
Abstract
Objective
The aims of this study were to determine the effects of the CYP2C8*3 and *4 polymorphisms on imatinib metabolism and plasma imatinib concentrations in chronic myeloid leukaemia (CML) patients.
Methods
We genotyped 210 CML patients from the TIDELII trial receiving imatinib 400–800 mg/day for CYP2C8*3 (rs11572080, rs10509681) and *4 (rs1058930). Steady-state trough total plasma N-desmethyl imatinib (major metabolite):imatinib concentration ratios (metabolic ratios) and trough total plasma imatinib concentrations were compared between genotypes (one-way ANOVA with Tukey post hoc).
Results
CYP2C8*3 (n = 34) and *4 (n = 15) carriers had significantly higher (P < 0.01) and lower (P < 0.01) metabolic ratios, respectively, than CYP2C8*1/*1 (n = 147) patients (median ± standard deviation: 0.28 ± 0.08, 0.18 ± 0.06 and 0.22 ± 0.08, respectively). Plasma imatinib concentrations were consequently > 50% higher for CYP2C8*1/*4 than for CYP2C8*1/*1 and CYP2C8*3 carriers (2.18 ± 0.66 vs. 1.45 ± 0.74 [P < 0.05] and 1.36 ± 0.98 μg/mL [P < 0.05], respectively).
Conclusions
CYP2C8 genotype significantly alters imatinib metabolism in patients through gain- and loss-of-function mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cytochrome P450 (CYP) 2C8 metabolism plays a role in imatinib clinical pharmacokinetics. |
The CYP2C8 genotype of a chronic myeloid leukaemia patient can significantly affect their imatinib metabolism, and consequently imatinib exposure for a given dose. |
1 Introduction
Imatinib is one of the most widely used first-line treatments for chronic myeloid leukaemia (CML), and is also indicated in Ph+ acute lymphoblastic leukaemia, c-KIT- and platelet-derived growth factor receptor (PDGFR)-positive gastrointestinal stromal tumours (GISTs), myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements, aggressive systemic mastocytosis (without D816V c-Kit mutation), hypereosinophilic syndrome and/or chronic eosinophilic leukaemia, and dermatofibrosarcoma protuberans [1–4]. Currently up to 50% of CML patients discontinue imatinib due to lack of efficacy or adverse effects [5]; a significant problem requiring switching to other treatments that may be more costly or may have significant toxicities. Imatinib response is significantly associated with plasma imatinib concentrations, which can vary more than 25-fold between patients for a given dose [6–9]. Therefore, imatinib dose individualisation will likely be a key to achieving treatment goals in more patients.
As a low hepatic extraction ratio drug, steady-state plasma imatinib concentrations are determined by variability in plasma protein binding and intrinsic clearance. Imatinib undergoes hepatic N-demethylation to the much less potent [10–13] major metabolite N-desmethyl imatinib (NDIM), with approximately 65% of systemic exposure corresponding to imatinib and 10–20% to NDIM [14, 15]. Both imatinib and NDIM undergo mostly hepatic excretion with very little renal contribution [16]. Therefore, imatinib biotransformation to NDIM is a clinically important inactivating process, with variable imatinib metabolism likely to be a major contributor to the large inter-patient variability in the dose–plasma concentration relationship; this may be partly genetically determined [17, 18].
Imatinib is metabolised to NDIM by cytochrome P450 (CYP) 2C8 and 3A4 in vitro, with evidence of dose- and time-dependent mechanism-based CYP3A4 inhibition [19–21]. Steady-state imatinib pharmacokinetics are unrelated to variable CYP3A activity (quinine 3-hydroxylation) in CML patients [22] (as in GIST patients [23, 24]), are not significantly influenced by CYP3A4 inducers or inhibitors [16, 25, 26], and there is no consistent evidence that CYP3A4 or CYP3A5 genetic polymorphisms alter imatinib metabolism [27–30]. We recently demonstrated that imatinib N-demethylation in human liver microsomes is mainly mediated by CYP2C8, for which the CYP2C8*3 (rs11572080 [R139K] and rs10509681 [K399R]) is a gain-of-function haplotype [31]. The other major polymorphism in Caucasians, CYP2C8*4 (rs1058930), has been associated with reduced paclitaxel, amodiaquine and fluoxetine metabolism [32–35]. Compared with other members of the CYP2C family, CYP2C8 metabolism and pharmacogenetics have not been the focus of much clinical pharmacology research in general, and CYP2C8 genotype effects on imatinib metabolism in vivo have not been investigated.
