
Abstract
The cytochrome P450 (CYP) superfamily plays a key role in the oxidative metabolism of a wide range of drugs and exogenous chemicals. CYP2C8 is the principal enzyme responsible for the metabolism of the anti-cancer drug paclitaxel in the human liver. Nearly all previous works about polymorphic variants of CYP2C8 were focused on unpurified proteins, either cells or human liver microsomes; therefore their structure–function relationships were unclear. In this study, two polymorphic enzymes of CYP2C8 (CYP2C8.4 (I264M) and CYP2C8 P404A) were expressed in E. coli and purified. Metabolic activities of paclitaxel by the two purified polymorphic enzymes were observed. The activity of CYP2C8.4 was 25% and CYP2C8 P404A was 30% of that of WT CYP2C8, respectively. Their structure–function relationships were systematically investigated for the first time. Paclitaxel binding ability of CYP2C8.4 increased about two times while CYP2C8 P404A decreased about two times than that of WT CYP2C8. The two polymorphic mutant sites of I264 and P404, located far from active site and substrate binding sites, significantly affect heme and/or substrate binding. This study indicated that two important nonsubstrate recognition site (SRS) residues of CYP2C8 are closely related to heme binding and/or substrate binding. This discovery could be valuable for explaining clinically individual differences in the metabolism of drugs and provides instructed information for individualized medication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The CYP superfamily, a group of hemoproteins present in all forms of life, plays a key role in the oxidative metabolism of a wide range of exogenous chemicals such as drugs, toxins, carcinogens, steroids, and fatty acids. The number of CYP families has been continuously growing and has now reached more than 200, with more than 57 known CYPs in humans. There is considerable variation in the way patients respond to the same drugs in the clinical treatment. Researches in “Human Genome Project” (HGP) suggested that these individual differences were caused by drug-related genetic polymorphism (Lee et al. 2001). Polymorphisms are gene mutations, which may change drug metabolism in the individuals. So whether a certain drug was effective or caused adverse reactions depended on the polymorphisms in the patient. Human CYPs are important drug metabolism enzymes to the detoxification and activation of drugs. Therefore, the polymorphisms of human CYPs are of great significance for clinicians to do individual medication. The polymorphic metabolizers phenotype can affect the clinical toxicity and the efficacy of drugs (Dai et al. 2001). Polymorphic variants in CYPs that metabolize endogenous compounds are also responsible for certain inherited diseases (Stoilov et al. 1998; Plasilova et al. 1999). So the researches about CYPs polymorphic variants are quite valuable for explaining clinically individual differences in the metabolism of drugs and provide instructed information for individualized medication. The CYP2Cs are a major subfamily of CYP enzymes that are found primarily in liver cells. The four human CYP2C enzymes: CYP2C8, CYP2C9, CYP2C18, and CYP2C19 are known to be polymorphic. Their functional polymorphic variants result in clinically relevant inter-individual differences in the metabolism of drugs. Polymorphisms in CYP2C8 were determined in 2001 as the last one to be known polymorphic, and were known least in the human CYP2C subfamily. CYP2C8, which makes up about 7% of total microsomal human hepatic CYP (Shimada et al. 1994; Rendic and Di Carlo 1997), carries out the oxidative metabolism of at least 5% of clinic drugs (Totah and Rettie 2005). The X-ray crystal structure of CYP2C8 (Schoch et al. 2004) shows a characteristic large active site cavity which can accommodate for reasonably large substrates (Schoch et al. 2004; Totah and Rettie 2005). Many kinds of therapeutically important drugs, including the anticancer drugs (Rahman et al. 1994; Sonnichsen et al. 1995), antiarrhythmic drugs (Ohyama et al. 2000), anti-diabetes drugs (Yamazaki et al. 1999), and antiepileptic drugs (Kerr et al. 1994) are the substrates mainly metabolized by CYP2C8. Thus polymorphisms of CYP2C8 are closely related to an individually variant response to drugs. Some cases about certain diseases causally related to polymorphisms of CYP2C8 have been reported as well (Yasar et al. 2003; Ishikawa et al. 2004). Up to now, 14 polymorphic variants in CYP2C8, designated as CYP2C8.2 to CYP2C8.14 (http://www.cypalleles.ki.se/cyp2c8.htm) and an unclassified form temporarily named CYP2C8 P404A (Soyama et al. 2002; Totah and Rettie 2005), have been reported. Polymorphic variants CYP2C8.2 to CYP2C8.4 have a wide range of distribution in various ethnic populations of white and black, while polymorphic variants CYP2C8.5 to CYP2C8.14 and CYP2C8 P404A have only been detected among the Japanese population.
Almost all the previous studies on polymorphic variants of CYP2C8 were not focused on the purified proteins, either cells or human liver microsomes (Soyama et al. 2001, 2002; Bahadur et al. 2002; Hichiya et al. 2005; Hanioka et al. 2010), while limited information about purified CYP2C8 polymorphic variants, except kinetics of paclitaxel and arachidonic acid metabolized by CYP2C8.2 and those of arachidonic acid metabolized by CYP2C8.3, have been reported by Dai et al. (2001). The structure–function relationships of CYP2C8 polymorphic variants were unclear. In this paper, we focused on the structure–function relationships of two purified polymorphic enzymes of CYP2C8, CYP2C8.4 (I264M) and CYP2C8 P404A (Fig. 1). The purified proteins were systematically characterized with UV and CD spectra. Their thermal stability, binding ability, and catalytic activity of paclitaxel were investigated, and the electron transfer kinetics was studied by stopped-flow. The results based on purified proteins indicated that I264 and P404 in CYP2C8 were important for heme binding and/or substrate binding even if they were non-SRS residues and located far from the active site and substrate binding sites.

