
Abstract
Metastasis is the main cause of cancer death. Metastatic foci are derived from tumor cells that detach from the primary tumor and then enter the circulation. Circulating tumor cells (CTCs) are generally associated with a high probability of distant metastasis and a negative prognosis. Most CTCs die in the bloodstream, and only a few cells form metastases. Such metastatic CTCs have a stem-like and hybrid epithelial-mesenchymal phenotype, can avoid immune surveillance, and show increased therapy resistance. Targeting metastatic CTCs and their progenitors in primary tumors and their descendants, particularly disseminated tumor cells, represents an attractive strategy for metastasis prevention. However, current therapeutic strategies mainly target the primary tumor and only indirectly affect metastasis-initiating cells. Here, we consider potential methods for preventing metastasis based on targeting molecular and cellular features of metastatic CTCs, including CTC clusters. Also, we emphasize current knowledge gaps in CTC biology that should be addressed to develop highly effective therapeutics and strategies for metastasis suppression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
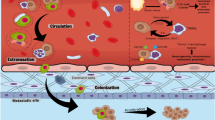
Circulating tumor cells (CTCs) originate from primary tumors. |
Only a small fraction of CTCs may reach distant organs and form metastases. |
Investigation of metastatic CTCs can identify new targets for metastasis-inhibiting therapy. |
1 Introduction
Metastasis is the most fatal feature of cancer, and is orchestrated by tumor cells detaching from the primary tumor and entering the bloodstream [1]. Tumor cells are able to penetrate the blood vessels even at the early stages of cancer [2] and exist in the blood as single cells or clustered together with platelets and leukocytes [3, 4]. A significant proportion of circulating tumor cells (CTCs) die in the bloodstream as a result of external factors [5], and only a small percentage (0.01%) are able to give rise to metastases [6]. Such metastasis-associated/initiating CTCs are characterized by specific features: stemness, hybrid epithelial-mesenchymal transition (EMT) state, escape from immune surveillance, and drug resistance [7]. CTC clusters also show a stem-like phenotype, immune escape, and low sensitivity to therapy, as well as anoikis and shear stress resistance, resulting in an increase of their metastatic potential [8,9,10]. The survival of CTCs in the bloodstream and their metastatic potential is also determined by their interaction with other blood cells: neutrophils, platelets, etc. [11, 12].
Metastasis is a complex process that consists of at least five stages: (1) invasion of tumor cells; (2) intravasation into the surrounding vascular network; (3) circulation and interaction with cells of the immune system; (4) extravasation into the secondary tissue, including adhesion to the vascular endothelium; and (5) growth of micro- and macrometastasis [13]. Indeed, each of the metastatic cascade stages is a critical point and can be targeted to prevent or reduce the risk of metastasis.
The primary tumor is usually a target in anti-cancer treatment [14]. However, even after the successful removal of the primary tumor, there is still a chance of metastasis due to the release of tumor cells into the bloodstream during a surgical operation and/or the presence of disseminated tumor cells (DTCs). Revealing the molecular features of metastasis-initiating cells, particularly metastatic CTCs, would allow the prevention of metastasis through targeting the primary tumor, for example, destroying the source of metastatic CTCs or preventing their release into the blood and killing DTCs. The opportunity to target metastatic CTCs themselves is not also excluded despite their short-term stay in the bloodstream and the constant feeding by the primary tumor.
Thus, the phenotype of metastatic CTCs can act as an indicator for the development of antimetastatic therapy simultaneously aimed at the primary tumor, circulation, and sites of dissemination. This review describes potential methods for the prevention of metastasis based on the molecular and cellular characteristics of metastatic CTCs, including CTC clusters (Fig. 1).

Potential approaches for metastasis prevention based on molecular features of metastatic circulating tumor cells (CTCs)
2 Conventional and Potential Targeted Therapy Against Metastatic Circulating Tumor Cells
Some molecules being suggested as targets for antitumor therapy have been described as potential markers of metastatic CTCs. Their targeting can be considered as one of the methods of preventing metastasis.
The epidermal growth factor receptors EGFR and HER2 are transmembrane glycoproteins that are involved in the regulation of cell growth and proliferation [15, 16]. EGFR- and HER2-targeted drugs are widely used for the treatment of patients with various cancers in clinical practice: lapatinib (EGFR and HER2)—breast cancer; pertuzumab (HER2)—breast cancer; trastuzumab (HER2)—breast and stomach cancers; cetuximab (EGFR)—head and neck and colorectal cancers, etc. [17]. These drugs inhibit tumor growth, reduce the tumor mass, and significantly increase patient survival. For example, overall survival (OS) is greater in HER2-overexpressing advanced breast cancer patients taking lapatinib in combination with capecitabine chemotherapy compared with capecitabine monotherapy (75.0 vs. 64.7 weeks) [18]. The time to death from brain metastases is significantly longer in HER2-positive breast cancer patients treated with trastuzumab than in cases without this therapy (median 14.9 vs. 4.0 months) [19]. OS of patients with HER2-positive stomach cancer is higher with trastuzumab deruxtecan (a conjugate consisting of trastuzumab and a cytotoxic topoisomerase I inhibitor) therapy than with irinotecan or paclitaxel chemotherapy (12.5 vs. 8.4 months) [20]. In patients with unresectable esophageal cancer, OS tends to be longer when treated with cetuximab in combination with radiochemotherapy compared to radiochemotherapy alone (49.1 vs. 24.1 months) [21].
EGFR-positive CTCs are detected in 90% of patients with prostate cancer and bone metastases [22]. The existence and frequency of HER2-positive CTCs correlate with a decrease in overall and relapse-free survival in patients with breast cancer, distant metastases occur in 22.9% of patients, and half of these patients have bone metastases [23, 24]. Trastuzumab and/or pertuzumab/lapatinib in combination with or without docetaxel, eribulin, and paclitaxel chemotherapy leads to the complete disappearance of HER2-positive CTCs and an increase in progression-free survival (PFS) (14.5 months) and OS (42.7 months) in patients with breast cancer compared to patients who do not receive anti-HER2 therapy (10.6 months and 26.8 months, respectively) [25]. PFS is also significantly higher (8.8 months) in breast cancer patients with HER2-positive CTCs treated with a combination of trastuzumab and lapatinib than in patients without this therapy (1.5 months) [26]. Interestingly, cetuximab had no significant effect on PFS and OS in colorectal cancer patients with EGFR-positive CTCs compared to cases with EGFR-negative CTCs [27].
The estrogen receptors (ERs) are proteins located in the cell nucleus (ERα and ERβ) and membrane (mERs), and regulate proliferation, migration, and survival of cells [28]. In prostate and breast cancer, ERs also affect the migration and invasion of tumor cells [29, 30]. Targeting ERs is one of the main strategies in breast cancer treatment [31]. Adjuvant tamoxifen reduces the incidence of breast cancer-related events and distant recurrence [32]. Nevertheless, the effect of endocrine treatment on CTCs is challenging due to the high probability of shifting ER positivity to negativity in CTCs compared to the primary tumor. As a result, ER expression is observed in CTCs of only 19% of patients with breast cancer [33]. Metastatic breast cancer patients with ER-positive tumors often lack ERs in CTCs, which may indicate a mechanism to escape endocrine therapy and lead to distant metastasis [34, 35]. The retention of ER positivity in CTCs after systemic therapy is tended to be related to better PFS and can reflect a favorable phenotype that still responds to therapy [35].
In breast cancer, metastatic CTCs also demonstrate expression of heparanase (HPSE) and Notch homolog 1 (Notch1) [36], mucin 16 (MUC16), and transmembrane protease serine 4 (TMPRSS4) [37], as well as neuroplastin (NPTN) and S100 calcium-binding proteins A4 and A9 (S100A4, S100A9) [38]. In hepatocellular carcinoma, epithelial cell adhesion molecule (EpCAM)-positive CTCs are associated with short disease-free survival (DFS) and OS and worse prognosis [39]. In pancreatic cancer, survivin (baculoviral inhibitor of apoptosis repeat-containing 5 or BIRC5) is one of the highest upregulated genes in CTCs during metastatic spread [40]. Targeting these molecules might be another promising strategy in anticancer therapy.
HPSE is an enzyme that cleaves heparin sulfate leading to extracellular matrix remodeling with the release of growth factors and cytokines and the induction of angiogenesis, immune cell migration, inflammation, wound healing, and metastasis [41]. A number of HPSE inhibitors (PI-88, PG545, SST0001, M402, etc.) showed tumor- and metastasis-suppressor activities in different types of cancer in vivo [42,43,44,45]. PI-88 (phosphomannopentaose sulfate) reduced both tumor volume and intrahepatic metastasis of hepatocellular carcinoma [46]. PG545 (heparan sulfate mimetic) inhibited angiogenesis and demonstrated anti-tumor and anti-metastatic effects in murine models of breast, prostate, liver, lung, colon, head and neck cancers, and melanoma [43].
