
Abstract
Hereditary forms of nephrotic syndrome (NS) have been considered as infrequent disorders; however, 3–6% of the cases with NS have an affected sibling (1–3). Over the last decade, screening of large cohorts of pediatric patients presenting with steroid-resistant nephrotic syndrome (SRNS) for gene mutations has revealed the importance of genetic disorders in the pathogenesis of proteinuric glomerulopathies. At least 66% of the cases presenting with SRNS during the first year of life have an underlying genetic disease (4). In cases with infantile and juvenile SRNS, the overall proportion of genetic forms appears significantly lower, although the precise frequency remains unknown. Because autosomal recessive diseases may present as sporadic cases, the incidence of hereditary forms of NS is certainly underestimated. From a clinical perspective, most patients with hereditary SRNS will be resistant to immunosuppressive agents and do not experience relapse after transplantation (5–7).
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Nephrotic Syndrome
- Glomerular Basement Membrane
- Epidermolysis Bullosa
- Slit Diaphragm
- Congenital Nephrotic Syndrome
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Hereditary forms of nephrotic syndrome (NS) have been considered as infrequent disorders; however, 3–6% of the cases with NS have an affected sibling (1–3). Over the last decade, screening of large cohorts of pediatric patients presenting with steroid-resistant nephrotic syndrome (SRNS) for gene mutations has revealed the importance of genetic disorders in the pathogenesis of proteinuric glomerulopathies. At least 66% of the cases presenting with SRNS during the first year of life have an underlying genetic disease (4). In cases with infantile and juvenile SRNS, the overall proportion of genetic forms appears significantly lower, although the precise frequency remains unknown. Because autosomal recessive diseases may present as sporadic cases, the incidence of hereditary forms of NS is certainly underestimated. From a clinical perspective, most patients with hereditary SRNS will be resistant to immunosuppressive agents and do not experience relapse after transplantation (5–7).
Gene discovery efforts aimed at unraveling the causes of Mendelian forms of nephrotic syndrome have resulted in the identification of mutations in novel genes that encode proteins crucial for the establishment and maintenance of the glomerular filtration barrier. These discoveries have helped decipher the pathophysiologic mechanisms of the glomerular filtration process. Mutations in six genes have been implicated in different forms of non-syndromic SRNS ( Table 27-1 , Fig. 27-1 ). Mutations in NPHS1, encoding nephrin, are responsible for most of the cases with congenital nephrotic syndrome (CNS) and might be found in infantile forms of SRNS (8, 9). Mutations in the NPHS2 gene, encoding podocin, are the most frequent cause of early-onset autosomal recessive SRNS (10), and account for 37.5% of the cases with NS presenting in the first year of life among European populations (4). Some dominant forms of juvenile and adult onset SRNS are due to mutations in ACTN4 and TRPC6 (11, 12), encoding the cytoskeletal protein α-actinin-4 and the transient receptor potential ion channel (TRPC) 6, respectively. More recently, mutations in PLCE1, which encodes for phospholipase C-epsilon-1, were found in patients with early-onset SRNS and diffuse mesangial sclerosis (DMS) (13).

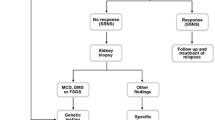
Clinical and genetic approach in patients with non-syndromic SRNS. An exhaustive investigation of extra-renal manifestations must be performed during the first clinical evaluation and subsequent follow-up. In cases in which mutations in NPHS1, NPHS2, NPHS3 and WT1 have been excluded, mutational screening of LAMB2, COQ2 and PDSS2 might be performed. A first approach genetic screening is proposed according to the phenotype; a negative result implies additional genetic testing if applicable MCD: minimal change disease, FSGS: focal segmental glomerulosclerosis, DMS: diffuse mesangial sclerosis, PT: proximal tubular, AR: autosomal recessive and AD: autosomal dominant.
Syndromic forms of SRNS are less common (Table 27-1 , Fig. 27-2 ) and may be due to mutations in several genes with varied functions including transcription factors, mitochondrial and lysosomal proteins or constituents of the glomerular basement membrane (GBM). Frasier syndrome (14–16), Denys-Drash syndrome (17–20) and WAGR syndrome (21) are caused by mutations in WT1, which encodes for a transcription factor, the Wilms’ tumor protein. Furthermore, isolated forms of SRNS may be due to mutations in WT1 (22). Most cases with Pierson syndrome carry mutations in the LAMB2 gene, encoding laminin β2, a main component of the GBM (23). Mutations in LMXB1, encoding the LIM homeobox transcription factor 1β, are associated with nail-patella syndrome (24). In addition, mutations in the ITGB4 (epidermolysis bullosa) (25), SMARCAL1 (Schimke syndrome) (26), MTTL1 (MELAS syndrome) (27, 28) and SCARB2 (action myoclonus-renal failure syndrome) genes (29) have been found in patients with diverse extrarenal manifestations associated with SRNS. Patients with primary coenzyme Q10 deficiency due to mutations in COQ2 and PDSS2 may develop nephrotic syndrome in addition to neuromuscular symptoms (30–32), although patients with isolated SRNS have been described as well.

Clinical and genetic approach in patients with syndromic nephrotic syndrome. Proposed algorithm to decide for directed mutational screening according with the main extra renal manifestations and histological findings in the kidney biopsy.
This chapter will review the available epidemiologic data, genotype-phenotype correlations and the mechanisms by which mutations in genes implicated in hereditary forms of isolated SRNS lead to proteinuric glomerular disease. A genetic overview on recently discovered genes responsible for rarer cases of hereditary syndromic SRNS and advances in the study of familial Steroid Sensitive Nephrotic Syndrome (SSNS) are presented, as well.
Isolated Steroid-Resistant Nephrotic Syndrome
Mutations in the NPHS2 Gene Encoding Podocin
Gene Identification and Protein Characterization
In 1995, Fuchshuber et al. mapped a genetic locus on chromosome 1q25-31 (SRN1, MIM #600995) in a group of patients from Europe and Northern Africa who presented with childhood onset SRNS, autosomal recessive inheritance, renal histologic findings of Focal Segmental Glomerular Sclerosis (FSGS) and absence of extra-renal disorders (33). These patients rapidly progressed to end-stage renal disease but no recurrence occurred after renal transplantation. Boute et al. used a positional cloning approach thereafter and identified mutations in the NPHS2 gene, encoding a novel protein podocin (10). Subsequent studies further defined the phenotype associated with mutations in the NPHS2 gene, revealing that patients usually develop NS from birth to 6 years of age, do not respond to immunosuppressive agents and reach ESRD before the end of the first decade of life (4–6, 34). Histologic findings range from minimal glomerular changes, in patients biopsied early, to FSGS at later stages (10).
Mutations in the NPHS2 gene are responsible for 39 to 48% of familial and for 10 to 28% of sporadic cases of SRNS (5, 6, 35–37). Interestingly, NPHS2 mutations have also been identified in patients presenting with congenital onset of NS (4–6, 38). Hinkes et al. demonstrated that among central European patients with NS presenting in the first 3 months of life, podocin mutations comprise 51.4% of all mutations identified (4). In addition, linkage to the chr 1q25-31 locus and mutations in the NPHS2 gene have been described in patients with late-onset FSGS; therein further broadening the spectrum of phenotypes attributable to podocin mutations (39, 40).
The NPHS2 gene spans a 25 kb region, consists of eight exons, and encodes podocin, a predicted 42-kDa protein with 383 amino acid residues. Podocin is a lipid raft-associated protein bearing strong homology with stomatin, an integral membrane protein of human erythrocytes which regulates monovalent cation transport and acts as a cytoskeletal anchor (41). Stomatin is also expressed in vertebrate sensory neurons where it plays a role in mechanotransduction (42–44). Moreover, the podocin orthologue in C. elegans is the protein MEC-2 (10), which links a neuronal mechanosensory channel involved in touch with the microtubular and cytoskeletal framework and leads to the opening of ion channels (45). Podocin is predicted to be an integral membrane protein with a single transmembrane domain and intracellular NH2- and COOH-terminal ends; thus, forming a hairpin-like structure (10). Immunolocalization studies have localized podocin exclusively in kidney at the slit diaphragm of podocytes (46, 47).
Podocin Expression and Interactions
At the mRNA level, podocin is expressed as early as the S-shaped stage, concomitant with vascularization of the interior cleft of the developing nephron (10); whereas protein expression has been documented beginning at the later capillary stage (47). Podocin was demonstrated to accumulate in an oligomeric form in lipid rafts of the slit diaphragm, in complex with nephrin and CD2AP (46), suggesting a potential role of podocin as a scaffolding protein. Nephrin-induced signaling is greatly enhanced by podocin, which binds to the C-terminus of nephrin (48). Interactions of podocin with nephrin, NEPH1, CD2AP and TRPC6 are crucial for structural organization and regulation of filtration function of the slit diaphragm, mechanosensory signaling, podocyte survival, cell polarity and cytoskeletal organization (46, 49–53). In addition, podocin binds cholesterol and creates large protein-cholesterol supercomplexes in the slit diaphragm, thereby regulating the activity of associated TRPC ion channels (51, 52).
Allelic Variants and Genotype/Phenotype Correlations
Mutations in the NPHS2 gene include a full spectrum of protein-truncating nonsense and frameshift mutations, splice-site variants and missense changes and involve all eight coding exons (6, 10, 35). To date, more than 90 pathogenic mutations and 25 variants of unknown significance have been reported. Mutations are frequently found in pediatric patients with SRNS originating from central Europe and North America, Turkey, Middle East, North Africa and South America (4, 6, 10, 35, 37, 38, 40, 54–61). However, NPHS2 mutations are rarely detected in cases from Japan (62–65), China (66, 67), Korea and sub-Saharan Africa (68, 69). Several founder mutations have been identified, including p.R138Q in Europe (10, 35), p.R138X in Israeli-Arab population (55), p.V260E in the Comoros island and p.A284V in South America (Antignac, unpublished data). Considering the two largest cohorts comprising pediatric patients with SRNS (5, 37), the most common mutation is p.R138Q, which represents up to 32% of mutant alleles (5).
Individuals bearing pathogenic NPHS2 mutations in the homozygous or compound heterozygous state manifested earlier than those in whom pathogenic mutations were not identified, for both familial and sporadic cases (5, 37). The mean age at onset of NS in patients carrying two pathogenic mutations ranges between 2.6 and 3.4 years of age (5, 37). Weber et al. also found that patients with frameshift or nonsense mutations in the homozygous or compound heterozygous states led to an earlier onset of nephrotic syndrome than those carrying missense mutations (5). In addition, individuals homozygous for the p.R138Q mutation present with early-onset disease (5, 37). At least two mutations, p.V180M and p.R238S, are associated with a milder phenotype, including later age at onset of NS and age at ESRD (5). The p.R138X mutation has been associated with a high incidence of cardiac abnormalities in children (70), although this finding has not been confirmed in patients carrying other mutations (71).
The p.R229Q variant is the most frequently reported non-synonymous NPHS2 variant in Caucasians (72), particularly among Europeans, in whom the observed frequency of heterozygotes ranges from 0.03 to 0.13 (5, 6, 40, 72–74). In African-Americans and sub-Saharan populations the p.R229Q allele is infrequent (72). In vitro studies demonstrated decreased binding of the p.R229Q mutant protein to nephrin, suggesting that this variant may be pathogenic (40). Indeed, the p.R229Q variant has been associated with microalbuminuria in a cohort of Brazilian individuals of mixed European and African ancestries (73). Furthermore, the frequency of the p.R229Q allele is significantly higher among individuals of European descent with FSGS compared with controls of similar origin (Machuca et al, in press) (40, 72). This observation has not been confirmed in cohorts with a high proportion of individuals of African-American origin or in patient cohorts with a presumed immune form of nephrotic syndrome (75, 76). In SRNS patients, the p.R229Q polymorphism is frequently found in a compound heterozygous state with a pathogenic NPHS2 mutation, whereas this association has never been detected among controls (5, 40). These patients present with nephrotic syndrome and ESRD in the second and third decades of life, respectively, markedly contrasting them from patients bearing two pathogenic NPHS2 mutations (Machuca et al, in press). By contrast, R229Q in the homozygous state has been reported in patients with NS as well as in controls (5, 37, 59), and more likely has a modulatory effect on the risk of developing renal disease. These observations support a pathogenic role of the p.R229Q variant.
Finally, tri-allelic inheritance of NPHS1 and NPHS2 mutations has been occasionally reported (36, 54, 77), but additional studies are needed to better understand the complex genetics of renal disease progression in the setting of nephrotic syndrome.