Other enzymes (CYP1A2, CYP2D6, CYP2C9 and CYP2C19) play little or no role in imatinib N-methylation to NDIM [20, 36], and consequently their genetics have shown no significant effect on imatinib pharmacokinetics [28, 29].
We hypothesised that the CYP2C8*3 polymorphisms would increase, and the CYP2C8*4 polymorphism would decrease, imatinib metabolism to NDIM in CML patients.
This study aimed firstly to determine CYP2C8 genotype differences in imatinib metabolism as indicated by steady-state metabolic ratio (trough plasma NDIM:imatinib concentration ratio) in CML patients treated with imatinib. It then aimed to identify any dose and time dependency of these genotype effects. Finally, the study aimed to investigate the potential clinically relevant consequences of any genotype effects on the metabolic ratio by determining genotype differences in steady-state plasma imatinib concentrations.
2 Methods
2.1 Patients, Data and Exclusions
The study was a retrospective analysis of the first 3 months of treatment for 210 predominantly Caucasian chronic-phase CML patients who participated in the TIDEL (Therapeutic Intensification in De Novo Leukaemia)-II study [37]. Briefly, all patients started treatment with imatinib 600 mg/day. If patient trough plasma imatinib concentrations at day 22 of treatment were <1000 ng/mL (based on previous studies indicating a correlation between the minimum plasma imatinib concentration achieved [>1000 ng/mL] and the likelihood of achieving complete cytogenetic remission and/or major molecular response) [7, 8], then the imatinib dose was increased to 800 mg/day. Imatinib dose reductions to 400 mg/day were allowed at any time for grade III/IV or persistent grade II toxicities. Patients were switched to nilotinib 400 mg twice daily if they were unable to dose escalate to imatinib 800 mg due to intolerance or had a loss of imatinib response at any time. Dose changes in response to failure to achieve pre-determined time-dependent treatment response targets [37] were made after the 90-day timepoint analysed in this retrospective analysis. The study was approved by the relevant Human Research Ethics Committees of participating study sites [37].
The following data from the TIDEL-II study were used in the main and supplementary analyses: imatinib dose and trough plasma imatinib and NDIM concentrations determined by HPLC with UV detection as described in Electronic Supplementary Material Online Resource 2 for days 8, 22 and 90 of treatment; and patient age and sex.
Three patients were excluded for all timepoints due to unclear dosing histories. Additional timepoint-specific exclusions (n = 5) were made due to documented deviations in time of blood sampling. Demographics of the 207 patients included in this study, and a summary of dose and timepoint data used, are shown in Table 1.
2.2 Genotyping
CYP2C8 rs11572080, rs10509681 and rs1058930 were determined by probe-based allelic discrimination assays as detailed in Online Resource 1.
2.3 Statistical Analysis
Chi squared analysis (GraphPad PRISM 5, GraphPad Software, San Diego, CA, USA) was used to test for genotype deviations from Hardy–Weinberg equilibrium. All other data analyses were conducted in the R statistical package (version 3.3.1) [38].
The metabolic ratio was calculated as plasma NDIM concentration (μmol/L) divided by plasma imatinib concentration (μmol/L). Histograms and quantile–quantile (Q–Q) plots indicated that plasma imatinib and NDIM concentrations (ng/mL) and the metabolic ratio were not normally distributed. Square-root-transformed plasma imatinib concentration, and log e -transformed plasma NDIM concentration and metabolic ratio, data were normally distributed. Transformed data were used in all statistical comparisons.
Day 8 metabolic ratios and plasma imatinib concentrations were compared between genotypes by one-way ANOVA with Tukey post hoc test (lm function in stats package [38], Anova function in car package [39], and glht function of the multcomp package [40]). Tests for linear trend between genotypes (gene–dose effect) were performed in GraphPad PRISM 5. The proportion of patients achieving the day 22 plasma imatinib 1000 ng/mL concentration target was also compared (Chi squared test) between genotypes.
Forward stepwise (inclusion criteria: type II ANOVA P < 0.05) linear mixed-effects modelling was used to investigate the main (fixed) effects of dose (factor: 400, 600 and 800 mg/day), time (factor: day 8, 22 and 90), age, sex and genotype on the metabolic ratio and plasma imatinib or NDIM concentrations, with random effect (intercept) for patient ID (lmer function of lme4 package [41]). To determine dose and time dependency of any significant genotype effects on metabolic ratios or plasma imatinib concentrations, genotype was held as a fixed effect and each patient held as a random intercept, whilst testing (type II ANOVA P < 0.05) random slopes and intercepts for dose and/or day. The effects of individual predictors from multiple regression analyses (averaging over other terms in the model) were determined using the allEffects function of the effects package [42]. Simultaneous Tukey post hoc tests on significant main effects were conducted using the glht function of the multcomp package [40].