3D structure of WT CYP2C8 (PDB ID: pdb1pq2a) showing polymorphic mutation sites of I264 and P404
Materials and methods
Materials
The pCWOri+ plasmid, encoding CYP2C8dH, was a kind gift from Prof. E.F. Johnson (The Scripps Research Institute, California, USA) (Schoch et al. 2004). The E. coli XL1-Blue strain and restriction enzymes were purchased from New England Biolabs in Shanghai, China. Ni-NTA Agarose was from Qiagen (Chatsworth, CA, USA). 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) was from Amresco (Solon, OH, USA). Paclitaxel was got from Fujian South Bio-Technology Co., Ltd (Fujian, China). β-NADPH and human cytochrome P450 reductase were obtained from Sigma-Aldrich (St. Louis, MO, USA). The oligonucleotide primer pairs listed below were synthesized, and DNA sequencing reactions were performed by Shanghai Invitrogen Biotech Co. Ltd.
Constructions of polymorphic mutations in CYP2C8
Mutants for CYP2C8.4 (I264M) and CYP2C8 P404A were constructed using Quik Change Site-Directed Mutagenesis Kit according to its protocol (Stratagene) with plasmid pCWOri+ encoding CYP 2C8dH as a template. The mutants for CYP2C8.4 and CYP2C8 P404A were made by replacing I264 with methionine using primer pair P1 and replacing P404 with alanine using primer pair P2, respectively. The mutated plasmids were isolated and sequenced by Shanghai Invitrogen Biotech Co. Ltd.
- P1:
-
[5′AACAATCCTCGGGACTTTATGGATTGCTTCCTGATCAAAATGG3′ (F), 5′CATTTTGATCAGGAAGCAATCCATAAAGTCCCGAGGATTGTT3′ (R)]
- P2:
-
[5′GATGACAAAGAATTTCCTAATGCAAATATCTTTGACCCTGGCC3′ (F), 5′GGCCAGGGTCAAAGATATTTGCATTAGGAAATTCTTTGTCATC3′ (R)]
Protein expression and purification
The expression and purification of WT CYP2C8 and its two variants were carried out based on several reported methods with some modifications (Wester et al. 2002, 2004; Schoch et al. 2004; Sun et al. 2009). Terrific broth media was seeded with an overnight culture of E. coli XL1-Blue containing the pCW2C8dH plasmid. When OD600 of the culture reached 0.5, 1 mM of isopropyl-β-d-thiogalactoside (IPTG) was added to induce CYP expression. The culture was incubated at 30°C for 48 h with shaking at 180 rpm. Cells were harvested by centrifugation at 4°C. The pellets were re-suspended in 20 mM KH2PO4–K2HPO4 (KPi) buffer containing 20% glycerol, 1 mM phenylmethanesulfonyl fluoride (PMSF), and 10 mM β-mercaptoethanol. Lysozyme was added and the cells were incubated at 4°C with stirring for 1 h. An equal volume of double distilled water of 4°C was added to the cell solution, and stirring was continued for an additional 30 min. The spheroplasts were pelleted by centrifugation at 5,000g for 15 min at 4°C; afterwards the supernatant was discarded, and the pellets were frozen in liquid nitrogen. The pellet was thawed, re-suspended in 500 mM KPi buffer, pH 7.4, containing 20% glycerol, 0.5 mM PMSF, 10 mM β-mercaptoethanol, and then was sonicated. Following centrifugation at 7,000g for 15 min at 4°C, 0.1% CHAPS was added and the material was incubated with stirring at 4°C for 30 min. Following the incubation, the material was centrifuged at 105,000g for 60 min at 4°C. The supernatant was incubated with Ni-NTA Agarose that was pre-equilibrated with buffer (I) containing 500 mM KPi, pH 7.4, 20% glycerol, 0.5 mM PMSF, and 10 mM β-mercaptoethanol. After 3 h, the Ni-NTA resin was loaded into a small chromatography column and was washed with buffer (I), buffer (II) containing 100 mM KPi, pH 7.4, 100 mM NaCl, 20% glycerol, 1 mM PMSF, 10 mM β-mercaptoethanol, 0.1% CHAPS, buffer (III) [buffer (II) with 50 mM glycine], and buffer (IV) [buffer (II) with 10 mM histidine] consecutively. The protein was eluted with buffer (V) containing 10 mM KPi, pH 7.4, 100 mM NaCl, 20% glycerol, 1 mM PMSF, 10 mM β-mercaptoethanol, 0.1% CHAPS, and 40 mM histidine. The fractions containing CYP2C8 as judged by its red-brown color were pooled. The protein was concentrated and the buffer exchanged with 5 mM KPi, pH 7.4, 20% glycerol, 1 mM EDTA, 1 mM PMSF, 0.2 mM DTT, and 0.1% CHAPS using an ultrafiltration device. The concentrated protein was then applied to a CM-Sepharose column that was pre-equilibrated with buffer (VI) containing 10 mM KPi, pH 7.4, 20% glycerol, 1 mM EDTA, 1 mM PMSF, 0.2 mM DTT, and 0.1% CHAPS, then was washed with the buffer (VI) without CHAPS. The CYP2C8 protein was eluted sharply in 50 mM KPi, 500 mM NaCl, 20% glycerol, 1 mM EDTA, and 0.2 mM DTT, at pH 7.4. The eluted protein was concentrated using an ultrafiltration device and then stored at −80°C.