The identification of NPHS2 mutations in children presenting with nephrotic syndrome may have important clinical implications. Screening of NPHS2 mutations in patients with SSNS (6), late steroid resistance (78), steroid-dependence or frequent relapses and those with sensitivity to cyclophosphamide have failed to identify pathogenic mutations (6, 36, 79). Patients with mutations in the NPHS2 gene do not respond to steroid or immunosuppressive therapy; although in a small number of cases a partial reduction of proteinuria has been reported with cyclosporine A (4, 6). Avoidance or withdrawal of immunosuppressive therapies in these patients would spare them from the potential risks and side-effects associated with these drugs.
NPHS2 mutations are rare in patients presenting with FSGS and relapse after transplantation (5). Indeed, patients with two pathogenic NPHS2 mutations have a significantly lower risk of relapse after transplantation than cases in whom mutations were not identified (8% vs. 30%) (5, 6, 80, 81). Patients bearing mutations in the heterozygous state have a risk comparable to those without mutations (82). In the few patients reported with two NPHS2 mutations who developed proteinuria post-transplant (5, 6, 83–86), the clinical evolution and renal histology did not correspond to the classic picture of NS relapse after transplantation (5, 83, 85). This potentially suggests de novo glomerulopathy or drug toxicity. In contrast with the mechanism of relapse in cases with NPHS1 mutations, there is no evidence to support a role for anti-podocin antibodies (5, 83, 87). To-date, the pathophysiologic mechanisms of recurrence of proteinuria in these patients are unclear.
Functional Studies
Functional studies have elucidated some of the mechanism by which missense podocin mutations lead to disease. In vitro studies have shown that podocin missense mutations may either maintain proper intracellular targeting to the plasma membrane or be retained in the endoplasmic reticulum (ER) (88). Interestingly, patients with missense mutations retained in the ER had an earlier onset of disease than patients with mutations that traffic to the membrane (20.8 ± 4 vs. 128.7 ± 9 months) (88). Plasma membrane localization of p.V180M and p.R238S mutations suggests that their deleterious effect could affect the function of the protein by directly modifying its signaling properties and/or altering its interaction with other proteins at the slit diaphragm (88). Moreover, the p.R138X podocin mutant is able to traffic to the plasma membrane (88); however, nephrin is not recruited to lipid rafts, from which downstream signaling events are generated (50). In cells expressing ER-retained podocin mutants, nephrin is similarly retained in the endoplasmic ER (89).
A potential therapeutic strategy that might delay the onset and ameliorate the severity of glomerular disease in patients with missense mutations in genes encoding proteins located in the plasma membrane (i.e., slit-diaphragm) relies on chemical chaperones. Several of these molecules have been used in in vitro systems, allowing for targeting of the mutant protein to the plasma membrane (90).
Animal Models
Podocin-null mice (Nphs2 −/−) mice are massively proteinuric at birth, NS progresses rapidly, and animals die in the first 5 weeks of life with end-stage renal failure (91). Interestingly, disease progression rate is strongly determined by genetic background and appear to be subject not only to genetic modification, but also to the effects of the maternal environment in which mice are nourished prior to weaning (92). Nephrogenesis appears to be normal in podocin null mice and kidney size at birth is similar to that in wild-type littermates. Unexpectedly, Nphs2 −/− mice do not show FSGS lesions, but display typical features of diffuse mesangial sclerosis (DMS). In addition, severe arteriolar lesions characterized by marked thickening of the arteriolar wall, endothelial cell hypertrophy, diffuse dilatation of peritubular capillaries, and multiple foci of interstitial hemorrhages predominating in the superficial cortex are observed. By electron microscopy, podocyte foot processes are only occasionally seen, are abnormal and lack slit diaphragms (91). Resembling the phenotype of a Nphs2 null mouse, a mouse model in which the p.R138Q mutant is expressed, leading to mislocalization of the mutant podocin in the ER, develops early-onset severe nephrotic syndrome, display features of DMS, progress rapidly to ESRD and dies at 5 weeks after birth (93). Similarly, mice deficient for CD2AP or NEPH1 develop progressive DMS as the one observed in mice lacking podocin (94, 95).
In addition to mouse models, the zebrafish pronephros has been used as a model of glomerular maturation and development of the filtration barrier. Zebrafish podocin shares 46% identity with the human protein, is specifically expressed in pronephric podocytes, and is required for the development of pronephric podocyte cell structure. Knockdown of podocin expression using antisense morpholino-oligonucleotides results in a loss of slit diaphragms, failure to form normal podocyte foot processes and loss of podocyte barrier function in the mature pronephros (96, 97).
Mutations in the NPHS1 Gene Encoding Nephrin
NPHS1 has been identified as the major gene involved in congenital nephrotic syndrome of the Finnish type (CNF) (8). However, recent findings have broadened the spectrum of renal disease related to nephrin mutations since patients with childhood-onset SRNS may have NPHS1 mutations (9, 98). The clinical aspects of CNF, the identification of the NPHS1 gene and characterization of nephrin are extensively described in chapter 25.
Epidemiologic Overview of NPHS1 Mutations
In the Finnish population, 94% of patients with CNF bear either of two protein-truncating mutations in the NPHS1 gene (8). The Fin-major mutation (c.121delCT; p.L41fsX91) leads to a frameshift deletion of two base pairs in exon two, resulting in a premature stop codon. The Fin-minor mutation (c.3325C>T; p.R1109X) generates a premature truncation of the terminal 132 amino acids of the protein. The Fin-major and Fin-minor mutations account for 78% and 16% of the mutated alleles in Finnish CNF patients, respectively (8); but are rare in other ethnic groups, therein suggesting founder effects (54). Other founder mutations have been sequenced among Old Order Mennonite patients from Lancaster, Pennsylvania (c.1481delC; p.S494fsX548) and patients from Malta (c.3478C > T; p.R1160X) (54, 99).
More than 100 NPHS1 mutations have been reported worldwide in patients with CNF, most of which are private mutations found in non-Finnish patients (4, 38, 54, 64, 100–106). In Europe, North Africa and North America, the NPHS1 mutation detection rate is estimated to be 66% (107). Among central European and Turkish patients presenting with NS in the first 3 months of life, NPHS1 mutations were found in 34.3% and 54.5% of cases, respectively (4). However, NPHS1 mutations were identified in only 2/13 patients in Japan (64). The lower frequency of congenital cases attributable to NPHS1 mutations in these ethnic groups compared to Finnish population points to the genetic heterogeneity of congenital nephrotic syndrome.
Milder and Unusual Phenotypes Associated with NPHS1 Mutations
The spectrum of NPHS1 mutations includes protein-truncating nonsense and frameshift insertion/deletion mutations, splice-site changes and missense variants. Most of these mutants are retained in the endoplasmic reticulum (ER), although Liu et al. have demonstrated that in vitro treatment with a chemical chaperone may allow for trafficking to the plasma membrane (108, 109). These mutations lead to a severe CNF phenotype, although some NPHS1 mutations have been reported in milder cases.
The p.R1160X mutation results in an unexpectedly milder phenotype in about 50% of cases, most of whom were females, suggesting a gender effect (54). This mutation is predicted to form a truncated protein lacking the C-terminal 82 amino acids implicated in the interaction with podocin. Surprisingly, all affected cases were homozygous for this mutation and, among those in which renal biopsy was performed, histologic findings were consistent with CNF. Nevertheless, these patients either had mild proteinuria or were in remission between the ages of 5 and 19 years.
Recently, NPHS1 mutations were identified in a cohort of 160 patients presenting with SRNS after 3 months of age (9). Mutations in the NPHS2 gene were excluded, as were mutations in exons 8 and 9 of the WT1 gene in phenotypically female patients (9). The mean age of onset of NS was 3 years (range 6 months to 8 years). Six patients had preserved renal function after 6 years of age, based on a normal serum creatinine. Renal biopsy performed at the time of presentation revealed that most cases had MCD or FSGS. All patients were resistant to corticosteroids, as well as other immunosuppressive agents when tried. Nine patients out of 98 with sporadic SRNS, and 1 family with 2 affected siblings among 44 families with familial SRNS, carried pathogenic NPHS1 mutations. Affected cases were compound heterozygotes for at least one “mild” missense mutation, which exhibited normal trafficking to the plasma membrane and maintained the abilities to form nephrin homodimers and to heterodimerize with NEPH1. These findings may explain the lesser severity of disease observed in these cases.
Finally, Kitamura et al. described the clinical course of two siblings bearing compound heterozygous NPHS1 missense mutations (98). The severe c.793T>C (p.C265R) mutation leads to ER retention, whereas the mild c.2464G>A (p.V822M) mutation encodes a protein that partially retains plasma membrane targeting. Both patients presented with mild to moderate persistent proteinuria detected from birth to 10 months of age, with several self-limited episodes of nephrotic syndrome triggered by upper airway infections. Kidney histology in both cases revealed minimal changes.
These recent studies highlight the importance of NPHS1 mutation screening in cases of childhood onset NS, particularly in those in whom mutations in podocin were not found. Studies of large patient cohorts with a broader range of disease onset and ethnic backgrounds are needed to better define the frequency and phenotypic spectrum of nephrin mutations.
Animal Models
Mouse models in which the Nphs1 gene has been inactivated revealed lesions reminiscent of histological changes observed in CNF patients and absence of slit diaphragms, corroborating the crucial role of nephrin in the establishment and maintenance of the glomerular filtration barrier. Nphs1 inactivation resulted in massive nonselective proteinuria, edema immediately after birth and death within 24 h (110–112). Histological characterization revealed slightly enlarged kidneys, dilated proximal and distal tubules, and microcysts in the cortex and medulla (110). No prominent changes in the branching morphogenesis of the developing collecting ducts could be found (112). Bowman spaces were enlarged and glomeruli were sclerotic and showed hypercellularity and excessive extracellular matrix deposition (112). Electron microscopy revealed effacement of podocyte foot processes and absence of slit diaphragms (110). The glomerular basement membrane appeared normal, and the expression of several basement membrane proteins including type IV collagen, laminin, nidogen, and perlecan, as well as podocyte-specific proteins such as podocin, CD2AP, α-actinin-4, synaptopodin, integrin α3, and α3, α4 and α5 chains of type IV collagen were normal (111).
The nephrin homologue in zebrafish shares only 36% identity with human nephrin; however, both have a similar predicted secondary structure. Nephrin is expressed in the zebrafish pronephros, specifically in the slit diaphragms of podocyte foot processes (96). Nephrin targeting with morpholino antisense oligonucleotides resulted in pericardial edema progressing to generalized edema (96). Nephrin morphant embryos demonstrated podocyte foot process effacement, lacked slit diaphragms and showed filtration barrier dysfunction in the mature pronephros (96). These findings resemble those found in podocin and CD2AP morphant embryos (96, 97).
Mutations in PLCE1 Encoding Phospholipase C Epsilon 1
The NPHS3 gene locus was identified on chromosome 10q23.32-q24.1 in seven consanguineous SRNS families (13). A positional cloning approach coupled to gene expression profiling in rat glomeruli identified the PLCE1 gene, encoding phospholipase C epsilon 1, as a good candidate. Mutational analysis subsequently revealed truncating and missense mutations in several of its 34 exons (13). In the 12 affected individuals carrying truncating mutations, proteinuria and edema, manifested at a median age of 0.8 years (range 0.2–4.0 yrs) and progressed to ESRD by 5 years of age (13). Furthermore, individuals bearing truncating mutations demonstrated lesions of DMS on renal biopsy, whereas FSGS was found in the affected cases homozygous for missense mutations. In the affected individuals presenting with DMS, immunofluorescence studies revealed that PLCE1 mutations may lead to an arrest of glomerular development at the S-shaped stage, suggesting a potential role not only in cell junction and signaling events, but in development, as well (13). Interestingly, two patients bearing truncating mutations achieved complete remission when treated early and remain free of proteinuria after several years of follow-up; hence potentially opening a window of opportunity for therapy of some forms of hereditary NS (13, 113). Subsequently these investigators have shown that mutations in the PLCE1 gene account for 28.6% of cases of isolated DMS in a cohort of 40 patients from 35 families mostly of Turkish origin (114). An additional report has described 4 patients with early-onset SRNS, DMS and PLCE1 mutations (38). In our cohort of patients with SRNS, we have identified several cases carrying truncating PLCE1 mutations presenting with early-onset SRNS and exhibiting FSGS on renal biopsy (Antignac, personal observation).
PLCε1 is a phospholipase enzyme that catalyzes the hydrolysis of phosphatidylinositol-4,5-biphosphate and generates two second messengers: inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) (115). IP3 releases Ca2+ from intracellular stores, and DAG stimulates protein kinase C. These products initiate a cascade of intracellular responses that result in cell growth and differentiation. Based on the observation that nephrin levels were diminished in the glomeruli of patients bearing mutations in PLCE1, it has been shown that PLCε1 interacts with the C-terminal half of IQGAP-1 (13), a cell junction-associated protein and binding partner of nephrin involved in cell morphology and adhesion (116).