The dose proportionality of trough plasma imatinib concentrations within patients decreasing to 400 mg/day, and increasing to 800 mg/day, from 600 mg/day before day 90 of treatment was determined as described in Online Resource 3.
Point-wise P-values (no correction for multiple testing) are presented unless specified otherwise.
3 Results
3.1 Genetic Variability
One hundred and sixty patients were CYP2C8*1/*1, 31 were *1/*3, 3 were *3/*3 and 16 were *1/*4. CYP2C8 rs11572080 and rs10509681 were in complete linkage disequilibrium. Given the low frequency of the CYP2C8*3/*3 genotype, these patients were grouped with CYP2C8*1/*3 for statistical analyses, and collectively referred to as CYP2C8*3 carriers. All CYP2C8*1/*4 patients were wildtype at both CYP2C8*3 loci.
CYP2C8 genotype distribution did not deviate significantly from Hardy–Weinberg Equilibrium (P = 1.0).
3.2 Metabolic Ratio
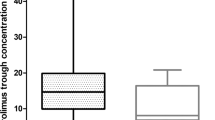
Day 8 plasma NDIM:imatinib concentration ratios ranged from 0.07 to 0.63 (median = 0.23). The metabolic ratio was significantly higher for CYP2C8*3 carriers compared to CYP2C8*1/*1 and CYP2C8*1/*4, and significantly lower for CYP2C8*1/*4 compared to CYP2C8*1/*1 (Table 2; Fig. 1a). CYP2C8 genotype accounted for 10% of variability in the metabolic ratio. There was no significant linear trend if CYP2C8*1/*3 and CYP2C8*3/*3 were separated (P = 0.4).

CYP2C8 genotype differences in steady-state trough total plasma N-desmethyl imatinib:imatinib concentration ratios and plasma imatinib concentrations in chronic myeloid leukaemia patients on day 8 of treatment
Linear mixed-effects analysis of day 8, 22 and 90 data identified significant dose (P = 0.0004), day (P = 5 × 10−6) and CYP2C8 genotype (P = 4 × 10−5) main effects (Fig. 2), but no significant effects of age or sex (P > 0.1).

Dose, day and CYP2C8 genotype effects on trough total plasma N-desmethyl imatinib:imatinib metabolic ratio in chronic myeloid leukaemia patients. Circles and bars are geometric means and 95% confidence intervals, respectively, holding other main effects to typical values (proportional distribution). CYP2C8*3 genotype group combines CYP2C8*1/*3 and *3/*3 genotypes. Tukey post hoc * P < 0.05
Accounting for other significant main effects, post hoc analyses showed that the imatinib 800 mg/day dose was associated with a significantly lower metabolic ratio than 600 (17% lower, P < 0.01) and 400 (25% lower, P < 0.01) mg/day, with no significant difference between 400 and 600 mg/day. Day 22 metabolic ratios were significantly higher than at day 8 (11% higher, P < 0.0001) and day 90 (7% higher, P < 0.05), with no significant difference between days 8 and 90.
The CYP2C8*3 carrier genotype was associated with a significantly higher metabolic ratio than CYP2C8*1/*1 (18% higher, P < 0.01) and CYP2C8*1/*4 (39% higher, P < 0.001); CYP2C8*1/*4 genotypes were 15% lower than CYP2C8*1/*1 (P = 0.09). Random slopes for genotype effect within dose and day did not significantly improve the model (P > 0.9).
3.3 Plasma Imatinib Concentration
Day 8 trough total plasma imatinib concentrations ranged from 280 to 5000 ng/mL (median = 1480 ng/mL) and were significantly (>50%) higher in CYP2C8*1/*4 patients than in CYP2C8*1/*1 and CYP2C8*3 carrier genotypes (Table 2; Figs. 1b and 3a).