TMPRSS4 is overexpressed in various cancers and is involved in tumor progression [47]. IMD-0354 and KRT1853, inhibitors TMPRSS4 and IKKβ (inhibitor of nuclear factor kappa-B kinase subunit beta), were shown to reduce migration, invasion, and proliferation of colon (SW480, HCT15, HCT116, and SW620 lines), prostate (DU145 and PC3), cervix (HeLa), stomach (SNU-638), and liver cancer cells (SNU-398) [48].
S100A4 and S100A9 are involved in the regulation of cell cycle progression and differentiation [49, 50]. Stable knockdown of S100A4 was found to suppress cell migration and metastasis of osteosarcoma [51]. A chimeric antibody (S100A8/A9 signal transfer inhibitor) consisting of mouse Ab45-Fab and human IgG2-FC reduced the mobility of tumor cells and suppressed melanoma metastasis to the lungs [52].
EpCAM is a cell adhesion molecule and is often expressed in CTCs in patients with breast, colon, pancreatic, prostate, and other cancers [53]. High-dose adecatumumab (recombinant human IgG1 monoclonal anti-EpCAM antibody) was shown to reduce the development of new metastases in breast cancer patients with high EpCAM expression [54].
Survivin is a regulator of mitosis and apoptosis. Treatment of a metastatic PDX model of pancreatic cancer by YM155 survivin inhibitor (sepantronium bromide) alone and in combination with nab-paclitaxel and danusertib chemotherapy hindered metastasis and improved survival [40].
3 Targeting CTC Plasticity
Tumor cells demonstrate high plasticity undergoing EMT and acquiring distinct stem phenotypes. EMT is a dynamic and reversible program that results in the loss of epithelial identity and morphology, and acquisition of mesenchymal characteristics and increased drug-resistant, invasive, and metastatic potentials [55, 56]. EMT is not a binary process and tumor cells can be found at any locale on the EMT spectrum, often sharing certain epithelial and mesenchymal characteristics [57]. Stemness is the ability of cells to proliferate in an asymmetric way, allowing maintenance of stem cell identity and generating differentiated cells. EMT and stemness are closely related to each other. Hybrid EMT cells show similarity with stem cells [58] and contribute to metastatic progression of different cancers [59]. As primary tumor cells, CTCs are highly heterogeneous in terms of EMT and stem phenotypes [57, 60,61,62]. CTCs considered to be in a hybrid epithelial/mesenchymal state are more apoptosis-resistant and have higher tumor-initiating potential [63, 64]. CTCs in partial EMT correlate with advanced disease, worse progression-free and overall survival, and earlier recurrence [65]. CTCs with stem properties display enhanced tumorigenicity and resistance to radiation and chemotherapy [66, 67]. Such plasticity provides CTCs with an increased potential to adapt to the different microenvironments encountered during metastatic spread and successful formation of metastases [68]. An increase in plasticity of CTCs, namely dynamic interconversion of diverse subsets of CTCs, is frequently observed upon anticancer therapy. CTC expressing EMT-inducing gene transcripts are enriched in primary breast cancer patients who receive neoadjuvant chemotherapy [69]. Taxane treatment of patients with castration-resistant prostate cancer results in changes of expression of EMT and neuroendocrine markers in CTCs [70]. A dynamic change of mesenchymal CTCs is detected in patients with colorectal and esophageal squamous cell cancers during chemo- and radiation therapy [71, 72].
3.1 Targeting Epithelial-Mesenchymal Transition-Positive CTCs
CTCs with an expression of EMT transcription factors (SLUG, SNAIL, TWIST1, and ZEB1) and associated genes (VIM, TGFβ1, ZEB2, FOXC1, and CXCR4) are related to negative prognosis and low survival in breast cancer patients [38, 73]. Upregulation of Notch activity in CTCs is associated with breast cancer metastasis to the brain [74]. The presence and quantity of N-cadherin-positive CTCs are linked to inferior PFS of patients with renal cell carcinoma [75].
The molecules and signaling pathways involved in the regulation of EMT are considered attractive therapeutic targets for the prevention of metastasis [76]. Several strategies have been proposed to target EMT: suppression, reversion, and terminal (mesenchymal) stage stimulation, or transdifferentiation, each of which is thoroughly discussed in Denisov et al. [77].
EMT suppression allows retaining the epithelial phenotype of tumor cells and thereby avoiding the appearance of the ability to migrate and invade. A wide variety of chemically synthesized and natural agents have been described to have a suppressive effect on EMT and metastasis (Table 1). Nimotuzumab, humanized anti-EGFR monoclonal antibody, inhibits KKU-214 and KKU-213 cholangiocarcinoma metastasis by suppressing SNAIL and vimentin (VIM) [78]. Ginsenoside Rg3, extracted from ginseng, upregulates E-cadherin, reduces the expression of SNAIL, N-cadherin, and VIM, blocks the MAPK and NF-kB signaling pathways, and suppresses lung cancer metastasis [79]. The flavonoid quercetin suppresses the metastatic ability of lung cancer by inhibiting the SNAIL-dependent Akt signaling pathway [80].
EMT reversal (mesenchymal-epithelial transition) means the loss of mesenchymal features in tumor cells and acquisition of the epithelial phenotype. A few EMT reversal drugs have been described. Inositol (cyclohexane polyol) reverses EMT in metastatic MDA-MB-231 breast cancer cell line by inhibiting EMT-inducing proteins (PI3K/Akt, NF-kB, COX-2, and SNAIL) and suppresses cell motility and invasiveness [89]. Genistein, one of the active ingredients of soy isoflavones, inhibits the HT-29 colon cancer cell migration by reversing EMT and increasing the E-cadherin and decreasing SNAIL/SLUG, ZEB1, ZEB2, FOXC1, FOXC2, TWIST1, and N-cadherin expression [90]. Liposomal delivery of organic disulfide and a glutathione derivative—glutathione disulfide (GSSG)—reverses EMT via downregulation of N-cadherin, β-catenin, and STAT3 and prevents lung metastasis in a murine melanoma model [91].
Stimulation of EMT to the terminal mesenchymal stage suggests a complete loss of epithelial characteristics and differentiation of tumor cells into other cells. For example, the antidiabetic drug rosiglitazone and MEK (mitogen-activated protein kinase) inhibitors, PD98059 and trametinib, induce the differentiation of EMT-undergoing breast cancer cells into adipocytes and reduce metastasis in vivo [92].
3.2 Targeting Stem-Like CTCs
Several markers have been described to identify tumor cells with the stem phenotype: cell adhesion molecule CD44, CD133 (prominin-1), aldehyde dehydrogenase (ALDH), etc. Some of these molecules are considered potential targets for therapeutic actions on CSCs. At this time, many agents have been identified to inhibit cancer cell stemness [93]. For example, withaferin A (a bioactive compound isolated from the plant Withania somnifera) in combination with cisplatin results in a 70–80% reduction in tumor growth and complete inhibition of metastasis in an orthotopic model of ovarian cancer [94]. P245 monoclonal antibody inhibits breast cancer growth and prevents relapse through targeting CD44+ CSCs in vivo [95]. These drugs may also be valid for targeting CTCs, especially metastasis-associated populations, which often show stem features. For example, the expression of CD44 in CTCs has been found to be related to the high probability of gastric cancer metastasis and relapse [96]. The number of metastatic foci positively correlates with the amount of EpCAM+CD44+CD47+MET+ CTCs [97]. Metastasis of colorectal cancer to the liver is associated with CD133+CD54+CD44+ CTCs [98]. Frequently, stem-like CTCs show the expression of mesenchymal markers that indicate a close relationship between stemness and EMT and the necessity for the development of combined therapy [99, 100].
4 Disaggregation of CTC Clusters
Most single CTCs die in the bloodstream during anoikis (apoptosis due to the loss of cell-matrix interactions), immune attack, and other causes. The probability of CTC survival increases if they interact with each other, forming clusters and with immune cells and platelets. These clusters usually consist of two to 50 tumor cells and account for 5–20% of the total amount of CTCs depending on tumor stage [101,102,103,104]. Compared to single CTCs, circulating clusters have a 50-fold higher metastatic potential [105] and, as shown in mouse models, are responsible for the formation of 50–97% of metastases [106]. Nevertheless, CTC clusters are more frequently detected in non-metastatic than in metastatic breast cancer patients, suggesting that their dissemination is an early event [107].