The PLCε1 zebrafish orthologue shares 65% identity with the human protein sequence, suggesting conserved function among evolutionarily distant organisms. Plce1 targeting using antisense morpholino oligonucelotides induced edema and glomerular filtration barrier dysfunction, similar to zebrafish nephrin and podocin loss-of-function morphants (96). Surprisingly, PLCε1 null mice have no obvious developmental defects or evidence of glomerular filtration barrier abnormalities (13, 117).
Mutations in CD2AP
CD2AP is a 70-kilodalton adaptor protein that was originally cloned as an interaction partner of CD2, a signaling protein expressed on the surface of T lymphocytes (118). It has been shown that CD2AP is involved several processes including the regulation of the actin cytoskeleton (119–121), endocytosis (122, 123), in the phosphatidylinositol 3-kinase/AKT survival pathway and in the repression of TGF-β induced apoptosis (50, 124).
In the kidney, CD2AP is expressed in podocytes, proximal tubules and collecting ducts. Cd2ap null mice developed proteinuria 2 weeks after birth and die by 6 to 7 weeks of age due to advanced renal insufficiency (94).Renal histology showed increased glomerular size and cellularity, foot-process effacement at 1 week, and subsequent abnormal mesangial matrix deposition and glomerular sclerosis 4 weeks after birth (94). Cd2ap +/− mice did not develop proteinuria when followed up to 1 year; however, exhibited variable degrees of increased mesangial expansion and hypercellularity at 9 months of age. Two out of 30 patients with idiopathic FSGS were found to carry a heterozygous truncating mutation in the CD2AP gene (125), which lead to a reduce expression at the mRNA and protein level. These results suggested that CD2AP could act as a determinant of human susceptibility to glomerular disease.
Recently, Löwik et al. described a patient presenting at 10 months of age with failure to thrive, anemia, hypoalbuminemia and massive proteinuria (126). Renal biopsy showed global glomerular sclerosis and was suggestive of collapsing FSGS. Mutation analysis of the CD2AP gene revealed a novel truncating mutation in the homozygous state, which displayed significantly decreased F-actin binding efficiency in vitro. The mutant allele was not expressed in the patient’s lymphocytes. At the age of 5 years the patient was transplanted, without relapse of proteinuria (126). Both parents were heterozygous for the mutation and had normal glomerular filtration rate and no proteinuria. An additional patient bearing a heterozygous missense mutation in CD2AP in association with a missense mutation in NPHS2 was recently reported (127). This patient presented with SRNS and relapse after transplantation. Both parents had normal serum creatinine and no proteinuria (127). Mutational screening of large cohorts of SRNS patients will be crucial to elucidate the frequency of CD2AP mutations and the spectrum of phenotypes.
Autosomal Dominant Forms of SRNS
Autosomal dominant (AD) forms of FSGS are infrequent and generally observed in adults. Variable degree of proteinuria is detected between the third and fourth decades of life and slowly progresses to ESRD (128, 129). So far, two loci have been mapped in cases with non-syndromic AD FSGS; nevertheless, genetic heterogeneity is likely, since no linkage to those loci has been found in several families with similar phenotype (129).
Mutations in ACTN4 Encoding α-actinin-4
A genome-wide scan performed in a 100-member kindred allowed Mathis et al. to map the first locus of AD FSGS on chromosome 19q13 (130, 131). Linkage analysis including additional families helped to reduce the size of the region and led to the identification of 3 nonconservative missense mutations in the ACTN4 gene. ACTN4 encodes for the actin-binding protein α-actinin-4, which is highly expressed in podocytes (11). Affected cases presented with proteinuria starting in the teenage years or later, slowly progressed to renal insufficiency and developed ESRD in the fifth decade of life (11, 132). Disease was incompletely penetrant, since several individuals from 2 out of 3 families carried a disease allele without clinical symptoms. In affected cases, kidney histology revealed lesions consistent with FSGS and no evidence of a primary basement membrane defect or of immune complex deposition.
Further mutation screening of the ACTN4 gene in cases with familial and sporadic forms of FSGS allowed the identification of several additional patients carrying mutations (133, 134). In one affected case, proteinuria was diagnosed at 5 years of age and rapidly progressed to ESRD. Unexpectedly, this patient presented recurrence of proteinuria after transplantation. A superimposed immune form of SRNS or a de novo glomerulopathy may better explain the outcomes in this patient. Additional screening of small cohorts of patients with sporadic adult-onset FSGS and congenital SRNS have failed to identify ACTN4 mutations (62, 64, 135). Overall, ACTN4 mutations seem to account for approximately 4% of familial FSGS (134), although the precise proportion of AD forms in which this gene is mutated is unknown.
The mechanisms by which α-actinin-4 mutations cause disease in humans have been partially elucidated through functional studies and the characterization of mouse models. These studies suggest that the phenotypes in mice and humans involve both gain-of-function and loss-of-function mechanisms (11, 134, 136, 137). Alpha-actinin-4 has a key role in the maintenance of podocyte architecture cross-linking and bundling of actin filaments (138). Disease-associated mutations occur in the actin-binding domain, increasing actin-binding activity in vitro and diverting its normal localization from actin stress fibers and focal adhesions in vivo (11, 134, 137). Moreover, over-expression of GFP-fusion mutant proteins in cultured podocytes led to the formation of aggregates adjacent to the nucleus, confirming the subcellular mislocalization of mutants (134). Further supporting the hypothesis of loss-of-function, Yao et al. showed increased degradation of α-actinin-4 in cells from knock-in mice carrying an Actn4 point missense mutation homologue, in comparison to that found in humans with FSGS (136). Finally, a β1-integrin-dependent, α-actinin-4 mediated adhesion is necessary to maintain podocyte attachment to the-glomerular basement membrane (139, 140). Consequently, podocytes from α-actinin-4 deficient mice showed reduced adherence to GBM components type IV collagen and laminin-10 and -11 (140).
The phenotype seen in Actn4 −/− mice is more aggressive than the human disease. Actn4 null mice exhibited abnormalities only in the kidneys. At 5 weeks of age, mice had only focal areas of podocyte foot process effacement, whereas FSGS was evident by 10 weeks. Proteinuria was observed with increasing age in most, but not all mice. Progressive renal insufficiency led to death at 12 weeks after birth. Mice heterozygous for the targeted allele (Actn4 +/−) showed no obvious phenotype up to 6 months of age (141).
Resembling human disease, a transgenic mouse developed by Michaud et al. (142), which expressed both endogenous wild-type and a K256E-mutant α-actinin-4 transgene, developed proteinuria at 10 weeks, elevated blood pressure and histological features consistent with FSGS. Interestingly, not all ACTN4 mutant mice were proteinuric, and only a few among those with proteinuria had reduced renal function. Detailed histological analysis revealed segmental sclerosis and tuft adhesion of some glomeruli, tubular dilatation, mesangial matrix expansion, podocyte vacuolization and foot process fusion (142).
A mouse model in which one Actn4 allele was replaced with a copy bearing a disease-associated mutation in humans (K256E) was developed by Yao et al. (136). Although this model is genetically closer to the human disease, homozygous mutant mice had no glomerular defects evident using light microscopy, although focal areas of foot process effacement and abnormal electron-dense structures in the podocyte cell bodies were observed at the electron microscopic level. Careful assessment of Actn4 heterozygous mutant mice confirmed that they do not develop evident FSGS, but exhibit focal glomerular hypertrophy and mild glomerular ultrastructural changes (143). The mechanisms underlying the differences between the human and mouse phenotypes remain unknown. Additional modulating factors appear to play a role in the development of ACTN4-mediated human disease.
Mutations in TRPC6 Encoding the Transient Receptor Potential Cation Channel 6
Winn et al. identified a second locus for autosomal dominant FSGS on chr 11q21-22 in a large family from New Zealand (144). Affected cases presented with nephrotic range proteinuria in their third or fourth decade and developed progressive renal insufficiency within 10 years after NS presentation. Using fine-mapping and candidate gene screening, the same group of investigators subsequently detected a missense mutation in the TRPC6 gene, encoding the transient receptor potential cation channel, subfamily C, member 6 (12). TRP channels are involved in mechanosensation (145), ion homeostasis, cell growth and PLC dependent calcium entry into cells (146). The proline to glutamine substitution at position 112 (p.P112Q) found in the index family, was shown to enhance TRPC6-mediated calcium signals in response to angiotensin II, suggesting that mutations in this gene disrupt glomerular cell function by amplifying injurious signals triggered by ligands, such as angiotensin II (12).
Subsequently, Reiser et al. identified TRPC6 mutations in five other unrelated families of diverse ethnic origin (53). Only two of the five mutations were associated with an increase in calcium influx, suggesting that diverse mechanisms may result in dysregulation of the ion channel or may affect the interaction with other slit diaphragm proteins (53). In addition, they demonstrated that TRPC6 is expressed in podocytes, specifically at the slit diaphragm where it interacts with podocin and nephrin (53).
In our cohort, we have found one patient bearing a de novo missense mutation in exon 13 of TRPC6, comprising a highly conserved region in the cytoplasmic tail of the protein (Antignac, unpublished data). This patient presented with NS at 6.5 years of age and reached ESRD a few months after diagnosis. Renal histology revealed advanced FSGS. No relapse was observed after transplantation. Mutation screening of large cohorts of patients is needed to evaluate the epidemiologic relevance of TRPC6 mutations and the phenotypic spectrum of renal disease attributable to the TRPC6 gene.
The contribution of animal models to understand the mechanisms underlying mutations in TRPC6 has been limited. Targeted deletion of Trpc6 in mice was not associated with a renal phenotype; although, mice exhibited an elevated blood pressure and enhanced agonist-induced contractility of isolated aortic rings, as well as cerebral arteries (147). An animal model carrying a missense point mutation in the Trpc6 gene, homologous to those found in humans, will be required to confirm a gain-of-function mechanism as the triggering event leading to glomerular disease. An evaluation of the role of TRPC6 in kidney disease using zebrafish has not been possible since TRPC6 was not detected at RNA level in developing and adult zebrafish podocytes (148).
Syndromic Steroid-Resistant Nephrotic Syndrome
Pierson Syndrome: Mutations in LAMB2 Encoding Laminin β2
In 1963, Pierson et al. described two patients with congenital nephrotic syndrome, unique ocular abnormalities and histopathological features of DMS (149). Subsequently, several isolated case reports appeared in the literature (150–154). Zenker et al. designated this disorder Pierson syndrome, refining the phenotype based on the description of eleven affected cases from two large consanguineous families and after reviewing previous case reports (155). Clinical findings include nephrotic syndrome and oliguria presenting at birth or within the first days of life, enlarged or large appearing cornea in some cases suggesting buphthalmos, extremely narrow, nonreactive pupils (microcoria) and DMS with an irregular basement membrane (155).
In two consanguineous families, a genome-wide scan and homozygosity mapping allowed the identification of a potential locus in chromosome 3p (23). Subsequent positional cloning was greatly facilitated by the previous description of the development of congenital nephrotic syndrome in Lamb2 null mice (156). Affected cases from five families had truncating or missense mutations of the LAMB2 gene, either in the homozygous or compound heterozygous states, leading to absent or reduced expression, respectively, of laminin β2 in the kidneys (23). Interestingly, mutation screening in additional families revealed patients bearing missense mutations in LAMB2 who presented with congenital nephrotic syndrome, minor or no ocular defects (transient fundus hypopigmentation, nistagmus and myopia) and normal psychomotor development (157–159). Childhood onset of Pierson syndrome has likewise been reported in a non-consanguineous family with seven affected individuals, with nephrotic syndrome and ESRD presenting between 5 and 10 years of age (160). Ocular problems paralleled or even preceded the renal symptoms. Visual impairment was progressive since affected individuals had no signs of impaired vision in early infancy, and they all developed blindness around 2 years of age (160).
Laminins are heterotrimeric extracellular matrix proteins that provide the basic scaffold for assembly of the other components of the glomerular basement membrane, including type IV collagen, nidogen/entactin and sulfated proteoglycans (161). The glomerular basement membrane is composed exclusively of laminin-521 (α5β2γ1) (162). The α5 chain is required for GBM integrity and glomerular vascularization (163), whereas the β2 chain is dispensable for glomerulogenesis; concordantly, Lamb2 null mice displayed no glomerular developmental abnormalities at birth (164).