CYP2C8 genotype effects on trough total plasma imatinib (a) and N-desmethyl imatinib (b) concentrations in chronic myeloid leukaemia patients over time in treatment. Circles and bars are back-transformed means and 95% confidence intervals, respectively, holding imatinib dose to typical values (proportional distribution) [linear mixed-effects model of √(plasma imatinib concentration) or log e (plasma N-desmethyl imatinib concentration) with dose and genotype × day interaction main effects, and patient ID random effect (intercept)]. NDIM N-desmethyl imatinib. Tukey post hoc ** P < 0.01 vs. day 8 and ## P < 0.01 vs. day 22 for one-way ANOVA in CYP2C8*1/*4 patients
Dose was the only significant (P = 1 × 10−11) main effect in linear mixed-effects analysis of day 8, 22 and 90 data; plasma imatinib concentrations were a median 52% higher at 600 mg/day than at 400 mg/day, and 149% higher (non-dose-proportional) at 800 mg/day than at 600 mg/day (see Online Resource 3).
Accounting for dose, the CYP2C8 genotype was not a significant predictor of plasma imatinib concentrations (P = 0.08), and nor were day (P = 0.6), age (P = 0.5) or sex (P = 0.2), when analysing day 8, 22 and 90 data together. Given the disparity in results for the CYP2C8 genotype between day 8 and multiple timepoint analyses (combined days 8, 22 and 90), an interaction between CYP2C8 genotype and day was examined. The genotype × day interaction was not significant (P = 0.18); however, Fig. 3a illustrates a decrease in CYP2C8 genotype differences over time in treatment and a significant decrease in trough plasma imatinib concentrations on day 90 among CYP2C8*1/*4 genotype patients (P = 0.002 for day main effect [post hoc P < 0.01 day 90 vs. day 8 and day 22]), none of whom had a prescribed dose reduction. There were no significant (P > 0.1) day effects on plasma imatinib concentrations within CYP2C8*1/*1 or CYP2C8*3 carrier patients.
Of 198 patients without a dose change prior to day 22, 162 (82%) reached the day 22 plasma imatinib concentration target of >1000 ng/mL. The proportion of patients achieving this target was significantly different between CYP2C8 genotypes (Table 2), with all CYP2C8*1/*4 patients above 1000 ng/mL on day 22 compared with 81% for CYP2C8*1/*1 and 78% for CYP2C8*3 carriers.
3.4 Plasma N-Desmethyl Imatinib Concentration
Dose (P = 7 × 10−12) and day (P = 2 × 10−5), but not genotype (P = 0.5), showed significant main effects in linear mixed-effects analysis of day 8, 22 and 90 plasma NDIM concentrations. Day 22 plasma NDIM concentrations were significantly higher than at day 8 (P < 1 × 10−4) and day 90 (P < 0.01), with no significant difference between days 8 and 90 (see Online Resource 3, Fig. S1).
As with plasma imatinib concentrations, Fig. 3b shows a significant decrease in trough plasma NDIM concentrations on day 90 among CYP2C8*1/*4 genotype patients (P = 0.002 for day main effect [post hoc P < 0.01 day 90 vs. day 22]).
4 Discussion
CYP2C8 makes up as little as 7% of total hepatic CYPs, and drug metabolism driven solely by the CYP2C8 enzyme is relatively uncommon [43]. Consequently, CYP2C8 metabolism and pharmacogenetics have not been focal points for much previous clinical pharmacology research. Where substrates are also metabolised by more abundant CYPs such as CYP3A4, the contribution of CYP2C8 is often discounted, and until recently this was the case for imatinib. However, recent in vitro studies [20, 31] combined with these clinical findings demonstrate that CYP2C8 and its genetic variability can still play a significant role in this context, or at least where the CYP3A4 pathway may be suppressed by mechanism-based inhibition.
Here we show that CML patients who carry the CYP2C8*3 haplotype have significantly higher trough total plasma metabolic ratios, reflecting CYP2C8*3 gain-of-function for imatinib N-demethylation reported in human liver microsomes [31]. The 27% increase in metabolic ratio on day 8 is an order of magnitude less than the in silico prediction (>200%) of Filppula and colleagues [20] for a genotype that doubles CYP2C8 activity, but is consistent with human liver microsome results where CYP2C8*3 increases intrinsic clearance by only approximately 20% [31]. Our data indicated a possible CYP2C8*3 allele–dose effect on the metabolic ratio (Table 2); however, this was not statistically significant, likely because of too few (n = 3) CYP2C8*3/*3 genotype patients.