Protein purity was analyzed by imaging Coomassie-Blue stained 15% SDS-PAGE gels. Heme concentrations were determined by the pyridine hemochromogen method, using an extinction coefficient of εR-O, 557–540 (22.1 mM−1 cm−1) (Berry and Trumpower 1987). The extinction coefficients for the substrate-free forms of the wild-type protein and its two variants at 416 nm were 108 mM−1 cm−1.
Spectroscopic characterization
Protein samples were prepared by incubating wild-type or mutant CYP2C8 in 50 mM KPi buffer, with 500 mM NaCl, at pH 7.4, containing 20% glycerol. The UV/Vis spectra of substrate-free ferric WT CYP2C8 and the two variants were recorded using a HP8453 UV/Vis spectrophotometer equipped with a temperature controller at 25°C. The Fe2+-CYP2C8-CO samples were prepared by bubbling the proteins with CO prior to reducing the proteins with sodium dithionite (Omura and Sato 1964). The vacuum far-UV CD spectra (190–270 nm) of substrate-free ferric WT CYP2C8 and the two variants were measured with CD spectrophotometer performed at the Beijing Synchrotron Radiation Facility (BSRF) beam line 4B8 in the Institute of High Energy Physics, Chinese Academy of Sciences. The cell chamber was 0.1 mm and filled with N2. The temperature was kept at 4°C. The near-UV and visible CD spectra (250–650 nm) of substrate-free ferric WT CYP2C8 and the two variants were measured on a Jasco J720 spectrometer at 25°C. The data were recorded every 0.2 nm at a rate of 200 nm/min. The data of CD spectra were represented as molar ellipticity, [θ].
Analysis of thermal stability
The Fe2+-CYP2C8-CO samples were prepared by bubbling the protein with CO prior to reducing the proteins with sodium dithionite (Omura and Sato 1964). Each sample of the Fe2+-CYP-CO form was incubated at 48°C for 0–15 min, and at various time points its percentage of P450 form relative to the zero time point value was recorded (Chen et al. 1997; Dickmann et al. 2004). All preparations were performed in triplicate. Half-lives for the thermally induced conversion of P450 to P420 states were determined according to first-order decay kinetics (Dickmann et al. 2004). Electronic absorption spectra were collected on a HP8453 UV/Vis spectrophotometer equipped with a temperature controller.
Paclitaxel binding study
UV/Vis spectra of paclitaxel binding upon WT CYP2C8 and the two variants were recorded on a HP8453A UV/Vis spectrophotometer equipped with a temperature controller. The protein samples in 50 mM KPi buffer, with 500 mM NaCl, at pH 7.4, containing 20% glycerol and 0.1% CHAPS, and stock paclitaxel in methanol were prepared. The concentration of purified CYP2C8 or its variant was 2 μM. The CYP2C8 solutions were incubated at 37°C for about 20 min until the absorbance at 416 nm was stable, then the substrate stock solution was titrated to less than 1% of the initial volume into a 1.0-cm quartz cuvette containing CYP2C8 solutions (sample cuvette), and equal amounts of the substrate stock solution added to another cuvette containing buffer only (reference cuvette). All preparations were performed in triplicate. The absorption spectra were recorded. The apparent dissociation constant (K d) for paclitaxel binding to CYP2C8s, and the extrapolated maximum spectral change (ΔA max), were estimated based on nonlinear least-squares regression fitting plots of ΔA against substrate concentration with the following quadratic equation (Koo et al. 2002; Schoch et al. 2008):
Where ΔA is the absorbance difference between the peak and trough of the difference spectrum; S is the total concentration of paclitaxel at any given point during the titration; P is the concentration of WT CYP2C8 or the two variants, and n is the substrate binding sites (n = 1 in the case of paclitaxel).
Assay of metabolic activity to paclitaxel
Paclitaxel metabolism assay with WT CYP2C8 and its two variants was performed, respectively, according to reported methods (Richardson et al. 1995; Dai et al. 2001). The assay mixture (100 μl), containing 10 pmol WT CYP2C8 or variant protein, 3 μg 1,2-Didodecanoyl-sn-glycero-3-phosphocholine (DLPC), 40 pmol cytochrome P450 reductase, 1 mM NADPH, and paclitaxel (from 0 to 200 μM), was prepared with 50 mM HEPES buffer, pH 7.4, which contained 30 mM MgCl2, 3 mM glutathione reduced, 0.01% CHAPS, and 10% glycerol. The mixture was pre-incubated at 37°C for 5 min and then initiated by the addition of 1 μl of 100 mM NADPH, and terminated after 15 min by four extractions with 60 μl of ethyl acetate. The extracts were evaporated to dryness under a gentle steam of nitrogen, and the residues were re-dissolved in 20 μl of 40% acetonitrile. The resulting solutions were then subjected to HPLC for analysis with a ZORBAX 300 SB-C18 column (4.6 mm × 25 cm, 5 μm particle size). The separation was performed under isocratic conditions using acetonitrile: water (40:60, v/v) as the mobile phase at a flow rate of 1.0 ml/min. The product was monitored at 235 nm. The identity of the metabolite was confirmed by chromatographic retention time and LCMS analysis. Unknown concentrations of metabolite were estimated by comparing the areas of metabolite peaks with linear calibration curves obtained for paclitaxel. The assumption was made that paclitaxel and 6α-hydroxy paclitaxel had similar absorption coefficients at 235 nm according to previous report (Richardson et al. 1995). All assays were performed at least in triplicates.