The clinical features observed in patients with Pierson syndrome are consistent with the phenotype of Lamb2 null mice, which present with failure to thrive, heavy proteinuria within the first weeks of life (156), and revealed foot-process effacement and increased GBM permeability (164). In addition, mice show aberrantly formed and functionally impaired neuromuscular junctions (165, 166), and both structural and functional abnormalities in the retina (167, 168). Indeed, these findings reflect the fact that laminin β2 is highly expressed in the glomerulus, the skeletal neuromuscular junction and the retina (167–169).
Denys-Drash, Frasier and WAGR Syndromes: Mutations in WT1 Encoding the Wilms’ Tumor Protein
Through a positional cloning approach, the WT1 gene was found to be inactivated in Wilms’ tumor (170–172). The WT1 gene is located on chromosome 11p13 and encodes a zinc finger transcription factor that functions both as a tumor suppressor and as a critical regulator of kidney and gonadal development (173–175). The key role of WT1 in kidney development has been highlighted by the development of animal and in vitro models showing the failure of, arrest in or delayed development of nephrogenesis in the absence of WT1 expression (174, 176, 177). Mutations in the WT1 gene are associated with varied syndromic forms of glomerular disease and genitourinary abnormalities, as well as isolated cases of SRNS.
Denys-Drash syndrome (DDS, MIM 194080) is a rare urogenital disorder comprising nephropathy due to DMS, associated with male pseudohermaphroditism and Wilms’ tumors (17, 18). Nephrotic syndrome presents in the first months of life, may be preceded by isolated proteinuria and is always resistant to steroid therapy. Progression to ESRD occurs before 4 years of age and no recurrence is observed after renal transplantation (178–180). Wilms’ tumor may be the first presentation of the disease or may be discovered later during the course of nephropathy by systematic ultrasound screening. Patients with Denys-Drash syndrome bear heterozygous mutations, mostly de novo, in exons 8 and 9 of the gene, encoding the second and third zinc finger domains (19, 20). In vitro studies have confirmed that missense mutations in the WT1 gene lead to a change in the structural organization of the zinc finger domains, leading to loss or alteration of their DNA-binding abilities (181). Isolated cases of DMS have also been attributed to mutations in WT1 (182–184).
Frasier syndrome (FS, MIM 136680) is characterized by male pseudohermaphroditism with normal female external genitalia, streak gonads and 46,XY karyotype. Patients have an increased susceptibility to gonadoblastomas, but do not develop Wilms’ tumors. FS is associated with childhood-onset proteinuria, usually between 2 and 6 years of age, slowly progressing to ESRD towards the adolescence or early adulthood, exhibiting histological finding of FSGS on renal biopsy (15, 16). As in Denys-Drash syndrome, inheritance is autosomal dominant, although most cases are sporadic due to de novo mutations. The WT1 gene encodes up to 36 different isoforms, which are products of alternative translation start sites, alternative splicing and RNA editing (185).
Of particular interest are WT1(+KTS) and WT1(-KTS) variants, which differ by the presence of the three amino acids KTS between zinc fingers 3 and 4. The presence of this insert influences the molecular and biochemical properties of the resulting protein. While WT1(−KTS) binds DNA efficiently and acts as a transcriptional activator, WT1(+KTS) seems to have higher affinity to RNA (186). Mutations in the donor splice site in intron 9 of the WT1 gene are causative of Frasier syndrome, and leads to alternative splicing and loss of the +KTS isoform of the protein (14). This results in an alteration of the normal ratio of +KTS/-KTS isoforms in the cell (187).
De novo deletion of the 11p13 locus leads to WAGR syndrome (MIM 194072), characterized by Wilms’ tumors, aniridia, genitourinary abnormalities and mental retardation (21). Aniridia is due to the deletion of the PAX6 gene, which resides in the same locus than WT1.
Mutations in WT1 have also been associated with the development of isolated SRNS with kidney histology consistent with FSGS in some phenotypic females (XX or XY karyotype). Disease onset varies between few months of age to the end of the first decade of life, with rapid progression to ESRD (22, 188–190). Most of the cases carry missense or splice-site mutations in exons 8 and 9 of the WT1 gene. In these patients, genetic counseling is essential, since a male child from an affected XX female, would either have Denys-Drash syndrome or Frasier syndrome, respectively (188, 189).
Nail-Patella Syndrome: Mutations in LMX1B Encoding the LIM Homeobox Transcription Factor 1 β
Nail-patella syndrome (NPS; MIM 161200) is an autosomal dominant disorder with complete penetrance and variable phenotypic expression, characterized by pleiotropic developmental defects of dorsal limb structures. The most characteristic finding is nail involvement. Nails may be absent, hypoplastic or dystrophic. Defects are often bilateral, symmetrical and may be observed at birth. An additional pathognomonic feature of NPS are iliac horns, which are bony processes that project posteriorly and laterally from the central part of the iliac bones of the pelvis (191). Frequently, patellae may be hypoplastic or absent; involvement of shoulders, elbows and ankles is less common, and may be asymmetrical (192). Nephropathy may occur in 25–50% of the cases (193–197), being more frequent in women (197). This manifests as microalbuminuria progressing to proteinuria, usually associated with hematuria. Proteinuria, which may be intermittent, may present at any age, diagnosed in most of the cases after the second decade of life. Overt nephrotic syndrome is not a common feature and progression to ESRD occurs in 5–14% of the cases, usually many years after proteinuria onset (197, 198). To-date, no recurrence of proteinuria after transplantation has been reported (193, 199). Light microscopy of renal tissue usually reveals no specific changes (200), while glomerular basement membrane exhibits ultrastructural abnormalities that are the most specific histological hallmark of NPS (193, 201–203). Typically, there is irregular thickening and splitting of the GBM glomerular basement membrane, with electron lucent areas, and the presence of clusters of fibrillar type III collagen within the GBM and the mesangial matrix. Finally, primary open angle glaucoma and sensorineural hearing impairment have been recognized as less frequent features of the disease (197, 204).
Chen et al. demonstrated that targeted disruption of the Lmx1b gene (LIM homeobox transcription factor 1 β) in mice resulted in distinctive skeletal defects including hypoplastic nails, absence of patellae, joint abnormalities and glomerular basement membrane defects, recapitulating the phenotype of NPS (205). The disease phenotype was observed only in homozygous mutant mice, whereas heterozygous littermates did not exhibit any evident abnormalities. Nevertheless, these results led to the identification of de novo heterozygous mutations in the LMX1B gene in patients with NPS (24).
Approximately 85% of families with NPS present mutations in LMX1B, which consistently segregate with disease in an autosomal dominant pattern with complete penetrance. The majority of mutations, including nonsense mutations, small intragenic insertions/deletions or splice-site mutations, results in protein truncation. Missense mutations generally involve substitutions in the homeodomain region critical for DNA binding (195–197, 206, 207). Recently, entire-gene deletions were reported by Bongers et al., confirming that haploinsufficiency of the LMX1B transcription factor underlies this disease (208).
The precise role of LMX1B in the kidney remains partially elucidated. Immunohistochemical studies in several patients bearing heterozygous mutations in the LMX1B gene revealed that the expression of the α3 and α4 chains of type IV collagen, as well as podocin and CD2AP are no different than normal controls (209). In mouse, Lmx1b is expressed exclusively in glomeruli. Podocyte-specific Lmx1b inactivation invariably leads to proteinuria, renal insufficiency and death at 2 weeks after birth (210). In addition, LMX1B may be critical for glomerular development, since mice with podocyte-specific inactivation of Lmx1b showed severely impaired glomerular development and podocyte differentiation (211). Potential targets of Lmx1b in the kidney have been demonstrated in the shared 5′ regulatory regions of Col4a3 and Col4a4 genes (212), and in the promoter regions of the Nphs2 and Cd2ap genes (211, 213). In Lmx1b-deficient mice, the abundance of α(3)IV and α(4)IV chains of collagen were markedly diminished (212), as were the levels of podocin (211, 213), CD2AP (213), synaptopodin and VEGF (211). Nevertheless, no downregulation in the expression of the α3 and α4 chains of type IV collagen, podocin and CD2AP, were observed in mice in which Lmx1b had been inactivated specifically in podocytes (210). Interestingly, immunohistochemical studies in two patients bearing heterozygous mutations in the LMX1B gene revealed no downregulation in the expression of the α3 and α4 chains of type IV collagen, and in podocin and CD2AP (209). The latter findings may be explained by the fact that these patients carry one functional and one mutated alleles, whereas mice have two mutant alleles.
Schimke Immuno-Osseus Dysplasia: Mutations in SMARCAL1, Encoding the swi/snf-Related Matrix-Associated Actin-Dependent Regulator of Chromatin, Subfamily-A-Like-1
Schimke immuno-osseus dysplasia (SIOD, OMIM 242900) is a rare autosomal recessive disorder characterized by spondyloepiphyseal dysplasia, progressive renal dysfunction due to focal segmental glomerulosclerosis and T-cell immunodeficiency (214). Other additional, although inconstant features include cerebral ischemia, migraine-like headaches, deficiency of other blood cell lineages, hyperpigmented macules, corneal opacities, microdontia, intellectual delay, recurrent infections, premature atherosclerosis, hypothyroidism, cerebellar atrophy and testicular hypoplasia with atrophy and azospermia (215–218). Kidney disease in patients with SIOD manifests typically with proteinuria evolving to overt nephrotic syndrome, which is diagnosed between the first year of life and 14 years of age (215, 216). No response to steroids has been documented in patients who have been treated; nevertheless, transient reductions in proteinuria using ACE inhibitors, NSAID or even cyclosporin A have been observed (216). Patients who survive infectious complications progress to ESRD between 5 and 15 years of age. Numerous cases have been transplanted without evidence of relapse in the allograft (216); however, the evolution of cerebrovascular and infectious complications do not seem to improve after transplantation.
A genome-wide scan, performed in four families, detected significant linkage at chromosome 2q35, and mutations were identified in the SMARCAL1 gene (26). This gene encodes a member of an SNF2 subfamily of proteins that mediate DNA-nucleosome restructuring during gene regulation and DNA replication, recombination, methylation and gene repair (swi/snf-related matrix-associated actin-dependent regulator of chromatin, subfamily-a-like-1 gene). The gene consists of 18 exons and encodes a 106-kDa protein with 954 amino acid residues. The majority of mutations identified involve nonsense and frameshifting mutations, likely leading to loss-of function (26). Recently, Clewing et al. showed that SMARCAL1 biallelic mutations accounted for the phenotype in 38 of 72 independent cases with SIOD (219), revealing the genetic heterogeneity of this syndrome. Patients with two missense mutations tended to have a milder course of disease, surviving beyond 15 years of age. The functional targets of SMARCAL1 remain unidentified.
Action Myoclonus-Renal Failure Syndrome: Mutations in SCARB2 Encoding the Lysosome Membrane Protein 2
Action myoclonus-renal failure syndrome (AMRF, MIM 254900) is a rare autosomal recessive disease characterized by progressive myoclonic epilepsy associated with renal failure. It typically presents at 15–25 years of age with neurological symptoms including tremor, action myoclonus, seizures and later ataxia, while cognitive function is preserved. Proteinuria is usually diagnosed concomitantly with the onset of neurologic symptoms at a median age of 19 years (220), although proteinuria may also be the first symptom. Progression of renal impairment to ESRD occurs generally within 5 years after onset of proteinuria (220, 221). The renal pathology is characterized by focal glomerulosclerosis, sometimes with features of glomerular collapse (220). In three unrelated families, Berkovic et al. identified a region on chromosome 4q13-21 linked to AMRF. Subsequently, using gene expression profiling to prioritize gene sequencing within the region, they identified homozygous truncating mutations in the SCARB2 gene (encoding LIMP-2). These mutations led to a downregulation of SCARB2 mRNA and undetectable protein levels in western blots of cell lysates from lymphoblastoid B cell lines from the two affected subjects (29).
LIMP-2 is a transmembrane protein of the CD36 superfamily, which is ubiquitously expressed and is mainly found in lysosomes and late endosomes, where it is required for their biogenesis and maintenance (222–224). It has been shown that LIMP-2 acts as a trafficking receptor for β-glucocerebrosidase (β-GCase) (225), a lysosomal enzyme deficient in most cases of Gaucher disease. Interestingly, a nonsense mutation involving the interaction domain of LIMP-2/β-GCase was recently identified in two patients with ARMF in which a severe β-GCase deficiency was detected in cultured skin fibroblast (221). The pathophysiologic events leading to NS and FSGS in patients with SCARB2 mutations remain to be elucidated.