Conversely, the CYP2C8*1/*4 genotype was associated with lower metabolic ratios, indicating reduced imatinib N-demethylation activity. Similar reduced function has been reported for other substrates in vitro and in vivo [32–35]. There are currently no corresponding published data on the in vitro metabolism of imatinib in CYP2C8*1/*4 human liver microsomes, although the 18% reduction in metabolic ratio on day 8 was less than the in silico prediction (43% reduction) of Filppula and colleagues [20] for a genotype that halved CYP2C8 activity.
Metabolic ratios were also influenced by dose, being significantly lower at imatinib 800 mg/day than at 600 or 400 mg/day, which was coupled with greater than dose-proportional increases in plasma imatinib concentrations. Whilst an interesting finding, this was not a focus of this study, and further discussion of the apparent dose effect on the metabolic ratio and consequent non-dose-proportionality is provided in Online Resource 3.
The clinical relevance of the CYP2C8 genetic effects on imatinib metabolism is less obvious. The magnitude of CYP2C8*1/*4 genotype effect on the day 8 plasma imatinib concentration (+50%) was similar to the within-patient difference between 400 and 600 mg/day (+52%) in this study, and the 50% increase predicted in silico for a genotype with low (half) CYP2C8 activity [20]. The CYP2C8*1/*4 genotype effect was also of similar or greater magnitude than other gene polymorphisms previously associated with changes in plasma imatinib concentrations clinically (25–50% reported for ABCB1, SLC22A1 and ABCG2 [27, 30, 44]). In addition, there was a significant CYP2C8 genotype effect on the likelihood of achieving day 22 target plasma imatinib concentrations of 1000 ng/mL, with all CYP2C8*1/*4 patients reaching this threshold that has been associated with significantly improved long-term treatment outcomes [6, 7].
Alternatively, whilst the CYP2C8*3 carriers had significantly higher metabolic ratios, they did not have significantly lower plasma imatinib concentrations. In addition, whilst CYP2C8*3 and *4 genotype effects on the metabolic ratio appeared relatively consistent across time and the doses investigated, genotype (in particular CYP2C8*4) differences in plasma imatinib concentrations were less apparent at later timepoints (Fig. 3) and non-significant when all timepoints were combined in linear mixed-effects analysis. Plasma imatinib concentrations are more confounded than the metabolic ratio by variability in patient adherence to the treatment regimen, dose-sample interval (e.g. due to variability in sampling time or error), and plasma protein binding (over 5-fold variability in plasma α1 acid glycoprotein concentrations within and between CML patients [45]). For example, plasma imatinib concentration results may have been confounded by increased variability in patient adherence as the amount of time in treatment progresses [46], particularly amongst CYP2C8*1/*4 patients (Fig. 3). Whilst every effort was made to identify and limit the effects of non-adherence by excluding data based on documented non-adherence, it is difficult to exclude occult non-adherence entirely. Based on the data presented in Fig. 3, it is speculated that significantly higher plasma imatinib concentrations in CYP2C8*1/*4 patients could have resulted in adverse effects (not captured under the intolerance criteria of the TIDELII study [37]) leading to patient non-adherence after the major plasma imatinib concentration checkpoint on day 22 of the study. Significant decreases in both plasma imatinib and NDIM concentrations on day 90 in CYP2C8*1/*4 patients, with no significant change in metabolic ratio (data not shown), suggest non-adherence rather than increased metabolism. However, there were no adverse effect data or other adherence markers (e.g. pill counts) available to test such a hypothesis.
Genotyping for the CYP2C8*4 polymorphism could foreseeably aid in decision making regarding whether to start patients on the standard imatinib 400 (CYP2C8*4) or higher 600 (wildtype CYP2C8) mg/day dose, if the hypothesis regarding adverse effects and non-adherence among CYP2C8*1/*4 patients at 600 mg/day can be proven. No CYP2C8*4/*4 patients were identified in this study, and so it is unknown whether this genotype would lead to more significant differences in metabolism and plasma imatinib exposure (and possibly adverse events). However, such patients would be rare in Caucasian (<1 in 150 patients), and rarer still in non-Caucasian (≤1 in 2500 patients), patient populations [47].
This study intentionally focused on the novel investigation of the CYP2C8 genotype; however, other potential contributors to variability in imatinib metabolism and pharmacokinetics are acknowledged. In addition to the aforementioned transporter genetics, variability in plasma protein binding, and thus the imatinib and NDIM unbound fraction, will influence total plasma concentrations and the metabolic ratio (unpublished unbound plasma imatinib and NDIM concentration data from a subset of TIDELII patients indicate CYP2C8 genotype effects on metabolic ratio and total plasma imatinib concentrations are similar after adjusting for unbound fraction). The relative and combined contributions, and thus importance, of CYP2C8 genotype (or CYP2C8 function more generally), transporter genetics and plasma protein binding to variability in imatinib pharmacokinetics in CML patients remain to be determined. Whether a pharmacogenetic approach to individualised imatinib dosing might be complementary or redundant in the context of potential therapeutic drug monitoring/target concentration intervention should also be considered [6].