Tumor cells in the circulating clusters have increased adhesion to the vascular endothelium that can be one of the reasons for their high metastatic potential [11]. Although traditional antitumor treatment focuses on the inhibition of tumor cell proliferation and induction of apoptosis, blocking the attachment of CTCs to the vessel walls may be another potential therapeutic strategy. For example, the antitumor drug temozolomide (TMZ) significantly weakens the adhesion of single-U87 cells (acted as CTCs) on the HUVEC cell (acting as endothelial cells) layer through the induction of DNA damage and apoptosis in tumor cells [108].
Targeting CTC clusters usually consists in their disaggregation and, consequently, the loss of metastatic capacity (Table 2). The size of CTC clusters can be reduced by at least 39 compounds, including inhibitors of Na+/K+ ATPase, histonedeacetylase (HDAC), nucleotide biosynthesis, kinases, and tubulin, as well as DNA-binding compounds, antibiotics, cardiac glycosides, etc. [109].
Cell adhesion molecules such as plakoglobin and keratin 14 (K14) are involved in the formation of CTC clusters and maintaining high metastatic potential [105]. Accordingly, their targeting may be another potential strategy for the disaggregation of CTC clusters. In particular, shRNA-mediated plakoglobin suppression was found to trigger disruption of cell-cell contacts in breast cancer lines. Placoglobin knockdown led to a decrease of CTC cluster formation and an 80% reduction of lung metastasis of breast cancer in vivo [105]. K14 knockdown resulted in a sevenfold reduction in the mean number of breast cancer metastases in a mouse model [106]. Another cell adhesion protein, CD44, is regulated by EGFR and drives the aggregation of triple-negative breast cancer cells. EGFR inhibition effectively blocks cell aggregation in vitro and reduces lung metastasis in vivo [110].
5 Targeting Neutrophils in CTC Clusters
Neutrophils are the most common type of leukocytes (50–70%), and as such a heterogeneous cell population [111]. In cancer, these cells play either an anti-tumor (N1) or a pro-tumor role (N2) [112]. N2 neutrophils contribute to CTC survival in the bloodstream and their subsequent extravasation. The total number of neutrophils correlates with the probability of CTC-neutrophil cluster formation. In patients with breast cancer, the frequency of CTC-leukocyte clusters reaches 3.4%, including 86–92% CTC-neutrophil clusters [11].
Neutrophils can induce an immunosuppressive environment by disrupting the CD8+ T cell activation [113] or preventing the activity of natural killer cells (NKs) during the circulation and extravasation of tumor cells [114]. The interaction of neutrophils with endothelium contributes to tumor cell adhesion to the hepatic sinusoids and subsequent extravasation [115]. Inflammatory factors released by the tumor stimulate neutrophils to form chromatin networks termed neutrophil extracellular traps (NET). NETs can trap CTCs and enhance metastasis by facilitating cancer cell adhesion, migration, invasion, and proliferation during the establishment of metastatic foci [116, 117].
There are a few agents that can target neutrophils and abrogate their interaction with CTCs (Table 2). For example, DNase I resolves NETs and reduces the number of breast cancer metastases in the lungs and metastases of lung cancer in the liver in vivo [117, 118]. Inhibition of the enzyme peptidylarginine deaminase, which is essential for the NET formation, abrogates the progression of metastatic colorectal cancer in vivo [116]. The decrease in CTC-neutrophil clusters may be another strategy to suppress metastasis. Neutralizing antibodies against Ly-6G (lymphocyte antigen 6 complex locus G6D) destroy CTC-neutrophil clusters and delay their shedding in a mouse model of breast cancer [11]. This study also shows that vascular cell adhesion molecule 1 (VCAM1) functionally mediates the interaction between CTCs and neutrophils, and its knockout blocks the appearance of CTC-neutrophil clusters [11]. Moreover, suppression of VCAM1 by resveratrol (phytopolyphenol found in grapes and other fruits and medicinal plants) remarkably inhibits metastatic growth of melanoma cells in vivo [119].
6 Targeting Platelets in CTC Clusters
Platelets produce various molecules that contribute to the inflammation, progression, and metastasis of cancer [120, 121]. When tumor cells leave the primary tumor and enter the bloodstream, they firstly meet with platelets [122]. Cancer-related thrombosis is one of the causes of death among cancer patients. Tumor cells aggregate with platelets by activating the coagulation cascade [123]. Platelets facilitate CTC retention in the bloodstream and promote their extravasation [124, 125]. Tumor cells and platelets generate a positive feedback loop: tumor cells secrete adenosine diphosphate and activate platelets, which in turn secrete factors promoting tumor cell proliferation [12]. Platelets also produce TGFβ and induce EMT in CTCs [126].
Antithrombotic drugs, such as aspirin, warfarin, etc. (Table 2), improve the survival of patients with different cancers [127,128,129]. The primary target of antithrombotic therapy is P-selectin, which is necessary for the effective formation of platelet-CTC aggregates and is one of the key factors of cancer metastasis [130]. P-selectin knockout reduces aggregations between platelets and tumor cells and colon cancer metastasis in a mouse model [131, 132]. The antithrombotic drug ticagrelor blocks the activation of platelet P2Y12 receptors and is widely used in the treatment of patients with cardiovascular diseases [133]. Patients with metastatic breast and colorectal cancers show an almost twofold decrease in spontaneous platelet aggregation and activation after taking ticagrelor [134].
Unfortunately, anticoagulants lead to an excessive decrease in platelet function. Considering the fact that the production of new platelets takes at least 7–10 days, patients who receive antithrombotic drugs have an increased risk of bleeding. Therefore, it would be optimal to maintain the properties of platelets, and at the same time avoid prothrombotic effects. The use of platelet decoys is considered the most promising therapeutic approach aimed to reduce platelet aggregation and, consequently, their aggregation with CTCs [135] (Table 2). Platelet decoys have a similar round shape but a smaller size and reduced granularity with decreased membrane adhesion receptors compared to normal platelets. They are not activated or aggregated with other cells during physiological conditions. The addition of platelet decoys to intact platelets at a ratio of 1:5 reduces their aggregation functions. The antiplatelet activity of these decoys is similar to that of the clinically approved antithrombotic drugs aspirin and abciximab. It is important that decoys do not disrupt the thrombin activation pathway and normal hemostasis, as well as do not damage the liver and spleen tissues [136]. Another potential strategy to block tumor-specific platelet functions can be use tumor microenvironment-responsive nitric oxide release nanoparticles (Ptx@AlbSNO), which suppress EMT, prevent platelet adhesion around CTCs, and reduce distant metastasis in vivo [137].
Platelets protect CTCs from immune surveillance, for example, by hiding them from natural killer (NK) cells [138]. Particularly, MHC class I molecules are transmitted by platelets to tumor cells leading to a decrease in reactivity and cytotoxicity of NK cells [139]. Platelets also induce the release of soluble NKG2D ligands from CTCs, such as MICA and MICB, which suppress NK cell degranulation and inflammatory cytokine production [140]. The increased content of soluble MICA and MICB in the blood serum in cancer patients is closely associated with metastasis [141, 142]. Targeting NKG2D ligands may be a promising strategy in preventing metastasis. For example, antibodies targeting the MICA α3 domain (7C6, 6F11, and 1C2) prevent MICA and MICB production and inhibit lung metastasis of B16F10 melanoma and CT26 colon cancer in the humanized mouse model through NKG2D- and CD16-related activation of NK cells [143]. An additional approach for the CTC destruction and reducing metastasis, especially if platelet protection is removed, can be an adoptive transfer of highly activated NK cells to cancer patients [144] and increase in their cytotoxic activity, for example through the injection of TLR4-agonist (glucopyranosyl lipid-A), oncolytic viruses, or suppression of CD96, a negative regulator of NK cells [145,146,147,148].
7 Other Methods for CTC Eradication
CTCs in the bloodstream can be also neutralized by different vascular devices, which are widely discussed in other reviews [7, 149, 150]. In recent years, other methods were developed to eradicate CTCs. Minimally invasive therapeutic intravenous catheters with anti-EpCAM antibodies and interiors filled with black phosphorus nanosheets kill CTCs by photothermal effect [151]. Curcumin loaded poly(lactic-co-glycolic acid) nanoparticles and laser irradiation (λ = 447 nm, P = 100 mW) result in photodynamic inactivation of CTCs [152]. Electrostatic charge stimulation (PPECS) of the whole blood selectively deactivates the invasive function of CTCs and suppresses pulmonary metastasis of 4T1 breast cancer cells and extended survival in an in vivo model [153]. Combining bionic NSK (nanosponges and nanokillers—platelet-neutrophil hybrid membrane (PNM)-camouflaging gold nanocages (AuNCs)) with chemo-photothermal therapy is effective for actively clearing CTCs, neutralizing migrating tumor-derived exosomes, activating the innate immune system, and inhibiting breast cancer metastasis in 4T1 xenograft and orthotopic breast tumor-bearing mice [154].