Stopped-flow kinetic studies
Pre-steady-state stopped-flow experiments were performed at 37°C with an SF-61 DX2 stopped-flow instrument (Hi-Tech, UK) installed in a glove box (MB 100, Braun, Germany) under pure N2 atmosphere. Electron transfer kinetics was monitored at 450 nm based on the formation of Fe2+-CYP2C8-CO. All protein samples and Na2S2O4 were in 10 mM KPi buffer, pH 7.4, containing 500 mM NaCl, and 20% glycerol. Before mixing, protein samples (10 μM) and Na2S2O4 (40 mM) were saturated with 1 atm CO. The rate constants (k) were obtained by fitting the absorbance change at 450 nm using KinetAsyst 2 software (Hi-Tech Co.) with first order equation.
Statistical analysis
Data were analyzed by Student’s t test with the SPSS statistical program (SPSS Inc. USA). Data were expressed as mean ± SD. P < 0.05 was considered statistically significant.
Results
Expression and purification
Recombinant WT CYP2C8 and its variants CYP2C8.4 and CYP2C8 P404A were successfully expressed in E. coli and purified by Ni-NTA column and CM-Sepharose column. SDS-PAGE gel analysis indicated high purity (~90% homogeneity) of these proteins (Fig. 2). The molecular mass of WT CYP2C8, measured by MALDI-TOF mass spectrometry, was 53,657 Da, which was close to the reported value of 53,979 Da.

15% SDS-PAGE gel stained with Coomassie blue. Lanes 1, 2, 3, 4 are CYP2C8 P404A, CYP2C8.4, WT CYP2C8, standard marker, respectively
Spectroscopic characterization
WT CYP2C8 and the two variants showed similar substrate-free ferric UV/Vis spectra with other CYPs. The Soret absorbance at 416 nm, α and β bands at 569 and 535 nm revealed the low-spin hexa-coordinated heme iron (Dawson et al. 1982; Martinis et al. 1996; Denisov et al. 2005). The Fe2+-CYP-CO form had the characteristic peak at 450 nm and α/β band at 551 nm similar to that of other CYPs (Haugen and Coon 1976; Dawson et al. 1982; McLean et al. 1998; Koo et al. 2000). CYP2C8.4 showed an obvious peak at 420 nm in the spectra of Fe2+-CYP-CO form indicating that part of this protein was in P420 state, an inactive form of CYP enzymes (Martinis et al. 1996; Chen et al. 1997) (SF1).
Far-UV CD spectra (Fig. 3) showed the secondary structures of CYP2C8.4 and CYP P404A, which were different from that of WT CYP2C8. The secondary structures of the proteins were analyzed by Jasco Secondary Structure Estimation [Version 1.54.03 (Build 1), JASCO Corporation]. Helix and sheet increased while turn and random decreased in the two variants compared to WT CYP2C8. Detailed data of changed structure were shown in Table 1.

The vacuum far-UV CD spectra of substrate-free ferric WT CYP2C8 and its variants. Buffer was 50 mM KPi, 500 mM NaCl, and 20% glycerol buffer, pH 7.4. Protein concentrations were 10 μM. The cell chamber was 0.1 mm and filled with N2. The temperature was kept at 4°C
Near-UV and visible CD spectra indicated some information of tertiary structure of the proteins. The two variants of CYP2C8 shared the similar profile of near-UV and visible CD spectra to that of WT CYP2C8; however, the CD Soret/delta (~[θ]420 nm/~[θ]350 nm) ratios were significantly different (Fig. 4) (WT CYP2C8, 4.67; CYP2C8.4, 2.30; CYP2C8 P404A, 3.20). Enhancement of the Soret trough relative to the delta band showed a more “open” active-site pocket in CYP, such as CYPLM-2 vs. CYPLM-4 (Wang and Kimura 1976; Andersson and Peterson 1995). These CD Soret/delta ratios of the two variants reflected the alteration of the protein tertiary structure by mutations.

The near-UV and visible CD spectra of substrate-free ferric WT CYP2C8 and its variants. Spectra were recorded in 50 mM KPi, 500 mM NaCl, and 20% glycerol buffer, pH 7.4, at 25°C in a 10 mm pathlength quartz cuvette at 17 μM protein concentration
Thermal stability
The P420 state is an inative form of these CYP enzymes, with a typical absorbance peak at 420 nm in the UV/Vis spectra of Fe2+-CYP-CO (Martinis et al. 1996; Chen et al. 1997). Various conditions, such as high temperature, can convert P450 state to P420 state, and the conversion reflects the generalized function loss of the enzyme (Dickmann et al. 2004). The conversion of P450 to P420 of WT CYP2C8 and its variants, CYP2C8.4 and CYP2C8 P404A, were investigated at 48°C, and the percentage of remaining P450 form as a function of time was shown in Fig. 5. The half-lives of these enzymes were ~8.3 ± 0.4, 6.0 ± 0.5 (P < 0.01), 6.8 ± 0.5 (P < 0.05) min for WT CYP2C8, CYP2C8.4, and CYP2C8 P404A, respectively. Thermal stability of CYP2C8.4 and CYP2C8 P404A were significantly different from that of WT CYP2C8. Both of the variants were less thermally stable than WT CYP2C8.