5 Conclusion
This study shows that CYP2C8*3 and *4 genotypes significantly alter imatinib metabolism clinically and confirms a role for CYP2C8 in imatinib pharmacokinetics, in addition to increasing our limited knowledge of CYP2C8 clinical pharmacogenetics per se. Future prospective studies would need to be specifically designed and sufficiently powered [48, 49] to test whether personalising imatinib treatment based on transporter and metabolism genetics and plasma protein binding can improve imatinib treatment outcomes.
References
Maino E, Sancetta R, Viero P, Imbergamo S, Scattolin AM, Vespignani M, et al. Current and future management of Ph/BCR-ABL positive ALL. Expert Rev Anticancer Ther. 2014;14(6):723–40. doi:10.1586/14737140.2014.895669.
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol. 2014;89(5):547–56. doi:10.1002/ajh.23691.
Linch M, Claus J, Benson C. Update on imatinib for gastrointestinal stromal tumors: duration of treatment. Onco Targets Ther. 2013;6:1011–23. doi:10.2147/ott.s31260.
Gleevec NDA 021588 label information. Revised Sep 2016. US Food and Drug Administration, Center for Drug Evaluation and Research; 2016. http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021588s047lbl.pdf. Accessed 13 Oct 2016.
Gotta V, Bouchet S, Widmer N, Schuld P, Decosterd LA, Buclin T, et al. Large-scale imatinib dose-concentration-effect study in CML patients under routine care conditions. Leuk Res. 2014;38(7):764–72. doi:10.1016/j.leukres.2014.03.023.
Widmer N, Bardin C, Chatelut E, Paci A, Beijnen J, Leveque D, et al. Review of therapeutic drug monitoring of anticancer drugs part two-targeted therapies. Eur J Cancer. 2014;50(12):2020–36. doi:10.1016/j.ejca.2014.04.015.
Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Bernard MA, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109(8):3496–9. doi:10.1182/blood-2006-07-036012.
Larson RA, Druker BJ, Guilhot F, O’Brien SG, Riviere GJ, Krahnke T, et al. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia. Blood. 2008;111(8):4022–8. doi:10.1182/blood-2007-10-116475.
Berglund E, Ubhayasekera SJ, Karlsson F, Akcakaya P, Aluthgedara W, Ahlen J, et al. Intracellular concentration of the tyrosine kinase inhibitor imatinib in gastrointestinal stromal tumor cells. Anticancer Drugs. 2014;25(4):415–22. doi:10.1097/cad.0000000000000069.
Mlejnek P, Dolezel P, Faber E, Kosztyu P. Interactions of N-desmethyl imatinib, an active metabolite of imatinib, with P-glycoprotein in human leukemia cells. Ann Hematol. 2011;90(7):837–42. doi:10.1007/s00277-010-1142-7.
Manley PW, Blasco F, Mestan J, Aichholz R. The kinetic deuterium isotope effect as applied to metabolic deactivation of imatinib to the des-methyl metabolite, CGP74588. Bioorg Med Chem. 2013;21(11):3231–9. doi:10.1016/j.bmc.2013.03.038.
Skoglund K, Boiso Moreno S, Jonsson JI, Vikingsson S, Carlsson B, Green H. Single-nucleotide polymorphisms of ABCG2 increase the efficacy of tyrosine kinase inhibitors in the K562 chronic myeloid leukemia cell line. Pharmacogenet Genomics. 2014;24(1):52–61. doi:10.1097/FPC.0000000000000022.
Skoglund K, Moreno SB, Baytar M, Jonsson JI, Green H. ABCB1 haplotypes do not influence transport or efficacy of tyrosine kinase inhibitors in vitro. Pharmgenomics Pers Med. 2013;6:63–72. doi:10.2147/PGPM.S45522.
Josephs DH, Fisher DS, Spicer J, Flanagan RJ. Clinical pharmacokinetics of tyrosine kinase inhibitors: implications for therapeutic drug monitoring. Ther Drug Monit. 2013;35(5):562–87. doi:10.1097/FTD.0b013e318292b931.