8 Challenges and Perspectives
Metastasis is the biggest threat to cancer patient survival. However, current therapeutic strategies mainly target the primary tumor, assuming that metastatic seeds will also be killed. Indeed, several drugs, for example EGFR tyrosine kinase inhibitors and anti-HER2 monoclonal antibodies, can suppress metastasis and improve the survival of cancer patients [19, 21]. Nevertheless, this is rather an exception than the rule; most anticancer therapies only indirectly affect metastasis-initiating cells. In addition, these cells, including metastatic CTCs, can be resistant to drug therapy a priori due to stem-like features, partial EMT, and the ability to escape from immune control [7]. Therefore, the identification of metastatic CTCs and revealing their molecular features may be effective in developing therapeutic approaches for targeting metastasis-initiating cells in both the primary tumor and circulation and disseminated sites.
Despite the abundance of the studies reviewed here, there are almost no data that would describe the comprehensive makeup of metastatic CTCs in different cancers and their potential targets. As a rule, most researches are limited by the description of CTC composition using a panel of specific membrane markers and the identification of cell phenotypes that are associated with cancer metastasis. Only recent studies have focused on the deep molecular analysis of CTCs, revealing the mechanisms of their survival and the colonization of distant organs, and the discovery of druggable targets. For example, CTC heterogeneity and metastatic phenotype are successfully deciphered by single-cell sequencing-based technologies [62, 158, 159]. Metastasis is significantly inhibited by targeting molecules that are critical for CTC transmigration and adaptation in the alien microenvironment of other organs [160]. The progress in understanding CTC biology is also challenged by their heterogeneity [30, 57, 60, 62, 159] and the absence of common methods to isolate all subsets of these cells for further molecular, in vitro, and in vivo analyses.
Approaches for metastasis prevention based on the molecular features of metastatic CTCs are mainly experimental and should be validated in future clinical studies. Some preventive strategies, particularly aimed at platelet disaggregation in CTCs, can be highly toxic, and alternative drugs need to be developed. Most methods for metastasis suppression are still not proposed due to gaps in the current knowledge of metastasis-initiating cells. In particular, little information is available regarding the phylogenetic paths of CTCs from their origin in the primary tumor to transformation into metastases. It is unclear how diverse CTCs are in clonal composition and how much genetic diversity correlates to phenotypic heterogeneity in CTCs. The question remains as to whether molecular features of metastatic CTCs are kept in primary tumor cells and presented in DTCs and their targeting could prevent the entrance of tumor cells into circulation and the formation of micrometastases. There are almost no results regarding how much CTC phenotype changes in the bloodstream. Studies have still not been able to reveal the relationships between single CTCs and circulating clusters and their common or distinct role in metastasis, especially in the same patients. In addition, complete phenotypic and functional information of CTCs is missing, although there are several approaches for single-cell analysis of proteins by mass spectrometry [161,162,163]. Thus, further studies should focus on the comprehensive multi-omic analysis of CTCs, including the identification of metastatic seeds and their molecular targets to develop new methods for metastasis prevention.
9 Conclusion
Prevention of cancer metastasis is a high priority in oncology. Detection of metastatic CTCs in different cancers and understanding their molecular profile could be an effective tool for identifying new targets and developing therapies targeting both these cells and their progenitors in primary tumors and descendants (disseminated tumor cells) that have been exposed in distant sites. Several specific markers are already known for metastatic CTCs, and their targeting reduces the metastasis probability in vivo. However, these therapeutic strategies have not been validated in clinical studies and many potential targets found in metastatic CTCs are not evaluated in terms of metastasis suppression. Moreover, metastatic CTCs are not described for the majority of cancers at all. So, further studies are needed to clarify these issues.
References
Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298–306. https://doi.org/10.1038/nature17038.
Jin XR, Zhu LY, Qian K, Feng YG, Zhou JH, Wang RW, et al. Circulating tumor cells in early stage lung adenocarcinoma: a case series report and literature review. Oncotarget. 2017;8(14):23130–41. https://doi.org/10.18632/oncotarget.15506.
Jiang X, Wong KHK, Khankhel AH, Zeinali M, Reategui E, Phillips MJ, et al. Microfluidic isolation of platelet-covered circulating tumor cells. Lab Chip. 2017;17(20):3498–503. https://doi.org/10.1039/c7lc00654c.
Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Methods. 2015;12(7):685–91. https://doi.org/10.1038/nmeth.3404.
Nolan J, Nedosekin DA, Galanzha EI, Zharov VP. Detection of apoptotic circulating tumor cells using in vivo fluorescence flow cytometry. Cytometry A. 2019;95(6):664–71. https://doi.org/10.1002/cyto.a.23642.
Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153(3):865–73. https://doi.org/10.1016/s0002-9440(10)65628-3.
Menyailo ME, Tretyakova MS, Denisov EV. Heterogeneity of circulating tumor cells in breast cancer: identifying metastatic seeds. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21051696.
Rossi T, Gallerani G, Angeli D, Cocchi C, Bandini E, Fici P, et al. Single-cell NGS-based analysis of copy number alterations reveals new insights in circulating tumor cells persistence in early-stage breast cancer. Cancers (Basel). 2020. https://doi.org/10.3390/cancers12092490.
Agnoletto C, Corrà F, Minotti L, Baldassari F, Crudele F, Cook WJJ, et al. Heterogeneity in circulating tumor cells: the relevance of the stem-cell subset. Cancers (Basel). 2019. https://doi.org/10.3390/cancers11040483.
Lowes LE, Allan AL. Circulating tumor cells and implications of the epithelial-to-mesenchymal transition. Adv Clin Chem. 2018;83:121–81. https://doi.org/10.1016/bs.acc.2017.10.004.
Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566(7745):553–7. https://doi.org/10.1038/s41586-019-0915-y.
Ward Y, Lake R, Faraji F, Sperger J, Martin P, Gilliard C, et al. Platelets promote metastasis via binding tumor CD97 leading to bidirectional signaling that coordinates transendothelial migration. Cell Rep. 2018;23(3):808–22. https://doi.org/10.1016/j.celrep.2018.03.092.
Hapach LA, Mosier JA, Wang W, Reinhart-King CA. Engineered models to parse apart the metastatic cascade. Npj Precis Oncol. 2019;3(1):20. https://doi.org/10.1038/s41698-019-0092-3.
Hansen E, Read AF. Cancer therapy: attempt cure or manage drug resistance? Evol Appl. 2020;13(7):1660–72.
Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–75. https://doi.org/10.1016/j.jconrel.2010.04.009.
Bethune G, Bethune D, Ridgway N, Xu Z. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2010;2(1):48–51.
Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov. 2010;9(6):427–8. https://doi.org/10.1038/nrd3186.
Cameron D, Casey M, Oliva C, Newstat B, Imwalle B, Geyer CE. Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: final survival analysis of a phase III randomized trial. Oncologist. 2010;15(9):924–34. https://doi.org/10.1634/theoncologist.2009-0181.
Park YH, Park MJ, Ji SH, Yi SY, Lim DH, Nam DH, et al. Trastuzumab treatment improves brain metastasis outcomes through control and durable prolongation of systemic extracranial disease in HER2-overexpressing breast cancer patients. Br J Cancer. 2009;100(6):894–900. https://doi.org/10.1038/sj.bjc.6604941.
Shitara K, Bang YJ, Iwasa S, Sugimoto N, Ryu MH, Sakai D, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med. 2020;382(25):2419–30. https://doi.org/10.1056/NEJMoa2004413.
Rades D, Bartscht T, Hunold P, Schmidberger H, König L, Debus J, et al. Radiochemotherapy with or without cetuximab for unresectable esophageal cancer: final results of a randomized phase 2 trial (LEOPARD-2). Strahlenther Onkol. 2020;196(9):795–804. https://doi.org/10.1007/s00066-020-01646-4.
Day KC, Lorenzatti Hiles G, Kozminsky M, Dawsey SJ, Paul A, Broses LJ, et al. HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Res. 2017;77(1):74–85. https://doi.org/10.1158/0008-5472.can-16-1656.
Wülfing P, Borchard J, Buerger H, Heidl S, Zänker KS, Kiesel L, et al. HER2-positive circulating tumor cells indicate poor clinical outcome in stage I to III breast cancer patients. Clin Cancer Res. 2006;12(6):1715–20. https://doi.org/10.1158/1078-0432.ccr-05-2087.