In mouse, the Limp2 gene is expressed in a range of tissues including brain and kidney. Interestingly, a Limp2-deficient mouse model presents with hearing impairment, demyelinating neuropathy, cerebral and cerebellar cytoplasmic inclusions, hydronephrosis caused by uretero-pelvic junction obstruction, mesangial proliferation and foot-process effacement, but does not recapitulate the glomerular lesions seen in humans (29, 223). Proteinuria is present, but occurs only with aged mice (29).
Mitochondropathies Manifesting with Nephrotic Syndrome
The mitochondropathies are a diverse group of disorders due to structural, biochemical, or genetic derangements of mitochondria (226). Renal dysfunction is a rare event, and may result from mutations in the mitochondrial or nuclear genomes. The mitochondrial genome encodes for 13 essential subunits of the mitochondrial respiratory chain, as well as the 22 transfer RNA (tRNA) and 2 ribosomal RNA (rRNA) genes (227). The c.3243A>G point mutation in the tRNALeu(UUR) gene is associated with MELAS syndrome (myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) (27). Some patients carrying tRNALeu(UUR) gene mutations may present with diabetes and deafness (228, 229), cardiomyopathy (230), progressive external ophthalmoplegia and FSGS (231–236), with or without the nephrotic syndrome (234–240). Although most of the affected cases are diagnosed in adulthood and have glomerular disease associated with other manifestations of mitochondrial disease, some patients present with isolated nephropathy or may have an earlier onset during the adolescence (235, 238, 240, 241).
Nephrotic syndrome has, likewise, been described in coenzyme Q10 (CoQ10) deficiency (MIM 607426), in association with encephalomyopathy and multisystemic involvement (30, 242, 243). The COQ2 gene is part of the coenzyme Q10 pathway, a component of the mitochondrial respiratory chain vital for the transport of electrons from complexes I and II to complex III. Mutations in the COQ2 gene were identified in several patients presenting with early-onset nephrotic syndrome, with or without neuromuscular symptoms (244). The clinical presentation varied from severe oliguric renal failure due to crescentic GN on the fifth day of life to development of SRNS at 18 months in association with collapsing glomerulopathy (244). In all renal biopsies dysmorphic mitochondria were characteristic (244).
Lopez et al. recently described an infant with fatal Leigh syndrome, CoQ10 deficiency in muscle and fibroblasts, nephrotic syndrome, and compound heterozygous mutations in the PDSS2 gene (245). The PDSS2 gene encodes a subunit of decaprenyl diphosphate synthase, the first enzyme of the CoQ10 biosynthetic pathway (245). Similarly, the kd/kd mouse, which develops collapsing glomerulopathy, carries mutations in the murine orthologue of the human PDSS2 gene (246). Moreover, mice in which the Pdss2 gene has been conditionally inactivated in podocytes exhibit proteinuria and foot process effacement (247). Recently, Saiki et al. showed that coenzyme Q10 supplementation rescues renal disease in Pdss2 kd/kd mice (248).
It is likely that mutations in other genes involved in the CoQ10 biosynthetic pathway are responsible for cases of NS with or without neuromuscular manifestations. In cases with CoQ10 deficiency, early ubiquinone supplementation may be crucial for the resolution of renal symptoms and for preventing neurologic damage, as demonstrated in patients and animal models (248, 249).
Finally, a deletion in the mitochondrial DNA has been associated with FSGS in a cohort of Japanese patients and in a Turkish patient (239, 250).
Hereditary Multisystemic Disorders of Unknown Cause Associated with Steroid-Resistant Nephrotic Syndrome
Galloway-Mowat syndrome
Galloway-Mowat syndrome is a rare disorder, of autosomal recessive inheritance, characterized by SRNS, microcephaly and severe neurological impairment (251). Disease frequency is unknown; however more than 70 cases have been reported since the original description in 1968 (251). Inconstant morphological defects include hiatus hernia, micrognathia, arachnodactyly and floppy ears. Proteinuria is usually discovered within the first year of life; although congenital onset is not unusual as are cases in which the onset of NS is close to the third year of life (251–259). Kidney histology may reveal either FSGS or DMS, the later more frequent in early onset forms (254, 260–263). In addition, a single patient with collapsing FSGS has been recently reported (264). The great majority of patients reach ESRD between 36 and 72 months after birth, although there are rarer cases with preserved renal function after this age (259, 263).
The distinctive neurological feature is marked microcephaly, which might be congenital (primary) or may develop after birth (secondary). Structural brain abnormalities include cortical and cerebellar atrophy, severe myelination deficiency and gyral defects (257, 263, 265–268). Profound mental retardation, hypotonia and seizures are the most recurrent neurological symptoms. In addition, choreoathetosis may develop later in the course of the disease (Antignac, personal observation). Sensorineural blindness and deafness have also been described. Patients may occasionally be able to walk, interact with their families and eventually develop a rudimentary monosyllabic language (Antignac, personal observation). The association with microphthalmia and corneal defects has been occasionally reported (252, 260, 261, 269–271). Undeniably, ocular malformations are a common feature of Pierson syndrome, in which microcephaly might be occasionally observed (272). Due to the overlapping phenotype with GMS, screening of mutations in laminin-β2 and several related proteins was performed by Dietrich et al. in 18 unrelated patients with GMS (273). Unfortunately they failed to find pathogenic mutations. Indeed, GMS represents a heterogeneous group of diseases and so far, the underlying genetic abnormalities have not been identified.
SRNS and Deafness
The association of SRNS and deafness has been described in patients with familial forms of SRNS with both autosomal dominant and recessive inheritance, revealing the genetic heterogeneity of this clinical association. Excluding patients carrying mutations in genes involved in the mitochondrial respiratory chain, in which deafness and nephrotic syndrome may be present in addition to neuromuscular symptoms, two loci have been identified to-date (274, 275).
Ruf et al. mapped the first locus on chr 14q24.2 in a consanguineous Palestinian family (275). Congenital sensorineural deafness was diagnosed in the four affected cases. The onset of NS ranged from 0.3 to 6.4 years and all the patients progressed to ESRD before 10 years of age. Kidney histology was compatible with FSGS. Three cases were transplanted, with no relapse of proteinuria. Prakash et al. described a 39-member kindred from India, consisting of 7 affected members, showing male-to-male transmission with an AD pattern of inheritance (274). Age at presentation varied between 8 and 44 years of age. Five of the affected cases also had sensorineural deafness. Renal biopsies revealed FSGS with irregular GBM. A genome-wide scan identified a novel locus on chr 11q24, after exclusion of linkage to currently known loci for Alport syndrome.
Epidermolysis Bullosa and FSGS
Nephrotic syndrome and renal failure may occur in some patients with epidermolysis bullosa (276). The most common histological finding is secondary amyloidosis (277–283). The association of FSGS and epidermolysis bullosa has been reported in a male infant with pyloric atresia, junctional epidermolysis bullosa and nephrotic range proteinuria diagnosed 6 weeks after birth (25). Renal biopsy revealed immature glomeruli, segmental sclerosis in the absence of microcystic tubular dilatation, atrophy and interstitial fibrosis. Ultrastructural changes included a thin glomerular basement membrane, extensive foot process effacement and microvillous transformation of podocytes. Mutation analysis of the β4- and α6-integrin genes ITGB4 and ITGA6 revealed a homozygous missense mutation in exon 31 of the ITGB4 gene, resulting in a substitution of tryptophan for arginine at codon 1281. This mutation affects the second fibronectin type III domain which is involved in the interaction with bullous pemphigoid antigen 1 (BPAG1) and plectin. Moreover, in one patient with pyloric atresia, epidermolysis bullosa and nephrotic proteinuria diagnosed at 5 months of age, mutation screening of the ITGB4 gene revealed a c.4851delCA truncating mutation (Dr. Françoise Broux, Rouen, France; personal communication). The mechanisms by which mutations in the ITGB4 gene induce glomerular disease remain unknown.
Familial Forms of Steroid-Sensitive Nephrotic Syndrome
The incidence of SSNS in pediatric population ranges between 2 and 7/100,000. Although most of the cases are sporadic, several reports have confirmed the existence of hereditary forms of this disease (1, 284–289). The exact incidence of familial forms of SSNS is unknown, but according to a single survey, it may represent up to 3% of the cases (1). Based on six cases reports describing 58 patients from 21 families, the most common pattern of inheritance was autosomal recessive. There was a male-to-female preponderance of 3 to 1 and the average age of onset was 4 years. Kidney histology revealed minimal change disease. Most of the cases presented with multiple episodes of relapse and achieved complete remission at the end of adolescence (284–289). Analysis of our own cohort consisting of 46 affected cases from 23 families, revealed similar results (Antignac, unpublished data).
At least two attempts to identify a putative disease locus have been performed (288, 289). Ruf et al. performed a genome-wide scan in a consanguineous SSNS kindred allowing the identification of a locus on chr 2p12-p13.2 between markers D2S292 and D2S289 (288). More recently, Landau et al. studied an extended SSNS Bedouin family with a high rate of consanguinity (289). A whole genome scan was performed, using 382 microsatellite markers; however, the index family was not linked to any of the presently known loci associated with nephrotic syndrome. It remains unanswered whether the primary defect in hereditary SSNS lies in a gene that plays a central role in the function of the immune system, or in a gene expressed in podocytes.
Conclusion
Hereditary forms of NS are far more common than previously thought 10 years ago, since the discovery of mutations in causative genes in cases with Mendelian inheritance, as well as in patients with sporadic disease. Most of the cases with hereditary forms of NS have a disease onset within early childhood, are resistant to immunosuppressive therapy, and do not relapse after kidney transplantation. The highest rates of mutation detection are in patients presenting with proteinuria in the first year of life and subsequently decrease among older patients.
It is plausible that more complex patterns of inheritance, as has been described in patients bearing bi- or tri-allelic variants, may be associated with an increased risk of developing NS. Indeed, disease predisposing mutations may lead to variable disease expression and penetrance depending upon unidentified environmental and genetic factors. Moreover, common variants in genes expressed in podocytes may account for an increased risk of FSGS and ESRD observed in selected ethnic groups, as has been described recently with the MYH9 gene, in which several haplotypes conferred a major-risk effect for FSGS in individuals of African ancestry (290–292).
The accessibility to custom genotyping chips and deep-sequencing techniques will facilitate the screening of mutations in a broader approach, including clusters of podocyte-specific genes. To-date, several fascinating disorders, such as Galloway-Mowat syndrome, involving brain and kidney development, and familial forms of SSNS, connecting podocyte physiology and the immune system, remain unsolved.
Non-syndromic forms of NS are frequently restricted to mutation in genes exclusively expressed in podocytes at the slit-diaphragm, while the association with extrarenal manifestations is observed in cases carrying mutations in ubiquitously expressed genes, mostly transcription factors or components of the mitochondrial respiratory chain. Nevertheless, individuals with mutations in genes associated with syndromic SRNS may present with a milder phenotype and only with SRNS; thus, making directed mutation screening a difficult task.
A promising therapy, still explored at a basic level, include protein chaperones. These drugs redirect the trafficking of missense mutant proteins to the plasma membrane when abnormally retained in subcellular organelles. Additional encouraging results have been obtained with drugs, which stabilize the podocyte actin cytoskeleton.
It is now clear that genetic diagnosis of cases with SRNS or familial NS is necessary to avoid ineffective therapies, to allow for accurate genetic counseling and, in the future, to offer specific mutation-based therapies.
References
White RH. The familial nephrotic syndrome. I. A European survey. Clin Nephrol 1973;1:215–219.
Moncrieff MW et al. The familial nephrotic syndrome. II. A clinicopathological study. Clin Nephrol 1973;1:220–229.
Mattoo TK, Mahmood MA, al-Harbi MS. Nephrotic syndrome in Saudi children clinicopathological study of 150 cases. Pediatr Nephrol 1990;4:517–519.
Hinkes BG et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007;119:e907–e919.
Weber S et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int 2004;66:571–579.
Ruf RG et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol 2004;15:722–732.
Winn MP. Young Investigator Award: TRP’ing into a new era for glomerular disease. J Am Soc Nephrol 2008;19:1071–1075.
Kestila M et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell 1998;1:575–582.
Philippe A et al. Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2008;19:1871–1878.
Boute N et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000;24:349–354.
Kaplan JM et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000;24:251–256.
Winn MP et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005;308:1801–1804.
Hinkes B et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006;38:1397–1405.
Barbaux S et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 1997;17:467–470.
Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr 1964;64:740–745.
Moorthy AV, Chesney RW, Lubinsky M. Chronic renal failure and XY gonadal dysgenesis: “Frasier” syndrome – a commentary on reported cases. Am J Med Genet Suppl 1987;3:297–302.