Percentage of remaining P450 state of WT CYP2C8 and its variants as a function of time. The Fe2+-CYP2C8-CO samples were prepared by bubbling the protein with CO in 50 mM KPi buffer, 500 mM NaCl, pH 7.4, containing 20% glycerol prior to reducing the proteins with sodium dithionite. Each sample of the Fe2+-CYP-CO form was incubated at 48°C for 0–15 min, and at various time points its percentage of P450 form (value of 450 nm) relative to the zero time point value was recorded
Paclitaxel binding ability
Paclitaxel was the prototypical substrate of CYP2C8, and was commonly used in vitro to study the metabolic activity of the enzyme. The difference spectrum analysis of substrate binding showed that paclitaxel induced the WT CYP2C8, CYP2C8.4, and CYP P404A to form a type I difference spectrum with a peak at near 380 nm and a trough at near 420 nm (Fig. 6) (Jefcoate 1978). The substrate binding was monitored by ΔA, the absorbance difference between the peak and trough of the difference spectrum (Estabrook and Werringloer 1978). The paclitaxel-binding ability was shown in Table 2. K d of WT CYP2C8, CYP2C8.4, and CYP P404A were 139.5 ± 10.3 μM, 67.9 ± 3.6 μM, and 245.1 ± 41.8 μM, respectively. These indicated that paclitaxel-binding ability of CYP2C8.4 was about two times stronger, while that of CYP P404A was about two times weaker than that of WT CYP2C8.

The difference spectrum of 30 μM paclitaxel binding to WT CYP2C8 and its variants
Paclitaxel metabolic activity
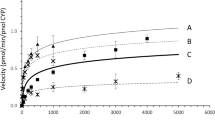
The paclitaxel metabolic activity of WT CYP2C8 and the two variants, CYP2C8.4 and CYP2C8 P404A, were assayed by monitoring the product 6α-hydroxypaclitaxel at 235 nm with 300 SB-C18 columns on HPLC. The retention times were ~18 min for paclitaxel and ~9 min for 6α-hydroxypaclitaxel. The identity of the metabolite was confirmed by chromatographic retention time and LCMS analysis (SF3-SF5). LCMS data (see supporting information) were consistent with the previous report of paclitaxel and 6α-hydroxy paclitaxel (Kumar et al. 1994; Poon et al. 1996). The concentrations of metabolite were estimated by comparing the areas of metabolite peaks with linear calibration curves obtained for paclitaxel because paclitaxel and 6α-hydroxypaclitaxel had similar absorption coefficients at 235 nm according to previous report (Richardson et al. 1995). LCMS data was consistent with the previous report of paclitaxel and 6α-hydroxypaclitaxel (SF4 and SF5) (Kumar et al. 1994; Poon et al. 1996). Ion transitions of m/z 854 → 569 for paclitaxel and m/z 870 → 585 for 6α-hydroxypaclitaxel. Representative Michaelis–Menten kinetics for 6α-hydroxylation catalyzed by these enzymes was shown in Fig. 7. The solid lines indicated the fitting of kinetic data with the Michaelis–Menten equation by nonlinear regression. The metabolic activity of paclitaxel by CYP2C8.4 and CYP2C8 P404A were about 25 and 30% of that of wild-type CYP2C8, respectively. The kinetic parameters are presented in Table 3.

Representative Michaelis–Menten kinetics of paclitaxel α-hydroxylation by WT CYP2C8 and its variants. The solid line indicates fitting of data to the Michaelis–Menten equation by nonlinear regression
Stopped-flow kinetic studies
Electron transfer from Na2S2O4 to the heme Fe3+ ion of WT CYP2C8 or its polymorphic enzymes was successfully monitored at 450 nm with Stopped-flow (SF2). The rate constants are 0.47, 0.55, and 0.45 s−1 for WT CYP2C8, CYP2C8.4, and CYP2C8 P404A, respectively. The maximum absorbance changes at 450 nm (ΔA max) are 0.36, 0.32, and 0.35 AU for WT CYP2C8, CYP2C8.4, and CYP 2C8 P404A, respectively. The rate constant and reduction extent of the variants are close to those of WT CYP2C8. The results indicated that the heme center of CYP2C8.4 or CYP2C8 P404A was probably not disturbed by mutation.
Discussion
The objective of this study was to investigate the structure–function relationship of two polymorphic enzymes, CYP2C8.4 and CYP2C8 P404A. Through genetics (site-directed mutagenesis), biochemistry, and biophysics approaches, we purified WT CYP2C8 and the two polymorphic enzymes, and then compared the properties and metabolic activities of the resulting polymorphic mutants to the WT CYP2C8. Based on the results above we try to decipher why the metabolic activity of paclitaxel by CYP2C8.4 and CYP2C8 P404A were just about 30% of that by WT CYP2C8.
CD spectra indicated that the secondary and tertiary structures of the two variants greatly changed due to their respective amino acid mutations. The mutant CYP2C8.4 (Ile264 M) on helix H altered residue hydropathy from 4.5 (Ile) to 1.9 (Met), while the variant Pro404A on the loop between helix K’ and L (Fig. 1) changed relative residue volume from 123.4 Å3 (Pro) to 91.5 Å3 (Ala), respectively (Kyte and Doolittle 1982; Pontius et al. 1996). The structural changes resulted in loss of function. The conversion half-life from P450 state to P420 state for both the polymorphic enzymes was shorter than that of WT CYP2C8, and the variants were less thermally stable than WT CYP2C8. CYP2C8.4 showed an obvious P420 state, as an inactive form of the enzyme, in reduced-CO spectrum at room temperature, and the P420 state was also observed in unpurified CYP2C8.4 in membrane of bacterial (Singh et al. 2008), most likely because of the weakening of iron-thiolate interaction between the heme and coordinated Cys435 in this polymorphic enzyme (Tian et al. 1995; Udit et al. 2006).