Apostolaki S, Perraki M, Pallis A, Bozionelou V, Agelaki S, Kanellou P, et al. Circulating HER2 mRNA-positive cells in the peripheral blood of patients with stage I and II breast cancer after the administration of adjuvant chemotherapy: evaluation of their clinical relevance. Ann Oncol. 2007;18(5):851–8. https://doi.org/10.1093/annonc/mdl502.
Deutsch TM, Riethdorf S, Fremd C, Feisst M, Nees J, Fischer C, et al. HER2-targeted therapy influences CTC status in metastatic breast cancer. Breast Cancer Res Treat. 2020;182(1):127–36. https://doi.org/10.1007/s10549-020-05687-2.
Liu Y, Liu Q, Wang T, Bian L, Zhang S, Hu H, et al. Circulating tumor cells in HER2-positive metastatic breast cancer patients: a valuable prognostic and predictive biomarker. BMC Cancer. 2013;13:202. https://doi.org/10.1186/1471-2407-13-202.
Kuboki Y, Matsusaka S, Minowa S, Shibata H, Suenaga M, Shinozaki E, et al. Circulating tumor cell (CTC) count and epithelial growth factor receptor expression on CTCs as biomarkers for cetuximab efficacy in advanced colorectal cancer. Anticancer Res. 2013;33(9):3905–10.
Saczko J, Michel O, Chwiłkowska A, Sawicka E, Mączyńska J, Kulbacka J. Estrogen receptors in cell membranes: regulation and signaling. Adv Anat Embryol Cell Biol. 2017;227:93–105. https://doi.org/10.1007/978-3-319-56895-9_6.
Lombardi APG, Vicente CM, Porto CS. Estrogen receptors promote migration, invasion and colony formation of the androgen-independent prostate cancer cells PC-3 through β-catenin pathway. Front Endocrinol (Lausanne). 2020;11:184. https://doi.org/10.3389/fendo.2020.00184.
Yan S, Dey P, Ziegler Y, Jiao X, Kim SH, Katzenellenbogen JA, et al. Contrasting activities of estrogen receptor beta isoforms in triple negative breast cancer. Breast Cancer Res Treat. 2021;185(2):281–92. https://doi.org/10.1007/s10549-020-05948-0.
Ahmad I. Tamoxifen a pioneering drug: an update on the therapeutic potential of tamoxifen derivatives. Eur J Med Chem. 2018;143:515–31. https://doi.org/10.1016/j.ejmech.2017.11.056.
Ekholm M, Bendahl PO, Fernö M, Nordenskjöld B, Stål O, Rydén L, et al. Effects of adjuvant tamoxifen over three decades on breast cancer-free and distant recurrence-free interval among premenopausal women with oestrogen receptor-positive breast cancer randomised in the Swedish SBII:2pre trial. Eur J Cancer. 2019;110:53–61. https://doi.org/10.1016/j.ejca.2018.12.034.
Aktas B, Müller V, Tewes M, Zeitz J, Kasimir-Bauer S, Loehberg CR, et al. Comparison of estrogen and progesterone receptor status of circulating tumor cells and the primary tumor in metastatic breast cancer patients. Gynecol Oncol. 2011;122(2):356–60. https://doi.org/10.1016/j.ygyno.2011.04.039.
Babayan A, Hannemann J, Spötter J, Müller V, Pantel K, Joosse SA. Heterogeneity of estrogen receptor expression in circulating tumor cells from metastatic breast cancer patients. PLoS ONE. 2013;8(9): e75038. https://doi.org/10.1371/journal.pone.0075038.
Forsare C, Bendahl PO, Moberg E, Jørgensen CLT, Jansson S, Larsson AM, et al. Evolution of estrogen receptor status from primary tumors to metastasis and serially collected circulating tumor cells. Int J Mol Sci. 2020;21(8):2885. https://doi.org/10.3390/ijms21082885.
Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5(180):180ra48. https://doi.org/10.1126/scitranslmed.3005109.
Kwan TT, Bardia A, Spring LM, Giobbie-Hurder A, Kalinich M, Dubash T, et al. A digital RNA signature of circulating tumor cells predicting early therapeutic response in localized and metastatic breast cancer. Cancer Discov. 2018;8(10):1286–99. https://doi.org/10.1158/2159-8290.cd-18-0432.
Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE. 2012;7(5): e33788. https://doi.org/10.1371/journal.pone.0033788.
Yu J-j, Xiao W, Dong S-l, Liang H-f, Zhang Z-w, Zhang B-x, et al. Effect of surgical liver resection on circulating tumor cells in patients with hepatocellular carcinoma. BMC Cancer. 2018;18(1):835. https://doi.org/10.1186/s12885-018-4744-4.
Dimitrov-Markov S, Perales-Patón J, Bockorny B, Dopazo A, Muñoz M, Baños N, et al. Discovery of new targets to control metastasis in pancreatic cancer by single-cell transcriptomics analysis of circulating tumor cells. Mol Cancer Ther. 2020;19(8):1751–60. https://doi.org/10.1158/1535-7163.mct-19-1166.
Jayatilleke KM, Hulett MD. Heparanase and the hallmarks of cancer. J Transl Med. 2020;18(1):453. https://doi.org/10.1186/s12967-020-02624-1.
Ferro V, Dredge K, Liu L, Hammond E, Bytheway I, Li C, et al. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Semin Thromb Hemost. 2007;33(5):557–68. https://doi.org/10.1055/s-2007-982088.
Dredge K, Hammond E, Handley P, Gonda TJ, Smith MT, Vincent C, et al. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br J Cancer. 2011;104(4):635–42. https://doi.org/10.1038/bjc.2011.11.
Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17(6):1382–93. https://doi.org/10.1158/1078-0432.ccr-10-2476.
Zhou H, Roy S, Cochran E, Zouaoui R, Chu CL, Duffner J, et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE. 2011;6(6): e21106. https://doi.org/10.1371/journal.pone.0021106.
Liao BY, Wang Z, Hu J, Liu WF, Shen ZZ, Zhang X, et al. PI-88 inhibits postoperative recurrence of hepatocellular carcinoma via disrupting the surge of heparanase after liver resection. Tumour Biol. 2016;37(3):2987–98. https://doi.org/10.1007/s13277-015-4085-8.
Li XM, Liu WL, Chen X, Wang YW, Shi DB, Zhang H, et al. Overexpression of TMPRSS4 promotes tumor proliferation and aggressiveness in breast cancer. Int J Mol Med. 2017;39(4):927–35. https://doi.org/10.3892/ijmm.2017.2893.
Kim S, Ko D, Lee Y, Jang S, Lee Y, Lee IY, et al. Anti-cancer activity of the novel 2-hydroxydiarylamide derivatives IMD-0354 and KRT1853 through suppression of cancer cell invasion, proliferation, and survival mediated by TMPRSS4. Sci Rep. 2019;9(1):10003. https://doi.org/10.1038/s41598-019-46447-7.
Kerkhoff C, Ghavami S (2011) S100A9 (S100 calcium binding protein A9). Atlas Genet Cytogenet Oncol Haematol. 15(9):746–57. https://doi.org/10.4267/2042/46028
Sherbet GV (2010) S100A4 (S100 Calcium Binding Protein A4). Atlas Genet Cytogenet Oncol Haematol. 14(10):877–86. https://doi.org/10.4267/2042/46036
Fujiwara M, Kashima TG, Kunita A, Kii I, Komura D, Grigoriadis AE, et al. Stable knockdown of S100A4 suppresses cell migration and metastasis of osteosarcoma. Tumour Biol. 2011;32(3):611–22. https://doi.org/10.1007/s13277-011-0160-y.
Kinoshita R, Sato H, Yamauchi A, Takahashi Y, Inoue Y, Sumardika IW, et al. Newly developed anti-S100A8/A9 monoclonal antibody efficiently prevents lung tropic cancer metastasis. Int J Cancer. 2019;145(2):569–75. https://doi.org/10.1002/ijc.31982.
Gires O, Pan M, Schinke H, Canis M, Baeuerle PA. Expression and function of epithelial cell adhesion molecule EpCAM: where are we after 40 years? Cancer Metastasis Rev. 2020;39(3):969–87. https://doi.org/10.1007/s10555-020-09898-3.
Schmidt M, Scheulen ME, Dittrich C, Obrist P, Marschner N, Dirix L, et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann Oncol. 2010;21(2):275–82. https://doi.org/10.1093/annonc/mdp314.
Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ, et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther. 2019;194:161–84. https://doi.org/10.1016/j.pharmthera.2018.09.007.
Bocci F, Mandal S, Tejaswi T, Jolly MK. Investigating epithelial-mesenchymal heterogeneity of tumors and circulating tumor cells with transcriptomic analysis and biophysical modeling. Comput Syst Oncol. 2021;1(2): e1015. https://doi.org/10.1002/cso2.1015.
