cordero A, Gallego MI, Dougall WC, Munoz P, Pujana MA, Gonzalez-Suarez E (2013) Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem Cells 31:1954–1965
Cross SS, Harrison RF, Balasubramanian SP, Lippitt JM, Evans CA, Reed MW, Holen I (2006) Expression of receptor activator nuclear factor kappa beta ligand (RANKL) and tumor necrosis factor related apoptosis inducing ligand (TRAIL) in breast cancer, and its relations with osteoprotegrin, estrogen receptor and clinicopathological variables. J Clin Pathol 59:716–720
Park HR, Min SK, Cho HD, Kim DH, Shin HS (2003) Expression of osteoprotegerin and RANK ligand in breast cancer bone metastasis. J Korean Med Sci 18(4):541–546
Sarink D, Schock H, Johnson T, Overvad K, Holm M, Tjønneland A, Boutron-Ruault MC, His M, Kvaskoff M, Boeing H, Lagiou P, Papatesta EM, Trichopoulou A, Palli D, Pala V, Mattiello A, Tumino R, Sacerdote C, Bueno-de-Mesquita HBA, van Gils CH, Peeters PH, Weiderpass E, Agudo A, Sánchez MJ, Chirlaque MD, Ardanaz E, Amiano P, Khaw KT, Travis R, Dossus L, Gunter M, Rinaldi S, Merritt M, Riboli E, Kaaks R, Fortner RT (2017) Circulating RANKL and RANKL/OPG and breast cancer risk by ER and PR subtype: results from the EPIC cohort. Cancer Prev Res 10:525–534. https://doi.org/10.1158/1940-6207.CAPR-17-0125
Tang ZN, Zhang F, Tang P, Qi XW, Jiang J (2011) Hypoxia induces RANK and RANKL expression by activating HIF-1 alpha in breast cancer cells. Biochem Biophys Res Commun 408:411–416
Zhang L, Teng Y, Fan Y, Wang Y, Li W, Shi J, Ma Y, Li C, Shi X, Qu X, Liu Y (2015) The E3 ubiquitin ligase Cbl-b improves the prognosis of RANK positive breast cancer patients by inhibiting RANKL-induced cell migration and metastasis. Oncotarget 6(26):22918–22933
Papanastasiou AD, Sirinian C, Plakoula E, Zolota V, Zarkadis IK, Kalofonos HP (2018) RANK and EGFR in invasive breast carcinoma. J Cancer Gen 216-217:61–66. https://doi.org/10.1016/j.cancergen.2017.07.004. Epub 2017 Jul 26
Liu Y, Wang J, Ni T, Wang L, Wang Y, Sun X (2016) CCL20 mediates RANK/RANKL-induced epithelial-mesenchymal transition in endometrial cancer cells. Oncotarget 18:25328–25339. https://doi.org/10.18632/oncotarget.8291
Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, Choi Y (1997) TRANCE, a new TNF family member predominantly expressed in T cells, is a dendritic cell specific survival factor. J Exp Med 186:2075–2080
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hofman RM, Karin M (2011) Tumour infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signaling. Nature 470:548–553
Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q (2009) Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 116:210–217
Goswami S, Sharma-Walia N (2016) Osteoprotegerin rich tumor microenvironment: implications in breast cancer. Oncotarget 7(27):42777–42791
Min JK, Kim YM, Kim YM, Kim EC, Gho YS, Kang IJ, LEE SY, Kong YY, Kwon YG (2003) Vascular endothelial growth factor upregulates expression of receptor activator of NF-kB (RANK) in endothelial cells: concomitant increase of angiogenic responses to RANK ligand. J Biol Chem 278:39548–39557
Scagliotti GV, Hirsh V, Siena S, Henry DH, Woll PJ, Manegold C, Solal-Celigny P, Rodriguez G, Krzakowski M, Mehta ND, Lipton L, García-Sáenz JA, Pereira JR, Prabhash K, Ciuleanu TE, Kanarev V, Wang H, Balakumaran A, Jacobs I (2012) Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: subgroup analysis from a randomized phase 3 study. J Thorac Oncol 7(12):1823–1829. https://doi.org/10.1097/JTO.0b013e31826aec2b