Gschwind HP, Pfaar U, Waldmeier F, Zollinger M, Sayer C, Zbinden P, et al. Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab Dispos. 2005;33(10):1503–12. doi:10.1124/dmd.105.004283.
Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet. 2011;50(9):551–603. doi:10.2165/11593320-000000000-00000.
Bouchet S, Titier K, Moore N, Lassalle R, Ambrosino B, Poulette S, et al. Therapeutic drug monitoring of imatinib in chronic myeloid leukemia: experience from 1216 patients at a centralized laboratory. Fundam Clin Pharmacol. 2013;27(6):690–7. doi:10.1111/fcp.12007.
de Wit D, Guchelaar HJ, den Hartigh J, Gelderblom H, van Erp NP. Individualized dosing of tyrosine kinase inhibitors: are we there yet? Drug Discov Today. 2015;20(1):18–36. doi:10.1016/j.drudis.2014.09.007.
Filppula AM, Laitila J, Neuvonen PJ, Backman JT. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165(8):2787–98. doi:10.1111/j.1476-5381.2011.01732.x.
Filppula AM, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism. Drug Metab Dispos. 2013;41(1):50–9. doi:10.1124/dmd.112.048017.
Nebot N, Crettol S, d’Esposito F, Tattam B, Hibbs DE, Murray M. Participation of CYP2C8 and CYP3A4 in the N-demethylation of imatinib in human hepatic microsomes. Br J Pharmacol. 2010;161(5):1059–69. doi:10.1111/j.1476-5381.2010.00946.x.
Skoglund K, Richter J, Olsson-Stromberg U, Bergquist J, Aluthgedara W, Ubhayasekera SJ, et al. In vivo cytochrome P450 3A isoenzyme activity and pharmacokinetics of imatinib in relation to therapeutic outcome in patients with chronic myeloid leukemia. Ther Drug Monit. 2016;38(2):230–8. doi:10.1097/FTD.0000000000000268.
Gurney H, Wong M, Balleine RL, Rivory LP, McLachlan AJ, Hoskins JM, et al. Imatinib disposition and ABCB1 (MDR1, P-glycoprotein) genotype. Clin Pharmacol Ther. 2007;82(1):33–40. doi:10.1038/sj.clpt.6100201.
Widmer N, Decosterd LA, Csajka C, Leyvraz S, Duchosal MA, Rosselet A, et al. Population pharmacokinetics of imatinib and the role of alpha-acid glycoprotein [published erratum appears in Br J Clin Pharmacol 2010; 70 (2):316]. Br J Clin Pharmacol. 2006;62(1):97–112. doi:10.1111/j.1365-2125.2006.02719.x.
Schmidli H, Peng B, Riviere GJ, Capdeville R, Hensley M, Gathmann I, et al. Population pharmacokinetics of imatinib mesylate in patients with chronic-phase chronic myeloid leukaemia: results of a phase III study. Br J Clin Pharmacol. 2005;60(1):35–44. doi:10.1111/j.1365-2125.2005.02372.x.
van Erp NP, Gelderblom H, Karlsson MO, Li J, Zhao M, Ouwerkerk J, et al. Influence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib. Clin Cancer Res. 2007;13(24):7394–400. doi:10.1158/1078-0432.CCR-07-0346.
Singh O, Chan JY, Lin K, Heng CC, Chowbay B. SLC22A1-ABCB1 haplotype profiles predict imatinib pharmacokinetics in asian patients with chronic myeloid leukemia. PLoS One. 2012;7(12):e51771. doi:10.1371/journal.pone.0051771.
Seong SJ, Lim M, Sohn SK, Moon JH, Oh SJ, Kim BS, et al. Influence of enzyme and transporter polymorphisms on trough imatinib concentration and clinical response in chronic myeloid leukemia patients. Ann Oncol. 2013;24(3):756–60. doi:10.1093/annonc/mds532.
Yamakawa Y, Hamada A, Nakashima R, Yuki M, Hirayama C, Kawaguchi T, et al. Association of genetic polymorphisms in the influx transporter SLCO1B3 and the efflux transporter ABCB1 with imatinib pharmacokinetics in patients with chronic myeloid leukemia. Ther Drug Monit. 2011;33(2):244–50. doi:10.1097/FTD.0b013e31820beb02.
Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H, et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet. 2010;55(11):731–7. doi:10.1038/jhg.2010.98.