Tashireva LA, Savelieva OE, Grigoryeva ES, Nikitin YV, Denisov EV, Vtorushin SV, et al. Heterogeneous manifestations of epithelial-mesenchymal plasticity of circulating tumor cells in breast cancer patients. Int J Mol Sci. 2021;22(5):2504.
Forte E, Chimenti I, Rosa P, Angelini F, Pagano F, Calogero A, et al. EMT/MET at the crossroad of stemness, regeneration and oncogenesis: the ying-yang equilibrium recapitulated in cell spheroids. Cancers. 2017;9(8):98. https://doi.org/10.3390/cancers9080098.
Simeonov KP, Byrns CN, Clark ML, Norgard RJ, Martin B, Stanger BZ, et al. Single-cell lineage and transcriptome reconstruction of metastatic cancer reveals selection of aggressive hybrid EMT states. Cancer Cell. 2021. https://doi.org/10.1016/j.ccell.2021.05.005.
Savelieva OE, Tashireva LA, Kaigorodova EV, Buzenkova AV, Mukhamedzhanov RK, Grigoryeva ES, et al. Heterogeneity of stemlike circulating tumor cells in invasive breast cancer. Int J Mol Sci. 2020;21(8):2780. https://doi.org/10.3390/ijms21082780.
Sun YF, Guo W, Xu Y, Shi YH, Gong ZJ, Ji Y, et al. Circulating tumor cells from different vascular sites exhibit spatial heterogeneity in epithelial and mesenchymal composition and distinct clinical significance in hepatocellular carcinoma. Clin Cancer Res. 2018;24(3):547–59. https://doi.org/10.1158/1078-0432.CCR-17-1063.
Brechbuhl HM, Vinod-Paul K, Gillen AE, Kopin EG, Gibney K, Elias AD, et al. Analysis of circulating breast cancer cell heterogeneity and interactions with peripheral blood mononuclear cells. Mol Carcinog. 2020;59(10):1129–39.
Polioudaki H, Agelaki S, Chiotaki R, Politaki E, Mavroudis D, Matikas A, et al. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer. 2015;15(1):1–10. https://doi.org/10.1186/s12885-015-1386-7.
Ou H, Huang Y, Xiang L, Chen Z, Fang Y, Lin Y, et al. Circulating tumor cell phenotype indicates poor survival and recurrence after surgery for hepatocellular carcinoma. Dig Dis Sci. 2018;63(9):2373–80. https://doi.org/10.1007/s10620-018-5124-2.
Semaan A, Bernard V, Kim DU, Lee JJ, Huang J, Kamyabi N, et al. Characterisation of circulating tumour cell phenotypes identifies a partial-EMT sub-population for clinical stratification of pancreatic cancer. Br J Cancer. 2021;124(12):1970–7. https://doi.org/10.1038/s41416-021-01350-9.
Nadal R, Ortega FG, Salido M, Lorente JA, Rodríguez-Rivera M, Delgado-Rodríguez M, et al. CD133 expression in circulating tumor cells from breast cancer patients: potential role in resistance to chemotherapy. Int J Cancer. 2013;133(10):2398–407. https://doi.org/10.1002/ijc.28263.
Papadaki MA, Stoupis G, Theodoropoulos PA, Mavroudis D, Georgoulias V, Agelaki S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol Cancer Ther. 2019;18(2):437–47. https://doi.org/10.1158/1535-7163.MCT-18-0584.
Francart ME, Lambert J, Vanwynsberghe AM, Thompson EW, Bourcy M, Polette M, et al. Epithelial–mesenchymal plasticity and circulating tumor cells: travel companions to metastases. Dev Dyn. 2018;247(3):432–50. https://doi.org/10.1002/dvdy.24506.
Mego M, Mani SA, Lee BN, Li C, Evans KW, Cohen EN, et al. Expression of epithelial–mesenchymal transition-inducing transcription factors in primary breast cancer: the effect of neoadjuvant therapy. Int J Cancer. 2012;130(4):808–16. https://doi.org/10.1002/ijc.26037.
Jiménez N, Reig Ò, Montalbo R, Milà-Guasch M, Nadal-Dieste L, Castellano G, et al. Cell plasticity-related phenotypes and taxanes resistance in castration-resistant prostate cancer. Front Oncol. 2020;10: 594023. https://doi.org/10.3389/fonc.2020.594023.
Shi DD, Yang CG, Han S, Wang SY, Xiong B. Dynamic evaluation of mesenchymal circulating tumor cells in patients with colorectal cancer: clinical associations and prognostic value. Oncol Rep. 2020;44(2):757–67. https://doi.org/10.3892/or.2020.7629.72.
Ujiie D, Matsumoto T, Endo E, Okayama H, Fujita S, Kanke Y, et al. Circulating tumor cells after neoadjuvant chemotherapy are related with recurrence in esophageal squamous cell carcinoma. Esophagus. 2021;18(3):566–73. https://doi.org/10.1007/s10388-021-00829-x.73.
Mego M, Karaba M, Minarik G, Benca J, Silvia J, Sedlackova T, et al. Circulating tumor cells with epithelial-to-mesenchymal transition phenotypes associated with inferior outcomes in primary breast cancer. Anticancer Res. 2019;39(4):1829–37. https://doi.org/10.21873/anticanres.13290.
Boral D, Vishnoi M, Liu HN, Yin W, Sprouse ML, Scamardo A, et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat Commun. 2017;8(1):196. https://doi.org/10.1038/s41467-017-00196-1.
Nel I, Gauler TC, Bublitz K, Lazaridis L, Goergens A, Giebel B, et al. Circulating tumor cell composition in renal cell carcinoma. PLoS ONE. 2016;11(4): e0153018. https://doi.org/10.1371/journal.pone.0153018.
Fontebasso Y, Dubinett SM. Drug development for metastasis prevention. Crit Rev Oncog. 2015;20(5–6):449–73. https://doi.org/10.1615/CritRevOncog.v20.i5-6.150.
Denisov EV, Jolly MK, Shubin VP, Tsukanov AS, Cherdyntseva NV. Critical steps in epithelial-mesenchymal transition as target for cancer treatment. In: Approaching complex diseases. Springer; 2020. p. 213–44.
Padthaisong S, Thanee M, Techasen A, Namwat N, Yongvanit P, Liwatthakun A, et al. Nimotuzumab inhibits cholangiocarcinoma cell metastasis via suppression of the epithelial-mesenchymal transition process. Anticancer Res. 2017;37(7):3591–7. https://doi.org/10.21873/anticanres.11729.
Tian L, Shen D, Li X, Shan X, Wang X, Yan Q, et al. Ginsenoside Rg3 inhibits epithelial-mesenchymal transition (EMT) and invasion of lung cancer by down-regulating FUT4. Oncotarget. 2016;7(2):1619–32. https://doi.org/10.18632/oncotarget.6451.
Chang JH, Lai SL, Chen WS, Hung WY, Chow JM, Hsiao M, et al. Quercetin suppresses the metastatic ability of lung cancer through inhibiting Snail-dependent Akt activation and Snail-independent ADAM9 expression pathways. Biochim Biophys Acta Mol Cell Res. 2017;1864(10):1746–58. https://doi.org/10.1016/j.bbamcr.2017.06.017.
Zhou C, Li J, Lin L, Shu R, Dong B, Cao D, et al. A targeted transforming growth factor-beta (TGF-β) blocker, TTB, inhibits tumor growth and metastasis. Oncotarget. 2018;9(33):23102–13. https://doi.org/10.18632/oncotarget.24562.
Thaiparambil JT, Bender L, Ganesh T, Kline E, Patel P, Liu Y, et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int J Cancer. 2011;129(11):2744–55. https://doi.org/10.1002/ijc.25938.
Rhodes LV, Tate CR, Segar HC, Burks HE, Phamduy TB, Hoang V, et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat. 2014;145(3):593–604. https://doi.org/10.1007/s10549-014-2979-6.
Ferrari-Amorotti G, Chiodoni C, Shen F, Cattelani S, Soliera AR, Manzotti G, et al. Suppression of invasion and metastasis of triple-negative breast cancer lines by pharmacological or genetic inhibition of slug activity. Neoplasia. 2014;16(12):1047–58. https://doi.org/10.1016/j.neo.2014.10.006.
Roccaro AM, Mishima Y, Sacco A, Moschetta M, Tai YT, Shi J, et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep. 2015;12(4):622–35. https://doi.org/10.1016/j.celrep.2015.06.059.