The paclitaxel-binding ability of CYP2C8.4 was about two times stronger than that of WT CYP2C8, while paclitaxel-binding ability of CYP P404A was about 50% weaker than that of WT CYP2C8. The K d of CYP2C8.4 is about half of that of WT CYP2C8, but K m of CYP2C8.4 is not significantly different from that of WT CYP2C8. The smaller K d does not correspond to the smaller K m, indicating that the enhanced substrate-binding ability does not help to increase enzyme activity. This is likely due to the product 6α-hydroxypaclitaxel hard dissociation from the transformed active-site pocket, which retarded the metabolic activity of CYP2C8.4. CYP P404A showed 30% 6α-hydroxylase activity of that by WT CYP2C8. The depressed paclitaxel metabolic activity of CYP2C8 P404A was mainly due to weaker paclitaxel binding ability. A possible turn-to-helix change in the secondary structure of CYP2C8 P404A was predicted by Chou-Fasman secondary structure prediction software (Soyama et al. 2001), and this prediction was confirmed by our CD studies. The far-UV CD spectra showed that in the secondary structure of CYP P404A turn decreased 5.4% and helix increased 5.7%.
The substrate recognition sites (SRS) predetermine CYP substrate specificity (Denisov et al. 2005). Both I264 and P404 are non-SRS residues, and they locate far from heme and substrate binding sites. Our study showed that residue substitution at I264 and P404 in CYP2C8 resulted in structural and functional changes. These two residues may involve in “long-range” interactions in the surrounding structure to alter protein motif(s) or the key sites in the motif(s). Numerous examples have been reported to demonstrate that non-SRS residues are important for proper functions and folding of enzymes (Krone et al. 2005; Zhang et al. 2002; Hlavica 2006).
In this study we used purified proteins, and found that the metabolic activities of paclitaxel by CYP2C8.4 and CYP2C8 P404A were about 25 and 30% of that of WT CYP2C8, respectively. Metabolism of paclitaxel by E. coli membrance fractions coexpressing CYP2C8 variants and P450 reductase has been studied. A significantly lower level (~15%) of metabolic activity of paclitaxel by CYP2C8.4 was reported by Rajinder Singh et al. (2008). In Rajinder’s study the substrate paclitaxel concentration was in the range 15–100 μM, and lower than 15 μM was not performed due to the limit of the HPLC system. However, using lower than 15 μM substrate paclitaxel, Rowbotham et al. (2010) did not observe significant activity difference for the CYP2C8.3 or CYP2C8.4 enzymes with respect to paclitaxel 6α-hydroxylase kinetics compared with WT CYP2C8. This kind of inconsistence took place on CYP2C8.3 too. Dai et al. (2001) reported its paclitaxel metabolic activity was at about 15% of that of WT CYP2C8, while Akiko Soyama et al. (2001) reported that it was about 75%. It is quite complicated to compare these results obtained with different experiment systems.
The two variants, CYP2C8.4 and CYP2C8 P404A, exhibited depressed metabolic activity of paclitaxel, the prototypical substrate of CYP2C8, emphasizing that the difference between the variants and WT CYP2C8 should be carefully considered in the treatment of patients with these polymorphic genes when using the drugs metabolized by CYP2C8.
References
Andersson LA, Peterson JA (1995) Active-site analysis of ferric P450 enzymes: hydrogen-bonding effects on the circular dichroism spectra. Biochem Biophys Res Commun 211:389–395
Bahadur N, Leathart JB, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R, Houdt JV, Hendrickx J, Mannens G, Bohets H, Williams FM, Armstrong M, Crespi CL, Daly AK (2002) CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol 64:1579–1589
Berry EA, Trumpower BL (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem 161:1–15
Chen CD, Doray B, Kemper B (1997) Efficient assembly of functional cytochrome P450 2C2 requires a spacer sequence between the N-terminal signal anchor and catalytic domains. J Biol Chem 272:22891–22897
Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, Goldstein JA (2001) Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11:597–607
Dawson JH, Andersson LA, Sono M (1982) Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J Biol Chem 257:3606–3617
Denisov IG, Makris TM, Sligar SG, Schlichting I (2005) Structure and chemistry of cytochrome P450. Chem Rev 105:2253–2277
Dickmann LJ, Locuson CW, Jones JP, Rettie AE (2004) Differential roles of Arg97, Asp293, and Arg108 in enzyme stability and substrate specificity of CYP2C9. Mol Pharmacol 65:842–850
Estabrook RW, Werringloer J (1978) The measurement of difference spectra: application to the cytochromes of microsomes. Methods Enzymol 52:212–220
Hanioka N, Matsumoto K, Saito Y, Narimatsu S (2010) Functional characterization of CYP2C8.13 and CYP2C8.14: catalytic activities toward paclitaxel. Basic Clin Pharmacol Toxicol. doi:10.1111/j.1742-7843.2010.00543.x