Denys P, Malvaux P, Van Den Berghe H, Tanghe W, Proesmans W. Association of an anatomo-pathological syndrome of male pseudohermaphroditism, Wilms’ tumor, parenchymatous nephropathy and XX/XY mosaicism. Arch Fr Pediatr 1967;24:729–739.
Drash A, Sherman F, Hartmann WH, Blizzard RM. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr 1970;76:585–593.
Hastie ND. Dominant negative mutations in the Wilms tumour (WT1) gene cause Denys-Drash syndrome – proof that a tumour-suppressor gene plays a crucial role in normal genitourinary development. Hum Mol Genet 1992;1:293–295.
Pelletier J et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991;67:437–447.
Miller RW, Fraumeni JF Jr, Manning MD. Association of Wilms’s tumor with aniridia, hemihypertrophy and other congenital malformations. N Engl J Med 1964;270:922–927.
Ruf RG et al. Prevalence of WT1 mutations in a large cohort of patients with steroid-resistant and steroid-sensitive nephrotic syndrome. Kidney Int 2004;66:564–570.
Zenker M et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 2004;13:2625–2632.
Dreyer SD et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 1998;19:47–50.
Kambham N et al. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am J Kidney Dis 2000;36:190–196.
Boerkoel CF et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 2002;30:215–220.
Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990;348:651–653.
Kobayashi Y et al. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem Biophys Res Commun 1990;173:816–822.
Berkovic SF et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 2008;82:673–684.
Salviati L et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 2005;65:606–608.
Quinzii C et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 2006;78:345–349.
Mollet J et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest 2007;117:765–772.
Fuchshuber A et al. Mapping a gene (SRN1) to chromosome 1q25-q31 in idiopathic nephrotic syndrome confirms a distinct entity of autosomal recessive nephrosis. Hum Mol Genet 1995;4:2155–2158.
Hinkes B et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2008;19:365–371.
Karle SM et al. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2002;13:388–393.
Caridi G et al. Broadening the spectrum of diseases related to podocin mutations. J Am Soc Nephrol 2003;14:1278–1286.
Hinkes B et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2008;19:365–371.
Ismaili K, Wissing KM, Janssen F, Hall M. Genetic forms of nephrotic syndrome: a single-center experience in Brussels. Pediatr Nephrol 2009;24(2):287–94.
Tsukaguchi H et al. A locus for adolescent and adult onset familial focal segmental glomerulosclerosis on chromosome 1q25-31. J Am Soc Nephrol 2000;11:1674–1680.
Tsukaguchi H et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest 2002;110:1659–1666.
Stewart GW. Stomatin. Int J Biochem Cell Biol 1997;29:271–274.
Mannsfeldt AG, Carroll P, Stucky CL, Lewin GR. Stomatin, a MEC-2 like protein, is expressed by mammalian sensory neurons. Mol Cell Neurosci 1999;13:391–404.
Martinez-Salgado C et al. Stomatin and sensory neuron mechanotransduction. J Neurophysiol 2007;98:3802–3808.
Wetzel C et al. A stomatin-domain protein essential for touch sensation in the mouse. Nature 2007;445:206–209.
Huang M, Gu G, Ferguson EL, Chalfie M. A stomatin-like protein necessary for mechanosensation in C. elegans. Nature 1995;378:292–295.
Schwarz K et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest 2001;108:1621–1629.
Roselli S et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol 2002;160:131–139.
Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem 2001;276:41543–41546.
Sellin L et al. NEPH1 defines a novel family of podocin interacting proteins. Faseb J 2003;17:115–117.
Huber TB et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol 2003;23:4917–4928.
Huber TB et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA 2006;103:17079–17086.
Huber TB, Schermer B, Benzing T. Podocin organizes ion channel-lipid supercomplexes: implications for mechanosensation at the slit diaphragm. Nephron Exp Nephrol 2007;106:e27–e31.
Reiser J et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 2005;37:739–744.
Koziell A et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet 2002;11:379–388.
Frishberg Y et al. Mutations in NPHS2 encoding podocin are a prevalent cause of steroid-resistant nephrotic syndrome among Israeli-Arab children. J Am Soc Nephrol 2002;13:400–405.
Caridi G et al. Podocin mutations in sporadic focal-segmental glomerulosclerosis occurring in adulthood. Kidney Int 2003;64:365.
Ardiles LG, Carrasco AE, Carpio JD, Mezzano SA. Late onset of familial nephrotic syndrome associated with a compound heterozygous mutation of the podocin-encoding gene. Nephrology (Carlton) 2005;10:553–556.
Ozcakar ZB et al. Analysis of NPHS2 mutations in Turkish steroid-resistant nephrotic syndrome patients. Pediatr Nephrol 2006;21:1093–1096.
Berdeli A et al. NPHS2 (podicin) mutations in Turkish children with idiopathic nephrotic syndrome. Pediatr Nephrol 2007;22:2031–2040.
Bakr A et al. NPHS2 mutations. Indian J Pediatr 2008;75:135–138.
Drozdz D et al. Heterozygotic mutation in NPHS2 gene as a cause of familial steroid resistant nephrotic syndrome in two siblings – case report. Przegl Lek 2006;63 Suppl 3:85–86.
Komatsuda A et al. Analysis of mutations in alpha-actinin 4 and podocin genes of patients with chronic renal failure due to sporadic focal segmental glomerulosclerosis. Ren Fail 2003;25:87–93.
Maruyama K et al. NPHS2 mutations in sporadic steroid-resistant nephrotic syndrome in Japanese children. Pediatr Nephrol 2003;18:412–416.
Sako M et al. Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients with congenital nephrotic syndrome. Kidney Int 2005;67:1248–1255.
Kitamura A et al. Genetics and clinical features of 15 Asian families with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant 2006;21:3133–3138.
Mao J et al. NPHS1 and NPHS2 gene mutations in Chinese children with sporadic nephrotic syndrome. Pediatr Res 2007;61:117–122.
Yu Z et al. Mutations in NPHS2 in sporadic steroid-resistant nephrotic syndrome in Chinese children. Nephrol Dial Transplant 2005;20:902–908.
Cho HY et al. WT1 and NPHS2 mutations in Korean children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 2008;23:63–70.
Chernin G et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 2008;23:1455–1460.
Frishberg Y et al. The heart of children with steroid-resistant nephrotic syndrome: is it all podocin? J Am Soc Nephrol 2006;17:227–231.
Caridi G et al. Lack of cardiac anomalies in children with NPHS2 mutations. Nephrol Dial Transplant 2007;22:1477–1479.
Franceschini N, North KE, Kopp JB, McKenzie L, Winkler C. NPHS2 gene, nephrotic syndrome and focal segmental glomerulosclerosis: a HuGE review. Genet Med 2006;8:63–75.
Pereira AC et al. NPHS2 R229Q functional variant is associated with microalbuminuria in the general population. Kidney Int 2004;65:1026–1030.
Kottgen A et al. The association of podocin R229Q polymorphism with increased albuminuria or reduced estimated GFR in a large population-based sample of US adults. Am J Kidney Dis 2008;52:868–875.
McKenzie LM et al. NPHS2 variation in sporadic focal segmental glomerulosclerosis. J Am Soc Nephrol 2007;18:2987–2995.
He N et al. Recessive NPHS2 (Podocin) mutations are rare in adult-onset idiopathic focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2007;2:31–37.
Schultheiss M et al. No evidence for genotype/phenotype correlation in NPHS1 and NPHS2 mutations. Pediatr Nephrol 2004;19:1340–1348.
Schwaderer P et al. Clinical course and NPHS2 analysis in patients with late steroid-resistant nephrotic syndrome. Pediatr Nephrol 2008;23:251–256.
Gbadegesin R et al. Mutational analysis of NPHS2 and WT1 in frequently relapsing and steroid-dependent nephrotic syndrome. Pediatr Nephrol 2007;22:509–513.
Jungraithmayr TC et al. Primary focal segmental glomerulosclerosis – long-term outcome after pediatric renal transplantation. Pediatr Transplant 2005;9:226–231.
Tejani A, Stablein DH. Recurrence of focal segmental glomerulosclerosis posttransplantation: a special report of the North American Pediatric Renal Transplant Cooperative Study. J Am Soc Nephrol 1992;2:S258–S263.
Weber S, Tonshoff B. Recurrence of focal-segmental glomerulosclerosis in children after renal transplantation: clinical and genetic aspects. Transplantation 2005;80:S128–S134.
Becker-Cohen R et al. Recurrent nephrotic syndrome in homozygous truncating NPHS2 mutation is not due to anti-podocin antibodies. Am J Transplant 2007;7:256–260.
Billing H et al. NPHS2 mutation associated with recurrence of proteinuria after transplantation. Pediatr Nephrol 2004;19:561–564.
Hocker B et al. Recurrence of proteinuria 10 years post-transplant in NPHS2-associated focal segmental glomerulosclerosis after conversion from cyclosporin A to sirolimus. Pediatr Nephrol 2006;21:1476–1479.
Bertelli R et al. Recurrence of focal segmental glomerulosclerosis after renal transplantation in patients with mutations of podocin. Am J Kidney Dis 2003;41:1314–1321.
Carraro M et al. Serum glomerular permeability activity in patients with podocin mutations (NPHS2) and steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2002;13:1946–1952.
Roselli S, Moutkine I, Gribouval O, Benmerah A, Antignac C. Plasma membrane targeting of podocin through the classical exocytic pathway: effect of NPHS2 mutations. Traffic 2004;5:37–44.
Nishibori Y et al. Disease-causing missense mutations in NPHS2 gene alter normal nephrin trafficking to the plasma membrane. Kidney Int 2004;66:1755–1765.
Ohashi T, Uchida K, Uchida S, Sasaki S, Nihei H. Intracellular mislocalization of mutant podocin and correction by chemical chaperones. Histochem Cell Biol 2003;119:257–264.
Roselli S et al. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol 2004;24:550–560.
Ratelade J et al. Maternal environment interacts with modifier genes to influence progression of nephrotic syndrome. J Am Soc Nephrol 2008;19:1491–1499.
Philippe A et al. A missense mutation in podocin leads to early and severe renal disease in mice. Kidney Int 2008;73:1038–1047.
Shih NY et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 1999;286:312–315.
Donoviel DB et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol 2001;21:4829–4836.
Kramer-Zucker AG, Wiessner S, Jensen AM, Drummond IA. Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein Mosaic eyes. Dev Biol 2005;285:316–329.
Hentschel DM et al. Rapid screening of glomerular slit diaphragm integrity in larval zebrafish. Am J Physiol Renal Physiol 2007;293:F1746–F1750.
Kitamura A et al. A familial childhood-onset relapsing nephrotic syndrome. Kidney Int 2007;71:946–951.
Bolk S, Puffenberger EG, Hudson J, Morton DH, Chakravarti A. Elevated frequency and allelic heterogeneity of congenital nephrotic syndrome, Finnish type, in the old order Mennonites. Am J Hum Genet 1999;65:1785–1790.
Aya K, Tanaka H, Seino Y. Novel mutation in the nephrin gene of a Japanese patient with congenital nephrotic syndrome of the Finnish type. Kidney Int 2000;57:401–404.
Beltcheva O, Martin P, Lenkkeri U, Tryggvason K. Mutation spectrum in the nephrin gene (NPHS1) in congenital nephrotic syndrome. Hum Mutat 2001;17:368–373.
Gigante M et al. Congenital nephrotic syndrome of the Finnish type in Italy: a molecular approach. J Nephrol 2002;15:696–702.
Gigante M et al. Congenital nephrotic syndrome of Finnish type: detection of new nephrin mutations and prenatal diagnosis in an Italian family. Prenat Diagn 2005;25:407–410.
Shi Y, Ding J, Liu JC, Wang H, Bu DF. NPHS1 mutations in a Chinese family with congenital nephrotic syndrome. Zhonghua Er Ke Za Zhi 2005;43:805–809.
Frishberg Y et al. Misleading findings of homozygosity mapping resulting from three novel mutations in NPHS1 encoding nephrin in a highly inbred community. Genet Med 2007;9:180–184.
Heeringa SF et al. Thirteen novel NPHS1 mutations in a large cohort of children with congenital nephrotic syndrome. Nephrol Dial Transplant 2008;23:3527–3533.
Lenkkeri U et al. Structure of the gene for congenital nephrotic syndrome of the finnish type (NPHS1) and characterization of mutations. Am J Hum Genet 1999;64:51–61.
Liu L et al. Defective nephrin trafficking caused by missense mutations in the NPHS1 gene: insight into the mechanisms of congenital nephrotic syndrome. Hum Mol Genet 2001;10:2637–2644.
Liu XL et al. Defective trafficking of nephrin missense mutants rescued by a chemical chaperone. J Am Soc Nephrol 2004;15:1731–1738.