Wu CX, Xu A, Zhang CC, Olson P, Chen L, Lee TK, et al. Notch Inhibitor PF-03084014 inhibits hepatocellular carcinoma growth and metastasis via suppression of cancer stemness due to reduced activation of Notch1-Stat3. Mol Cancer Ther. 2017;16(8):1531–43. https://doi.org/10.1158/1535-7163.mct-17-0001.
Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, et al. Activating NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to notch1 inhibitors. J Clin Oncol. 2017;35(3):352–60. https://doi.org/10.1200/jco.2016.67.5264.
Qu H, Ma B, Yuan H-F, Wang Z-Y, Guo S-J, Zhang J. Effect of salinomycin on metastasis and invasion of bladder cancer cell line T24. Asian Pac J Trop Med. 2015;8(7):578–82. https://doi.org/10.1016/j.apjtm.2015.06.004.
Dinicola S, Fabrizi G, Masiello MG, Proietti S, Palombo A, Minini M, et al. Inositol induces mesenchymal-epithelial reversion in breast cancer cells through cytoskeleton rearrangement. Exp Cell Res. 2016;345(1):37–50. https://doi.org/10.1016/j.yexcr.2016.05.007.
Zhou P, Wang C, Hu Z, Chen W, Qi W, Li A. Genistein induces apoptosis of colon cancer cells by reversal of epithelial-to-mesenchymal via a Notch1/NF-κB/slug/E-cadherin pathway. BMC Cancer. 2017;17(1):813. https://doi.org/10.1186/s12885-017-3829-9.
Sadhu SS, Wang S, Dachineni R, Averineni RK, Seefeldt T, Xie J, et al. In vitro and in vivo antimetastatic effect of glutathione disulfide liposomes. Cancer Growth Metastasis. 2017;10:1179064417695255. https://doi.org/10.1177/1179064417695255.
Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, et al. Gain fat-lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell. 2019;35(1):17-32.e6. https://doi.org/10.1016/j.ccell.2018.12.002.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8. https://doi.org/10.1038/s41392-020-0110-5.
Kakar SS, Ratajczak MZ, Powell KS, Moghadamfalahi M, Miller DM, Batra SK, et al. Withaferin a alone and in combination with cisplatin suppresses growth and metastasis of ovarian cancer by targeting putative cancer stem cells. PLoS ONE. 2014;9(9): e107596. https://doi.org/10.1371/journal.pone.0107596.
Marangoni E, Lecomte N, Durand L, de Pinieux G, Decaudin D, Chomienne C, et al. CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. Br J Cancer. 2009;100(6):918–22. https://doi.org/10.1038/sj.bjc.6604953.
Li M, Zhang B, Zhang Z, Liu X, Qi X, Zhao J, et al. Stem cell-like circulating tumor cells indicate poor prognosis in gastric cancer. Biomed Res Int. 2014;2014: 981261. https://doi.org/10.1155/2014/981261.
Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31(6):539–44. https://doi.org/10.1038/nbt.2576.
Fang C, Fan C, Wang C, Huang Q, Meng W, Yu Y, et al. CD133+CD54+CD44+ circulating tumor cells as a biomarker of treatment selection and liver metastasis in patients with colorectal cancer. Oncotarget. 2016;7(47):77389–403. https://doi.org/10.18632/oncotarget.12675.
Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9(8):997–1007. https://doi.org/10.1158/1541-7786.mcr-10-0490.
Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012;14(1):R15. https://doi.org/10.1186/bcr3099.
Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–4. https://doi.org/10.1126/science.1228522.
Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci USA. 2010;107(43):18392–7. https://doi.org/10.1073/pnas.1012539107.
Cho EH, Wendel M, Luttgen M, Yoshioka C, Marrinucci D, Lazar D, et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys Biol. 2012;9(1): 016001. https://doi.org/10.1088/1478-3975/9/1/016001.
Wendel M, Bazhenova L, Boshuizen R, Kolatkar A, Honnatti M, Cho EH, et al. Fluid biopsy for circulating tumor cell identification in patients with early-and late-stage non-small cell lung cancer: a glimpse into lung cancer biology. Phys Biol. 2012;9(1): 016005. https://doi.org/10.1088/1478-3967/9/1/016005.
Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22. https://doi.org/10.1016/j.cell.2014.07.013.106.
Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci USA. 2016;113(7):E854–63. https://doi.org/10.1073/pnas.1508541113.
Reduzzi C, Di Cosimo S, Gerratana L, Motta R, Martinetti A, Vingiani A, et al. Circulating tumor cell clusters are frequently detected in women with early-stage breast cancer. Cancers. 2021;13(10):2356. https://doi.org/10.3390/cancers13102356.
Mao S, Zhang Q, Li H, Zhang W, Huang Q, Khan M, et al. Adhesion analysis of single circulating tumor cells on a base layer of endothelial cells using open microfluidics. Chem Sci. 2018;9(39):7694–9. https://doi.org/10.1039/c8sc03027h.
Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 2019;176(1):98-112.e14. https://doi.org/10.1016/j.cell.2018.11.046.
Liu X, Adorno-Cruz V, Chang YF, Jia Y, Kawaguchi M, Dashzeveg NK, et al. EGFR inhibition blocks cancer stem cell clustering and lung metastasis of triple negative breast cancer. Theranostics. 2021;11(13):6632–43. https://doi.org/10.7150/thno.57706.
Sionov RV, Fridlender ZG, Granot Z. The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron. 2015;8(3):125–58. https://doi.org/10.1007/s12307-014-0147-5.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94. https://doi.org/10.1016/j.ccr.2009.06.017.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8. https://doi.org/10.1038/nature14282.
Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils suppress intraluminal nk cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 2016;6(6):630–49. https://doi.org/10.1158/2159-8290.cd-15-1157.
Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, et al. Neutrophils promote liver metastasis via Mac-1–mediated interactions with circulating tumor cells. Cancer Res. 2012;72(16):3919–27. https://doi.org/10.1158/0008-5472.can-11-2393.
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76(6):1367–80. https://doi.org/10.1158/0008-5472.can-15-1591.
Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123(8):3446–58. https://doi.org/10.1172/jci67484.
Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138. https://doi.org/10.1126/scitranslmed.aag1711.
Salado C, Olaso E, Gallot N, Valcarcel M, Egilegor E, Mendoza L, et al. Resveratrol prevents inflammation-dependent hepatic melanoma metastasis by inhibiting the secretion and effects of interleukin-18. J Transl Med. 2011;9:59. https://doi.org/10.1186/1479-5876-9-59.
Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–90. https://doi.org/10.1016/j.ccr.2011.09.009.
Olsson AK, Cedervall J. The pro-inflammatory role of platelets in cancer. Platelets. 2018;29(6):569–73. https://doi.org/10.1080/09537104.2018.1453059.
Schlesinger M. Role of platelets and platelet receptors in cancer metastasis. J Hematol Oncol. 2018;11(1):125. https://doi.org/10.1186/s13045-018-0669-2.
Razak NBA, Jones G, Bhandari M, Berndt MC, Metharom P. Cancer-associated thrombosis: an overview of mechanisms, risk factors, and treatment. Cancers (Basel). 2018. https://doi.org/10.3390/cancers10100380.
Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA. 2001;98(6):3352–7. https://doi.org/10.1073/pnas.061615598.
Schwarz S, Gockel LM, Naggi A, Barash U, Gobec M, Bendas G, et al. Glycosaminoglycans as tools to decipher the platelet tumor cell interaction: a focus on P-selectin. Molecules. 2020. https://doi.org/10.3390/molecules25051039.
Thiery JP, Lim CT. Tumor dissemination: an EMT affair. Cancer Cell. 2013;23(3):272–3. https://doi.org/10.1016/j.ccr.2013.03.004.
Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376(9754):1741–50. https://doi.org/10.1016/s0140-6736(10)61543-7.
Nakchbandi W, Müller H, Singer MV, Löhr M, Nakchbandi IA. Effects of low-dose warfarin and regional chemotherapy on survival in patients with pancreatic carcinoma. Scand J Gastroenterol. 2006;41(9):1095–104. https://doi.org/10.1080/00365520600575720.129.
Bruno A, Dovizio M, Tacconelli S, Contursi A, Ballerini P, Patrignani P. Antithrombotic agents and cancer. Cancers (Basel). 2018. https://doi.org/10.3390/cancers10080253.
Li J, King MR. Adhesion receptors as therapeutic targets for circulating tumor cells. Front Oncol. 2012;2:79. https://doi.org/10.3389/fonc.2012.00079.
Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci USA. 1998;95(16):9325–30. https://doi.org/10.1073/pnas.95.16.9325.