Khan MS, Barratt DT, Somogyi AA. Impact of CYP2C8*3 polymorphism on in vitro metabolism of imatinib to N-desmethyl imatinib. Xenobiotica. 2015:1–10. doi: 10.3109/00498254.2015.1060649.
Bahadur N, Leathart JB, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R, et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol. 2002;64(11):1579–89.
Gao Y, Liu D, Wang H, Zhu J, Chen C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug-drug interactions. Xenobiotica. 2010;40(7):467–75. doi:10.3109/00498254.2010.487163.
Jiang H, Zhong F, Sun L, Feng W, Huang ZX, Tan X. Structural and functional insights into polymorphic enzymes of cytochrome P450 2C8. Amino Acids. 2011;40(4):1195–204. doi:10.1007/s00726-010-0743-8.
Wang Z, Wang S, Huang M, Hu H, Yu L, Zeng S. Characterizing the effect of cytochrome P450 (CYP) 2C8, CYP2C9, and CYP2D6 genetic polymorphisms on stereoselective N-demethylation of fluoxetine. Chirality. 2014;26(3):166–73. doi:10.1002/chir.22289.
Rochat B, Zoete V, Grosdidier A, von Grunigen S, Marull M, Michielin O. In vitro biotransformation of imatinib by the tumor expressed CYP1A1 and CYP1B1. Biopharm Drug Dispos. 2008;29(2):103–18. doi:10.1002/bdd.598.
Yeung DT, Osborn MP, White DL, Branford S, Braley J, Herschtal A, et al. TIDEL-II: first-line use of imatinib in CML with early switch to nilotinib for failure to achieve time-dependent molecular targets. Blood. 2015;125(6):915–23. doi:10.1182/blood-2014-07-590315.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013.
Fox J, Weisberg S. An R companion to applied regression. 2nd ed. Thousand Oaks: Sage; 2011.
Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50(3):346–63.
Bates D, Maechler M, Bolker B, Wlaker S. lme4: linear mixed-effects models using Eigen and S4. R package version 1.0-5. 2013. http://CRAN.R-project.org/package=lme4. Accessed 19 Apr 2016.
Fox J. Effect displays in R for generalised linear models. J Stat Softw. 2003;8(15):1–27.
Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270(1):414–23.
Di Paolo A, Polillo M, Capecchi M, Cervetti G, Barate C, Angelini S, et al. The c.480C > G polymorphism of hOCT1 influences imatinib clearance in patients affected by chronic myeloid leukemia. Pharmacogenomics J. 2014;14(4):328–35. doi:10.1038/tpj.2014.7.
Gandia P, Arellano C, Lafont T, Huguet F, Malard L, Chatelut E. Should therapeutic drug monitoring of the unbound fraction of imatinib and its main active metabolite N-desmethyl-imatinib be developed? Cancer Chemother Pharmacol. 2013;71:531–6. doi:10.1007/s00280-012-2035-3.
Gater A, Heron L, Abetz-Webb L, Coombs J, Simmons J, Guilhot F, et al. Adherence to oral tyrosine kinase inhibitor therapies in chronic myeloid leukemia. Leuk Res. 2012;36(7):817–25. doi:10.1016/j.leukres.2012.01.021.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi:10.1038/nature11632.
Stingl Kirchheiner JC, Brockmoller J. Why, when, and how should pharmacogenetics be applied in clinical studies? Current and future approaches to study designs. Clin Pharmacol Ther. 2011;89(2):198–209. doi:10.1038/clpt.2010.274.
Ritchie MD. The success of pharmacogenomics in moving genetic association studies from bench to bedside: study design and implementation of precision medicine in the post-GWAS era. Hum Genet. 2012;131(10):1615–26. doi:10.1007/s00439-012-1221-z.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the School of Medical Sciences (now Adelaide Medical School), University of Adelaide and a Royal Adelaide Hospital Research Foundation Clinical Project Grant.
Conflicts of Interest
DLW received research funding and honoraria from Novartis and BMS. TPH acted on the advisory board of, and received research funding and honoraria from, Novartis, BMS and Ariad. DTY received research funding and honoraria from, Novartis, BMS and Ariad. DTB, HKC, AM and AAS have no conflicts of interest to declare.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Barratt, D.T., Cox, H.K., Menelaou, A. et al. CYP2C8 Genotype Significantly Alters Imatinib Metabolism in Chronic Myeloid Leukaemia Patients. Clin Pharmacokinet 56, 977–985 (2017). https://doi.org/10.1007/s40262-016-0494-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0494-0