Putaala H, Soininen R, Kilpelainen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet 2001;10:1–8.
Hamano Y et al. Determinants of vascular permeability in the kidney glomerulus. J Biol Chem 2002;277:31154–31162.
Rantanen M et al. Nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli and cystic tubular lesions. J Am Soc Nephrol 2002;13:1586–1594.
Quaggin SE. A new piece in the nephrotic puzzle. Nat Genet 2006;38:1360–1361.
Gbadegesin R et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol Dial Transplant 2008;23:1291–1297.
Wing MR, Bourdon DM, Harden TK. PLC-epsilon: a shared effector protein in Ras-, Rho-, and G alpha beta gamma-mediated signaling. Mol Interv 2003;3:273–280.
Lehtonen S et al. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc Natl Acad Sci USA 2005;102:9814–9819.
Wang H et al. Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 2005;97:1305–1313.
Dustin ML et al. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell 1998;94:667–677.
Lehtonen S, Zhao F, Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol 2002;283:F734–F743.
Bruck S et al. Identification of a novel inhibitory actin-capping protein binding motif in CD2-associated protein. J Biol Chem 2006;281:19196–19203.
Hutchings NJ, Clarkson N, Chalkley R, Barclay AN, Brown MH. Linking the T cell surface protein CD2 to the actin-capping protein CAPZ via CMS and CIN85. J Biol Chem 2003;278:22396–22403.
Cormont M et al. CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic 2003;4:97–112.
Kobayashi S, Sawano A, Nojima Y, Shibuya M, Maru Y. The c-Cbl/CD2AP complex regulates VEGF-induced endocytosis and degradation of Flt-1 (VEGFR-1). FASEB J 2004;18:929–931.
Schiffer M, Mundel P, Shaw AS, Bottinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem 2004;279:37004–37012.
Kim JM et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 2003;300:1298–1300.
Lowik MM et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int 2007;72:1198–1203.
Lowik M et al. Bigenic heterozygosity and the development of steroid-resistant focal segmental glomerulosclerosis. Nephrol Dial Transplant 2008;23:3146–3151.
Conlon PJ et al. Clinical and pathologic features of familial focal segmental glomerulosclerosis. Am J Kidney Dis 1995;26:34–40.
Rana K et al. Clinical, histopathologic, and genetic studies in nine families with focal segmental glomerulosclerosis. Am J Kidney Dis 2003;41:1170–1178.
Mathis BJ, Calabrese KE, Slick GL. Familial glomerular disease with asymptomatic proteinuria and nephrotic syndrome: a new clinical entity. J Am Osteopath Assoc 1992;92:875–880, 883–884.
Mathis BJ et al. A locus for inherited focal segmental glomerulosclerosis maps to chromosome 19q13. Kidney Int 1998;53:282–286.
Pollak MR, Alexander MP, Henderson JM. A case of familial kidney disease. Clin J Am Soc Nephrol 2007;2:1367–1374.
Vats A et al. Familial nephrotic syndrome: clinical spectrum and linkage to chromosome 19q13. Kidney Int 2000;57:875–881.
Weins A et al. Mutational and Biological Analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol 2005;16:3694–3701.
Aucella F et al. Molecular analysis of NPHS2 and ACTN4 genes in a series of 33 Italian patients affected by adult-onset nonfamilial focal segmental glomerulosclerosis. Nephron Clin Pract 2005;99:c31–c36.
Yao J et al. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2004;2:e167.
Weins A et al. Disease-associated mutant alpha-actinin-4 reveals a mechanism for regulating its F-actin-binding affinity. Proc Natl Acad Sci USA 2007;104:16080–16085.
Lachapelle M, Bendayan M. Contractile proteins in podocytes: immunocytochemical localization of actin and alpha-actinin in normal and nephrotic rat kidneys. Virchows Arch B Cell Pathol Incl Mol Pathol 1991;60:105–111.
Otey CA, Pavalko FM, Burridge K. An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J Cell Biol 1990;111:721–729.
Dandapani SV et al. Alpha-actinin-4 is required for normal podocyte adhesion. J Biol Chem 2007;282:467–477.
Kos CH et al. Mice deficient in alpha-actinin-4 have severe glomerular disease. J Clin Invest 2003;111:1683–1690.
Michaud JL et al. Focal and segmental glomerulosclerosis in mice with podocyte-specific expression of mutant alpha-actinin-4. J Am Soc Nephrol 2003;14:1200–1211.
Henderson JM, Al-Waheeb S, Weins A, Dandapani SV, Pollak MR. Mice with altered alpha-actinin-4 expression have distinct morphologic patterns of glomerular disease. Kidney Int 2008;73:741–750.
Winn MP et al. Linkage of a gene causing familial focal segmental glomerulosclerosis to chromosome 11 and further evidence of genetic heterogeneity. Genomics 1999;58:113–120.
Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA 2006;103:16586–16591.
Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev 2007;87:165–217.
Dietrich A et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol 2005;25:6980–6989.
Moller CC, Mangos S, Drummond IA, Reiser J. Expression of trpC1 and trpC6 orthologs in zebrafish. Gene Expr Patterns 2008;8:291–296.
Pierson M, Cordier J, Hervouuet F, Rauber G. An unusual congenital and familial congenital malformative combination involving the eye and kidney. J Genet Hum 1963;12:184–213.
Beale MG, Strayer DS, Kissane JM, Robson AM. Congenital glomerulosclerosis and nephrotic syndrome in two infants. Speculations and pathogenesis. Am J Dis Child 1979;133:842–845.
Schneller M, Braga SE, Moser H, Zimmermann A, Oetliker O. Congenital nephrotic syndrome: clinico-pathological heterogeneity and prenatal diagnosis. Clin Nephrol 1983;19:243–249.
Braga S, Monn E, Zimmermann A, Oetliker O. Congenital nephrotic syndrome with congenital buphthalmos: a new genetic entity? Prog Clin Biol Res 1989;305:205–209.
Glastre C et al. Familial infantile nephrotic syndrome with ocular abnormalities. Pediatrie 1989;44:555–558.
Nielsen KF, Steffensen GK. Congenital nephrotic syndrome associated with Lowe’s syndrome. Child Nephrol Urol 1990;10:92–95.
Zenker M et al. Congenital nephrosis, mesangial sclerosis, and distinct eye abnormalities with microcoria: an autosomal recessive syndrome. Am J Med Genet A 2004;130:138–145.
Noakes PG et al. The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet 1995;10:400–406.
Hasselbacher K et al. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int 2006;70:1008–1012.
Choi HJ et al. Variable phenotype of Pierson syndrome. Pediatr Nephrol 2008;23:995–1000.
Kagan M, Cohen AH, Matejas V, Vlangos C, Zenker M. A milder variant of Pierson syndrome. Pediatr Nephrol 2008;23:323–327.
Matejas V, Al-Gazali L, Amirlak I, Zenker M. A syndrome comprising childhood-onset glomerular kidney disease and ocular abnormalities with progressive loss of vision is caused by mutated LAMB2. Nephrol Dial Transplant 2006;21:3283–3286.
Sasaki T, Fassler R, Hohenester E. Laminin: the crux of basement membrane assembly. J Cell Biol 2004;164:959–963.
Miner JH. Building the glomerulus: a matricentric view. J Am Soc Nephrol 2005;16:857–861.
Miner JH, Li C. Defective glomerulogenesis in the absence of laminin alpha5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol 2000;217:278–289.
Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities in Lamb2 −/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest 2006;116:2272–2279.
Knight D, Tolley LK, Kim DK, Lavidis NA, Noakes PG. Functional analysis of neurotransmission at beta2-laminin deficient terminals. J Physiol 2003;546:789–800.
Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature 1995;374:258–262.
Libby RT et al. Laminin expression in adult and developing retinae: evidence of two novel CNS laminins. J Neurosci 2000;20:6517–6528.
Libby RT, Xu Y, Selfors LM, Brunken WJ, Hunter DD. Identification of the cellular source of laminin beta2 in adult and developing vertebrate retinae. J Comp Neurol 1997;389:655–667.
Hunter DD, Shah V, Merlie JP, Sanes JR. A laminin-like adhesive protein concentrated in the synaptic cleft of the neuromuscular junction. Nature 1989;338:229–234.
Rose EA et al. Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms’ tumor gene. Cell 1990;60:495–508.
Call KM et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990;60:509–520.
Gessler M et al. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990;343:774–778.
Pritchard-Jones K et al. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 1990;346:194–197.
Kreidberg JA et al. WT-1 is required for early kidney development. Cell 1993;74:679–691.
Rivera MN, Haber DA. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer 2005;5:699–712.
Moore AW et al. YAC transgenic analysis reveals Wilms’ tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech Dev 1998;79:169–184.
Davies JA et al. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet 2004;13:235–246.
Habib R, Gubler MC, Antignac C, Gagnadoux MF. Diffuse mesangial sclerosis: a congenital glomerulopathy with nephrotic syndrome. Adv Nephrol Necker Hosp 1993;22:43–57.
Habib R et al. The nephropathy associated with male pseudohermaphroditism and Wilms’ tumor (Drash syndrome): a distinctive glomerular lesion – report of 10 cases. Clin Nephrol 1985;24:269–278.
Jadresic L et al. Clinicopathologic review of twelve children with nephropathy, Wilms tumor, and genital abnormalities (Drash syndrome). J Pediatr 1990;117:717–725.
Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat 1997;9:209–225.
Jeanpierre C et al. Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet 1998;62:824–833.
Ito S et al. Isolated diffuse mesangial sclerosis and Wilms tumor suppressor gene. J Pediatr 2001;138:425–427.
Hahn H, Cho YM, Park YS, You HW, Cheong HI. Two cases of isolated diffuse mesangial sclerosis with WT1 mutations. J Korean Med Sci 2006;21:160–164.
Haber DA et al. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc Natl Acad Sci USA 1991;88:9618–9622.
Hammes A et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 2001;106:319–329.
Klamt B et al. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/−KTS splice isoforms. Hum Mol Genet 1998;7:709–714.
Demmer L et al. Frasier syndrome: a cause of focal segmental glomerulosclerosis in a 46,XX female. J Am Soc Nephrol 1999;10:2215–2218.
Denamur E et al. Mother-to-child transmitted WT1 splice-site mutation is responsible for distinct glomerular diseases. J Am Soc Nephrol 1999;10:2219–2223.
Mucha B et al. Mutations in the Wilms’ tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr Res 2006;59:325–331.
Williams HJ, Hoyer JR. Radiographic diagnosis of osteo-onychodysostiosis in infancy. Radiology 1973;109:151–154.
Guidera KJ, Satterwhite Y, Ogden JA, Pugh L, Ganey T. Nail patella syndrome: a review of 44 orthopaedic patients. J Pediatr Orthop 1991;11:737–742.
Bennett WM et al. The nephropathy of the nail-patella syndrome. Clinicopathologic analysis of 11 kindred. Am J Med 1973;54:304–319.
Looij BJ Jr, te Slaa RL, Hogewind BL, van de Kamp JJ. Genetic counselling in hereditary osteo-onychodysplasia (HOOD, nail-patella syndrome) with nephropathy. J Med Genet 1988;25:682–686.
McIntosh I et al. Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am J Hum Genet 1998;63:1651–1658.
Knoers NV et al. Nail-patella syndrome: identification of mutations in the LMX1B gene in Dutch families. J Am Soc Nephrol 2000;11:1762–1766.
Bongers EM et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet 2005;13:935–946.
Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 2003;40:153–162.
Chan PC, Chan KW, Cheng IK, Chan MK. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron 1988;50:164–166.
Gubler MC. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol 2008;4:24–37.
Ben-Bassat M, Cohen L, Rosenfeld J. The glomerular basement membrane in the nail-patella syndrome. Arch Pathol 1971;92:350–355.
Hoyer JR, Michael AF, Vernier RL. Renal disease in nail-patella syndrome: clinical and morphologic studies. Kidney Int 1972;2:231–238.
Gubler MC, Levy M, Naizot C, Habib R. Glomerular basement membrane changes in hereditary glomerular diseases. Ren Physiol 1980;3:405–413.
Lichter PR et al. Cosegregation of open-angle glaucoma and the nail-patella syndrome. Am J Ophthalmol 1997;124:506–515.
Chen H et al. Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet 1998;19:51–55.
Vollrath D et al. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet 1998;7:1091–1098.
Hamlington JD, Jones C, McIntosh I. Twenty-two novel LMX1B mutations identified in nail patella syndrome (NPS) patients. Hum Mutat 2001;18:458.