Köhler S, Ullrich S, Richter U, Schumacher U. E-/P-selectins and colon carcinoma metastasis: first in vivo evidence for their crucial role in a clinically relevant model of spontaneous metastasis formation in the lung. Br J Cancer. 2010;102(3):602–9. https://doi.org/10.1038/sj.bjc.6605492.
Husted S, Van Giezen J. Ticagrelor: the first reversibly binding oral P2Y12 receptor antagonist. Cardiovasc Ther. 2009;27(4):259–74.
Wright JR, Chauhan M, Shah C, Ring A, Thomas AL, Goodall AH, et al. The TICONC (Ticagrelor-Oncology) Study: implications of P2Y12 inhibition for metastasis and cancer-associated thrombosis. JACC CardioOncol. 2020;2(2):236–50. https://doi.org/10.1016/j.jaccao.2020.04.009.
Yu L, Zhao ZJ. Engineered platelets with antithrombotic and antimetastatic properties. Stem Cell Investig. 2019;6:27. https://doi.org/10.2137/sci.2019.08.02.
Papa AL, Jiang A, Korin N, Chen MB, Langan ET, Waterhouse A, et al. Platelet decoys inhibit thrombosis and prevent metastatic tumor formation in preclinical models. Sci Transl Med. 2019. https://doi.org/10.1126/scitranslmed.aau5898.
Xu Y, Liu J, Liu Z, Ren H, Yong J, Li W, et al. Blockade of platelets using tumor-specific NO-releasing nanoparticles prevents tumor metastasis and reverses tumor immunosuppression. ACS Nano. 2020;14(8):9780–95. https://doi.org/10.1021/acsnano.0c01687.
Nieswandt B, Hafner M, Echtenacher B, Männel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999;59(6):1295–300.
Placke T, Örgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, et al. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012;72(2):440–8. https://doi.org/10.1158/0008-5472.can-11-1872.
Cluxton CD, Spillane C, O’Toole SA, Sheils O, Gardiner CM, O’Leary JJ. Suppression of natural killer cell NKG2D and CD226 anti-tumour cascades by platelet cloaked cancer cells: implications for the metastatic cascade. PLoS ONE. 2019;14(3): e0211538. https://doi.org/10.1371/journal.pone.0211538.
Zhao YK, Jia CM, Yuan GJ, Liu W, Qiu Y, Zhu QG. Expression and clinical value of the soluble major histocompatibility complex class I-related chain A molecule in the serum of patients with renal tumors. Genet Mol Res. 2015;14(2):7233–40. https://doi.org/10.4238/2015.June.29.16.
Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICB in malignant diseases: analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol Immunother. 2006;55(12):1584–9. https://doi.org/10.1007/s00262-006-0167-1.
de Andrade LF, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science. 2018;359(6383):1537–42. https://doi.org/10.1126/science.aao0505.
Qin Z, Chen J, Zeng J, Niu L, Xie S, Wang X, et al. Effect of NK cell immunotherapy on immune function in patients with hepatic carcinoma: a preliminary clinical study. Cancer Biol Ther. 2017;18(5):323–30. https://doi.org/10.1080/15384047.2017.1310346.
Matzner P, Sorski L, Shaashua L, Elbaz E, Lavon H, Melamed R, et al. Perioperative treatment with the new synthetic TLR-4 agonist GLA-SE reduces cancer metastasis without adverse effects. Int J Cancer. 2016;138(7):1754–64. https://doi.org/10.1002/ijc.29885.
Tai LH, de Souza CT, Bélanger S, Ly L, Alkayyal AA, Zhang J, et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013;73(1):97–107. https://doi.org/10.1158/0008-5472.can-12-1993.
Zhang J, Tai LH, Ilkow CS, Alkayyal AA, Ananth AA, de Souza CT, et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol Ther. 2014;22(7):1320–32. https://doi.org/10.1038/mt.2014.60.
Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016;6(4):446–59. https://doi.org/10.1158/2159-8290.cd-15-0944.
Zhang Z, King MR. Nanomaterials for the capture and therapeutic targeting of circulating tumor cells. Cell Mol Bioeng. 2017;10(4):275–94. https://doi.org/10.1007/s12195-017-0497-4.
Gribko A, Künzel J, Wünsch D, Lu Q, Nagel SM, Knauer SK, et al. Is small smarter? Nanomaterial-based detection and elimination of circulating tumor cells: current knowledge and perspectives. Int J Nanomed. 2019;14:4187–209. https://doi.org/10.2147/IJN.S198319.
Wang D, Ge C, Liang W, Yang Q, Liu Q, Ma W, et al. In vivo enrichment and elimination of circulating tumor cells by using a black phosphorus and antibody functionalized intravenous catheter. Adv Sci. 2020;7(17):2000940. https://doi.org/10.1002/advs.202000940.
Raschpichler M, Preis E, Pinnapireddy SR, Baghdan E, Pourasghar M, Schneider M, et al. Photodynamic inactivation of circulating tumor cells: an innovative approach against metastatic cancer. Eur J Pharm Biopharm. 2020;157:38–46. https://doi.org/10.1016/j.ejpb.2020.10.003.
Ghaderinia M, Khayamian MA, Abadijoo H, Shalileh S, Faramarzpour M, Zandi A, et al. Capture-free deactivation of CTCs in the bloodstream; a metastasis suppression method by electrostatic stimulation of the peripheral blood. Biosens Bioelectron. 2021;183: 113194. https://doi.org/10.1016/j.bios.2021.113194.
Ye H, Wang K, Lu Q, Zhao J, Wang M, Kan Q, et al. Nanosponges of circulating tumor-derived exosomes for breast cancer metastasis inhibition. Biomaterials. 2020;242: 119932. https://doi.org/10.1016/j.biomaterials.2020.119932.
Restivo A, Cocco IM, Casula G, Scintu F, Cabras F, Scartozzi M, et al. Aspirin as a neoadjuvant agent during preoperative chemoradiation for rectal cancer. Br J Cancer. 2015;113(8):1133–9. https://doi.org/10.1038/bjc.2015.336.
Khoo BL, Grenci G, Lim JSY, Lim YP, Fong J, Yeap WH, et al. Low-dose anti-inflammatory combinatorial therapy reduced cancer stem cell formation in patient-derived preclinical models for tumour relapse prevention. Br J Cancer. 2019;120(4):407–23. https://doi.org/10.1038/s41416-018-0301-9.
Uluçkan O, Eagleton MC, Floyd DH, Morgan EA, Hirbe AC, Kramer M, et al. APT102, a novel adpase, cooperates with aspirin to disrupt bone metastasis in mice. J Cell Biochem. 2008;104(4):1311–23. https://doi.org/10.1002/jcb.21709.
D’Avola D, Villacorta-Martin C, Martins-Filho SN, Craig A, Labgaa I, von Felden J, et al. High-density single cell mRNA sequencing to characterize circulating tumor cells in hepatocellular carcinoma. Sci Rep. 2018;8(1):11570. https://doi.org/10.1038/s41598-018-30047-y.
Cheng Y-H, Chen Y-C, Lin E, Brien R, Jung S, Chen Y-T, et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat Commun. 2019;10(1):2163. https://doi.org/10.1038/s41467-019-10122-2.
Klotz R, Thomas A, Teng T, Han SM, Iriondo O, Li L, et al. Circulating tumor cells exhibit metastatic tropism and reveal brain metastasis drivers. Cancer Discov. 2020;10(1):86–103. https://doi.org/10.1158/2159-8290.cd-19-0384.
Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021;22(1):50. https://doi.org/10.1186/s13059-021-02267-5.
Cong Y, Liang Y, Motamedchaboki K, Huguet R, Truong T, Zhao R, et al. Improved single-cell proteome coverage using narrow-bore packed nanoLC columns and ultrasensitive mass spectrometry. Anal Chem. 2020;92(3):2665–71. https://doi.org/10.1021/acs.analchem.9b04631.
Cong Y, Motamedchaboki K, Misal SA, Liang Y, Guise AJ, Truong T, et al. Ultrasensitive single-cell proteomics workflow identifies > 1000 protein groups per mammalian cell. Chem Sci. 2021;12(3):1001–6.
Acknowledgements
We thank Ms. Ekaterina Khitrinskaya for the preparation of the figure.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by the Russian Science Foundation (grant #19-75-30016).
Conflict of Interest
The authors declare no conflicts of interest.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author contributions
All authors drafted and critically revised the manuscript.
Rights and permissions
About this article

Cite this article
Menyailo, M.E., Bokova, U.A., Ivanyuk, E.E. et al. Metastasis Prevention: Focus on Metastatic Circulating Tumor Cells. Mol Diagn Ther 25, 549–562 (2021). https://doi.org/10.1007/s40291-021-00543-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-021-00543-5