Haugen DA, Coon MJ (1976) Properties of electrophoretically homogeneous phenobarbital-inducible and beta-naphthoflavone-inducible forms of liver microsomal cytochrome P-450. J Biol Chem 251:7929–7939
Hichiya H, Tanaka-Kagawa T, Soyama A, Jinno H, Koyano S, Katori N, Matsushima E, Uchiyama S, Tokunaga H, Kimura H, Minami N, Katoh M, Sugai K, Goto Y, Tamura T, Yamamoto N, Ohe Y, Kunitoh H, Nokihara H, Yoshida T, Minami H, Saijo N, Ando M, Ozawa S, Saito Y, Sawada J (2005) Functional characterization of five novel CYP2C8 variants, G171S, R186X, R186G, K247R, and K383 N, found in a Japanese population. Drug Metab Dispos 33:630–636
Hlavica P (2006) Functional interaction of nitrogenous organic bases with cytochrome P450: a critical assessment and update of substrate features and predicted key active-site elements steering the access, binding, and orientation of amines. Biochim Biophys Acta 1764:645–670
Ishikawa C, Ozaki H, Nakajima T, Ishii T, Kanai S, Anjo S, Shirai K, Inoue I (2004) A frameshift variant of CYP2C8 was identified in a patient who suffered from rhabdomyolysis after administration of cerivastatin. J Hum Genet 49:582–585
Jefcoate CR (1978) Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol 52:258–279
Kerr BM, Thummel KE, Wurden CJ, Klein SM, Kroetz DL, Gonzalez FJ, Levy RH (1994) Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10, 11-epoxide formation. Biochem Pharmacol 47:1969–1979
Koo LS, Tschirret-Guth RA, Straub WE, Moenne-Loccoz P, Loehr TM, Ortiz de Montellano PR (2000) The active site of the thermophilic CYP119 from Sulfolobus solfataricus. J Biol Chem 275:14112–14123
Koo LS, Immoos CE, Cohen MS, Farmer PJ, Ortiz de Montellano PR (2002) Enhanced electron transfer and lauric acid hydroxylation by site-directed mutagenesis of CYP119. J Am Chem Soc 124:5684–5691
Krone N, Riepe FG, Grotzinger J, Partsch CJ, Bramswig J, Sippell WG (2005) The residue E351 is essential for the activity of human 21-hydroxylase: evidence from a naturally occurring novel point mutation compared with artificial mutants generated by single amino acid substitutions. J Mol Med 83:561–568
Kumar GN, Oatis JE Jr, Thornburg KR, Heldrich FJ, Hazard ES 3rd, Walle T (1994) 6 alpha-Hydroxytaxol: isolation and identification of the major metabolite of taxol in human liver microsomes. Drug Metab Dispos 22:177–179
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132
Lee VH, Sporty JL, Fandy TE (2001) Pharmacogenomics of drug transporters: the next drug delivery challenge. Adv Drug Deliv Rev 50 (Suppl 1):S33–S40
Martinis SA, Blanke SR, Hager LP, Sligar SG, Hoa GH, Rux JJ, Dawson JH (1996) Probing the heme iron coordination structure of pressure-induced cytochrome P420cam. Biochemistry 35:14530–14536
McLean MA, Maves SA, Weiss KE, Krepich S, Sligar SG (1998) Characterization of a cytochrome P450 from the acidothermophilic archaea Sulfolobus solfataricus. Biochem Biophys Res Commun 252:166–172
Ohyama K, Nakajima M, Nakamura S, Shimada N, Yamazaki H, Yokoi T (2000) A significant role of human cytochrome P450 2C8 in amiodarone N-deethylation: an approach to predict the contribution with relative activity factor. Drug Metab Dispos 28:1303–1310
Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378
Plasilova M, Stoilov I, Sarfarazi M, Kadasi L, Ferakova E, Ferak V (1999) Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J Med Genet 36:290–294
Pontius J, Richelle J, Wodak SJ (1996) Deviations from standard atomic volumes as a quality measure for protein crystal structures. J Mol Biol 264:121–136
Poon GK, Wade J, Bloomer J, Clarke SE, Maltas J (1996) Rapid screening of taxol metabolites in human microsomes by liquid chromatography/electrospray ionization-mass spectrometry. Rapid Commun Mass Spectrom 10:1165–1168
Rahman A, Korzekwa KR, Grogan J, Gonzalez FJ, Harris JW (1994) Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Res 54:5543–5546
Rendic S, Di Carlo FJ (1997) Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev 29:413–580
Richardson TH, Jung F, Griffin KJ, Wester M, Raucy JL, Kemper B, Bornheim LM, Hassett C, Omiecinski CJ, Johnson EF (1995) A universal approach to the expression of human and rabbit cytochrome P450s of the 2C subfamily in Escherichia coli. Arch Biochem Biophys 323:87–96
Rowbotham SE, Boddy AV, Redfern CP, Veal GJ, Daly AK (2010) Relevance of nonsynonymous CYP2C8 polymorphisms to 13-cis retinoic acid and paclitaxel hydroxylation. Drug Metab Dispos 38:1261–1266
Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF (2004) Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J Biol Chem 279:9497–9503
Schoch GA, Yano JK, Sansen S, Dansette PM, Stout CD, Johnson EF (2008) Determinants of cytochrome P450 2C8 substrate binding: structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J Biol Chem 283:17227–17237
Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423
Singh R, Ting JG, Pan Y, Teh LK, Ismail R, Ong CE (2008) Functional role of Ile264 in CYP2C8: mutations affect haem incorporation and catalytic activity. Drug Metab Pharmacokinet 23:165–174
Sonnichsen DS, Liu Q, Schuetz EG, Schuetz JD, Pappo A, Relling MV (1995) Variability in human cytochrome P450 paclitaxel metabolism. J Pharmacol Exp Ther 275:566–575
Soyama A, Saito Y, Hanioka N, Murayama N, Nakajima O, Katori N, Ishida S, Sai K, Ozawa S, Sawada JI (2001) Non-synonymous single nucleotide alterations found in the CYP2C8 gene result in reduced in vitro paclitaxel metabolism. Biol Pharm Bull 24:1427–1430
Soyama A, Hanioka N, Saito Y, Murayama N, Ando M, Ozawa S, Sawada J (2002) Amiodarone N-deethylation by CYP2C8 and its variants, CYP2C8*3 and CYP2C8 P404A. Pharmacol Toxicol 91:174–178
Stoilov I, Akarsu AN, Alozie I, Child A, Barsoum-Homsy M, Turacli ME, Or M, Lewis RA, Ozdemir N, Brice G, Aktan SG, Chevrette L, Coca-Prados M, Sarfarazi M (1998) Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am J Hum Genet 62:573–584
Sun L, Wang ZH, Ni FY, Tan XS, Huang ZX (2009) The role of Ile476 in the structural stability and substrate binding of human cytochrome P450 2C8. Protein J. doi:10.1007/s10930-009-9218-8
Tian WD, Wells AV, Champion PM, Di Primo C, Gerber N, Sligar SG (1995) Measurements of CO geminate recombination in cytochromes P450 and P420. J Biol Chem 270:8673–8679
Totah RA, Rettie AE (2005) Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin Pharmacol Ther 77:341–352
Udit AK, Hagen KD, Goldman PJ, Star A, Gillan JM, Gray HB, Hill MG (2006) Spectroscopy and electrochemistry of cytochrome P450 BM3-surfactant film assemblies. J Am Chem Soc 128:10320–10325
Wang HP, Kimura T (1976) Purification and characterization of adrenal cortex mitochondrial cytochrome P-450 specific for cholesterol side chain cleavage activity. J Biol Chem 251:6068–6074
Wester MR, Stout CD, Johnson EF (2002) Purification and crystallization of N-terminally truncated forms of microsomal cytochrome P450 2C5. Methods Enzymol 357:73–79
Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF (2004) The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem 279:35630–35637
Yamazaki H, Shibata A, Suzuki M, Nakajima M, Shimada N, Guengerich FP, Yokoi T (1999) Oxidation of troglitazone to a quinone-type metabolite catalyzed by cytochrome P-450 2C8 and P-450 3A4 in human liver microsomes. Drug Metab Dispos 27:1260–1266
Yasar U, Bennet AM, Eliasson E, Lundgren S, Wiman B, De Faire U, Rane A (2003) Allelic variants of cytochromes P450 2C modify the risk for acute myocardial infarction. Pharmacogenetics 13:715–720
Zhang X, Su T, Zhang QY, Gu J, Caggana M, Li H, Ding X (2002) Genetic polymorphisms of the human CYP2A13 gene: identification of single-nucleotide polymorphisms and functional characterization of an Arg257Cys variant. J Pharmacol Exp Ther 302:416–423
Acknowledgments
We thank Prof. E.F. Johnson (The Scripps Research Institute, California, USA) for the generous gift of the pCWOri + plasmid, encoding CYP 2C8dH and Prof. J.M. Chen (Fudan University, Shanghai, China) for the generous support of paclitaxel. This work is supported in part by Shanghai Pujiang Talent Project (08PJ14017), the National Science Foundation of China (20771029), the Shanghai Leading Academic Discipline Project (B108), the Program for the Platform of new medicine creation (NO. 2009ZX09301-011), the Specialized Research Fund for the Doctoral Program of Higher Education, and Beijing Synchrotron Radiation Facility (BSRF) in the Institute of High Energy Physics, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
SF1
. UV/Vis spectra of the ferrous-CO states of WT CYP2C8 and its variants. Spectra were recorded in 50 mM KPi, 500 mM NaCl, and 20% glycerol buffer, pH 7.4 at 25°C, proteins concentration were 5 μM (TIFF 54 kb)
SF2
. Stopped-flow kinetics of electron transfer of WT CYP2C8 and its variants when reduced by Na2S2O4 (TIFF 490 kb)
SF3
. Reversed-phase HPLC of paclitaxel and its metabolite. (A) There was no NADPH in the reaction system. (B) Reaction started by the addition of NADPH. Conditions: see experimental section (TIFF 455 kb)
SF4
. The mass spectra of a band (paclitaxel) from the positive-ion MS analysis (TIFF 92 kb)
SF5
. The mass spectra of b band (6α-OH-paclitaxel) from the positive-ion MS analysis (TIFF 84 kb)
SF6
. Eadie-Hofstee plot of paclitaxel α-hydroxylation by WT CYP2C8 and its variants. The solid line indicates fitting of data to the Eadie-Hofstee equation by linear regression (TIFF 490 kb)
Rights and permissions
About this article
Cite this article
Jiang, H., Zhong, F., Sun, L. et al. Structural and functional insights into polymorphic enzymes of cytochrome P450 2C8. Amino Acids 40, 1195–1204 (2011). https://doi.org/10.1007/s00726-010-0743-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0743-8