Peters S, Danson SJ, Hasan B, Reinmuth N, Majem M, Tournoy KG et al (2018) A randomized phase III trial evaluating the addition of denosumab to standard first-line treatment in advanced NSCLC: the ETOP and EORTC SPLENDOUR trial (abstract 1385). Ann Oncol 29(mdy292):008
Gnant M, Pfeiler G, Steger G, Egle D, Greil R, Fitzal F, Wette V, Haslbauer F, Melbiner E et al (2018) Adjuvant denosumab in early breast cancer: disease-free survival analysis of 3425 postmenopausal patients in the ABCSG-18 trial. J Clin Oncol 36:S500
Coleman RE, Finkelstein D, Martin M, Iwata H, Glaspy JA, Zhou Y, Jandial D, Chan A (2018) Adjuvant denosumab in early breast cancer: first results from the international multicenter randomized phase III placebo controlled study. J Clin Oncol 36:S501
Nguyen B, Maetens M, Salgado R, Venet D, Vuylsteke P, Polastro L, Wildiers H, Simon P, Lindeman G, Larsimont D, Van de Eynden G, Velgh C, Rothe F, Garaud S, Michiels S, Willard Gallo K, Azi HA, Loi S, Piccart M, Sotoriou C (2018) D-BEYOND: a window of opportunity trial evaluating denosumab, a RANK ligand inhibitor and its biological effects in young pre-menopausal women diagnosed with early breast cancer. Cancer Res 78(13 Suppl):abstarct nr CT101
Bakhru P, Zhu ML, Wang HH, Hong LK, Khan I, Mouchess M, Gulati AS, Starmer J, Hou Y, Sailer D, Lee S, Zhoa F, Kirkwood JM, Moschos S, Fong L, Anderson MS, Su M (2017) Combination central tolerance and peripheral checkpoint blockade unleashes antimelanoma immunity. Immunology 93265. https://doi.org/10.1172/jci.insight.93265
Smyth MJ, Yagita H, McArthur GA (2016) Combination of anti-CTL-4 and anti-RANKL in metastatic melanoma. J Clin Oncol 34:e104–e106. https://doi.org/10.1200/JCO.2013.51.3572
Afzal MZ, Shirai K (2018) Immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy alone versus immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy in combination with anti-RANKL denosumuab in malignant melanoma: a retrospective analysis at a tertiary care center. Melanoma Res. https://doi.org/10.1097/CMR.0000000000000459. [Epub ahead of print]
Ahern E, Harjunpää H, Barkauskas D, Allen S, Takeda K, Yagita H, Wyld D, Dougall WC, Teng MWL, Smyth MJ (2017) Co-administration of RANKL and CTLA4 antibodies enhances lymphocyte-mediated antitumor immunity in mice. Clin Cancer Res 23(19):5789–5801. https://doi.org/10.1158/1078-0432.CCR-17-0606
Tsubaki M, Takeda T, Yoshizumi M, Ueda E, Itoh T, Imano M, Satou T, Nishida S (2016) RANK-RANKL interactions are involved in cell adhesion-mediated drug resistance in multiple myeloma cell lines. Tumour Biol 37(7):9099–9110. https://doi.org/10.1007/s13277-015-4761-8. Epub 2016 Jan 14
Timotheadou E, Kalogeras KT, Koliou GA, Wirtz RM, Zagouri F, Koutras A, Veltrup E, Christodoulou C, Pentheroudakis G, Tsiftsoglou A, Papakostas P, Aravantinos G, Venizelos V, Pectasides D, Kosmidis P, Karanikiotis C, Markopoulos C, Gogas H, Fountzilas G (2017) Evaluation of the prognostic value of RANK, OPG, and RANKL mRNA expression in early breast cancer patients treated with anthracycline-based adjuvant chemotherapy. Transl Oncol 10(4):589–598. https://doi.org/10.1016/j.tranon.2017.05.006
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
van Dam, P.A., Verhoeven, Y., Trinh, X.B. (2020). The Non-Bone-Related Role of RANK/RANKL Signaling in Cancer. In: Birbrair, A. (eds) Tumor Microenvironment . Advances in Experimental Medicine and Biology, vol 1277. Springer, Cham. https://doi.org/10.1007/978-3-030-50224-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-50224-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50223-2
Online ISBN: 978-3-030-50224-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)