Bongers EM, de Wijs IJ, Marcelis C, Hoefsloot LH, Knoers NV. Identification of entire LMX1B gene deletions in nail patella syndrome: evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur J Hum Genet 2008;16:1240–1244.
Heidet L et al. In vivo expression of putative LMX1B targets in nail-patella syndrome kidneys. Am J Pathol 2003;163:145–155.
Suleiman H et al. The podocyte-specific inactivation of Lmx1b, Ldb1 and E2a yields new insight into a transcriptional network in podocytes. Dev Biol 2007;304:701–712.
Rohr C et al. The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J Clin Invest 2002;109:1073–1082.
Morello R et al. Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat Genet 2001;27:205–208.
Miner JH et al. Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation. J Clin Invest 2002;109:1065–1072.
Schimke RN, Horton WA, King CR. Chondroitin-6-sulphaturia, defective cellular immunity, and nephrotic syndrome. Lancet 1971;2:1088–1089.
Saraiva JM et al. Schimke immuno-osseous dysplasia: case report and review of 25 patients. J Med Genet 1999;36:786–789.
Boerkoel CF et al. Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur J Pediatr 2000;159:1–7.
Clewing JM et al. Schimke immuno-osseous dysplasia: a clinicopathological correlation. J Med Genet 2007;44:122–130.
Lucke T et al. Cerebellar atrophy in Schimke-immuno-osseous dysplasia. Am J Med Genet A 2007;143A:2040–2045.
Clewing JM et al. Schimke immunoosseous dysplasia: suggestions of genetic diversity. Hum Mutat 2007;28:273–283.
Badhwar A et al. Action myoclonus-renal failure syndrome: characterization of a unique cerebro-renal disorder. Brain 2004;127:2173–2182.
Balreira A et al. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum Mol Genet 2008;17:2238–2243.
Fukuda M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem 1991;266:21327–21330.
Gamp AC et al. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum Mol Genet 2003;12:631–646.
Kuronita T et al. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J Cell Sci 2002;115:4117–4131.
Reczek D et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007;131:770–783.
DiMauro S, Moraes CT. Mitochondrial encephalomyopathies. Arch Neurol 1993;50:1197–1208.
Anderson S et al. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457–465.
van den Ouweland JM et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet 1992;1:368–371.
Chinnery PF, Turnbull DM. Mitochondrial DNA and disease. Lancet 1999;354 Suppl 1:SI17–S121.
Yoshida R et al. Congestive heart failure in mitochondrial diabetes mellitus. Lancet 1994;344:1375.
Jansen JJ et al. Mutation in mitochondrial tRNA(Leu(UUR)) gene associated with progressive kidney disease. J Am Soc Nephrol 1997;8:1118–1124.
Kurogouchi F et al. A case of mitochondrial cytopathy with a typical point mutation for MELAS, presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation. Am J Nephrol 1998;18:551–556.
Nakamura S et al. Renal complications in patients with diabetes mellitus associated with an A to G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Diabetes Res Clin Pract 1999;44:183–189.
Doleris LM et al. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy. Kidney Int 2000;58:1851–1858.
Hotta O et al. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int 2001;59:1236–1243.
Hirano M et al. Renal complications in a patient with A-to-G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Intern Med 2002;41:113–118.
Lowik MM, Hol FA, Steenbergen EJ, Wetzels JF, van den Heuvel LP. Mitochondrial tRNALeu(UUR) mutation in a patient with steroid-resistant nephrotic syndrome and focal segmental glomerulosclerosis. Nephrol Dial Transplant 2005;20:336–341.
Cheong HI et al. Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatr Nephrol 1999;13:477–480.
Yamagata K et al. Mitochondrial DNA mutations in focal segmental glomerulosclerosis lesions. J Am Soc Nephrol 2002;13:1816–1823.
Guery B et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol 2003;14:2099–2108.
Ueda Y et al. A boy with mitochondrial disease: asymptomatic proteinuria without neuromyopathy. Pediatr Nephrol 2004;19:107–110.
Rotig A et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000;356:391–395.
Rahman S, Hargreaves I, Clayton P, Heales S. Neonatal presentation of coenzyme Q10 deficiency. J Pediatr 2001;139:456–458.
Diomedi-Camassei F et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 2007;18:2773–2780.
Lopez LC et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 2006;79:1125–1129.
Barisoni L, Madaio MP, Eraso M, Gasser DL, Nelson PJ. The kd/kd mouse is a model of collapsing glomerulopathy. J Am Soc Nephrol 2005;16:2847–2851.
Peng M et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet 2008;4:e1000061.
Saiki R et al. Coenzyme Q10 Supplementation Rescues Renal Disease in Pdss2kd/kd mice with mutations in Prenyldiphosphate synthase subunit 2. Am J Physiol Renal Physiol 2008;295:F1535–F1544.
Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 2008;358:2849–2850.
Unal S et al. Four-month-old infant with focal segmental glomerulosclerosis and mitochondrial DNA deletion. J Child Neurol 2005;20:83–84.
Galloway WH, Mowat AP. Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs. J Med Genet 1968;5:319–321.
Shapiro LR, Duncan PA, Farnsworth PB, Lefkowitz M. Congenital microcephaly, hiatus hernia and nephrotic syndrome: an autosomal recessive syndrome. Birth Defects Orig Artic Ser 1976;12:275–278.
Roos RA, Maaswinkel-Mooy PD, vd Loo EM, Kanhai HH. Congenital microcephaly, infantile spasms, psychomotor retardation, and nephrotic syndrome in two sibs. Eur J Pediatr 1987;146:532–536.
Garty BZ et al. Microcephaly and congenital nephrotic syndrome owing to diffuse mesangial sclerosis: an autosomal recessive syndrome. J Med Genet 1994;31:121–125.
Yalcinkaya F et al. Congenital microcephaly and infantile nephrotic syndrome – a case report. Pediatr Nephrol 1994;8:72–73.
Yu CH et al. Congenital nephrotic syndrome with microcephaly: report of a case. J Formos Med Assoc 1994;93:528–530.
Hashimoto K et al. Three siblings of fatal infantile encephalopathy with olivopontocerebellar hypoplasia and microcephaly. Brain Dev 1998;20:169–174.
de Vries BB, van’tHoff WG, Surtees RA, Winter RM. Diagnostic dilemmas in four infants with nephrotic syndrome, microcephaly and severe developmental delay. Clin Dysmorphol 2001;10:115–121.
Steiss JO, Gross S, Neubauer BA, Hahn A. Late-onset nephrotic syndrome and severe cerebellar atrophy in Galloway-Mowat syndrome. Neuropediatrics 2005;36:332–335.
Cohen AH, Turner MC. Kidney in Galloway-Mowat syndrome: clinical spectrum with description of pathology. Kidney Int 1994;45:1407–1415.
Mildenberger E et al. Diffuse mesangial sclerosis: association with unreported congenital anomalies and placental enlargement. Acta Paediatr 1998;87:1301–1303.
Nakazato H et al. Another autosomal recessive form of focal glomerulosclerosis with neurological findings. Pediatr Nephrol 2002;17:16–19.
Shiihara T et al. Microcephaly, cerebellar atrophy, and focal segmental glomerulosclerosis in two brothers: a possible mild form of Galloway-Mowat syndrome. J Child Neurol 2003;18:147–149.
Sartelet H et al. Collapsing glomerulopathy in Galloway-Mowat syndrome: a case report and review of the literature. Pathol Res Pract 2008;204:401–406.
Robain O, Deonna T. Pachygyria and congenital nephrosis disorder of migration and neuronal orientation. Acta Neuropathol 1983;60:137–141.
Palm L et al. Nephrosis and disturbances of neuronal migration in male siblings – a new hereditary disorder? Arch Dis Child 1986;61:545–548.
Albrecht S, Schneider MC, Belmont J, Armstrong DL. Fatal infantile encephalopathy with olivopontocerebellar hypoplasia and micrencephaly. Report of three siblings. Acta Neuropathol 1993;85:394–399.
Abbas RM, Kingo MB, Solimano A, Phang M, McGillivray B. Further case of Galloway-Mowat syndrome of abnormal gyral patterns and glomerulopathy. Am J Med Genet 1997;69:431.
Lin CC et al. Galloway-Mowat syndrome: a glomerular basement membrane disorder? Pediatr Nephrol 2001;16:653–657.
Kang L et al. Late-onset growth restriction in Galloway-Mowat syndrome: a case report. Prenat Diagn 2005;25:159–162.
Hou JW, Wang TR. Galloway-Mowat syndrome in Taiwan. Am J Med Genet 1995;58:245–248.
Wuhl E et al. Neurodevelopmental deficits in Pierson (microcoria-congenital nephrosis) syndrome. Am J Med Genet A 2007;143:311–319.
Dietrich A et al. Analysis of genes encoding laminin beta2 and related proteins in patients with Galloway-Mowat syndrome. Pediatr Nephrol 2008;23:1779–1786.
Prakash S et al. Autosomal dominant progressive nephropathy with deafness: linkage to a new locus on chromosome 11q24. J Am Soc Nephrol 2003;14:1794–1803.
Ruf RG et al. A gene locus for steroid-resistant nephrotic syndrome with deafness maps to chromosome 14q24.2. J Am Soc Nephrol 2003;14:1519–1522.
Fine JD et al. Inherited epidermolysis bullosa and the risk of death from renal disease: experience of the National Epidermolysis Bullosa Registry. Am J Kidney Dis 2004;44:651–660.
Sams WM Jr, Smith JG Jr. Epidermolysis bullosa acquisita, dermal elastosis, amyloidosis. Arch Dermatol 1964;90:137–142.
Thivolet J, Moulin G, Durand B, Pellerat J. Dystrophic bullous epidermolysis complicated by visceral amylosis. Bull Soc Fr Dermatol Syphiligr 1965;72:315–319.
Bureau Y, Barriere H, Litoux P, Bureau B, Bouhour. Hepatic and renal amylosis associated with epidermolysis bullosa. Bull Soc Fr Dermatol Syphiligr 1968;75:360–362.
Malaga S, Fernandez Toral J, Santos F, Riesgo I, Crespo M. Renal amyloidosis complicating a recessive epidermolysis bullosa in childhood. Helv Paediatr Acta 1983;38:167–170.
Mann JF et al. The spectrum of renal involvement in epidermolysis bullosa dystrophica hereditaria: report of two cases. Am J Kidney Dis 1988;11:437–441.
Kaneko K et al. Renal amyloidosis in recessive dystrophic epidermolysis bullosa. Dermatology 2000;200:209–212.
Gunduz K, Vatansever S, Turel A, Sen S. Recessive dystrophic epidermolysis bullosa complicated with nephrotic syndrome due to secondary amyloidosis. Int J Dermatol 2000;39:151–153.
Bensman A, Vasmant D, Mougenot B, Baudon JJ, Muller JY. Steroid-responsive nephrotic syndrome in infants: 2 familial case reports. Arch Fr Pediatr 1982;39:381–383.
Mallmann R. Idiopathic nephrotic syndrome and hexadactyly in two brothers. Pediatr Nephrol 1998;12:417–419.
Fuchshuber A et al. Clinical and genetic evaluation of familial steroid-responsive nephrotic syndrome in childhood. J Am Soc Nephrol 2001;12:374–378.
Kari JA, Sinnott P, Khan H, Trompeter RS, Snodgrass GJ. Familial steroid-responsive nephrotic syndrome and HLA antigens in Bengali children. Pediatr Nephrol 2001;16:346–349.
Ruf RG et al. Identification of the first gene locus (SSNS1) for steroid-sensitive nephrotic syndrome on chromosome 2p. J Am Soc Nephrol 2003;14:1897–1900.
Landau D et al. Familial steroid-sensitive nephrotic syndrome in Southern Israel: clinical and genetic observations. Pediatr Nephrol 2007;22:661–669.
Freedman BI, Sedor JR. Hypertension-associated kidney disease: perhaps no more. J Am Soc Nephrol 2008;19:2047–2051.
Kao WH et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 2008;40:1185–1192.
Kopp JB et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 2008;40:1175–1184.
Acknowledgments
This work has been made possible through an International Society of Nephrology Fellowship awarded to E.M. ELE was supported by EuReGene, an integrated project (5085) of the 6th Framework Program of the European Commission.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Machuca, E., Esquivel, E.L., Antignac, C. (2009). Idiopathic Nephrotic Syndrome: Genetic Aspects. In: Avner, E., Harmon, W., Niaudet, P., Yoshikawa, N. (eds) Pediatric Nephrology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-76341-3_27
Download citation
DOI: https://doi.org/10.1007/978-3-540-76341-3_27
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-76327-7
Online ISBN: 978-3-540-76341-3
eBook Packages: MedicineReference Module Medicine