
Abstract
Background
A simple method to predict drug–drug interactions mediated by cytochrome P450 enzymes (CYPs) on the basis of in vivo data has been previously applied for several CYP isoforms but not for CYP1A2. The objective of this study was to extend this method to drug interactions caused by CYP1A2 inhibitors and inducers.
Methods
First, initial estimates of the model parameters were obtained using data from the literature. Then, an external validation of these initial estimates was performed by comparing model-based predicted area under the concentration–time curve (AUC) ratios with observations not used in the initial estimation. Third, refined estimates of the model parameters were obtained by Bayesian orthogonal regression using Winbugs software, and predicted AUC ratios were compared with all available observations. Finally, predicted AUC ratios for all possible substrates–inhibitors and substrates–inducers were computed.
Results
A total of 100 AUC ratios were retrieved from the literature. Model parameters were estimated for 19 CYP1A2 substrate drugs, 26 inhibitors and seven inducers, including tobacco smoking. In the external validation, the mean prediction error of the AUC ratios was −0.22, while the mean absolute error was 0.97 (37 %). After the Bayesian estimation step, the mean prediction error was 0.11, while the mean absolute error was 0.43 (22 %). The AUC ratios for 625 possible interactions were computed.
Conclusion
This analysis provides insights into the interaction profiles of drugs poorly studied so far and can help to identify and manage significant interactions in clinical practice. Those results are now available to the community via a web tool (http://www.ddi-predictor.org).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In this study, a quantitative approach was applied to predict drug interactions mediated by cytochrome P450 (CYP) 1A2, and provided good predictive performance. |
The sensitivity of 19 CYP1A2 substrate drugs and the potencies of 26 inhibitors and seven inducers were quantified. |
The magnitudes of 625 possible drug interactions mediated by CYP1A2 were computed. |
1 Introduction
The human cytochrome P450 (CYP) enzyme CYP1A2 plays an important role in the metabolism of drugs, as well as other xenobiotics and endogenous compounds. It is one of the major CYP enzymes and accounts for approximately 13 % of the total content of this enzyme group in the human liver [1]. CYP1A2 and other members of the CYP1A family are involved in the metabolism of about 9 % of marketed drugs [2].
Its contribution to caffeine metabolism is well documented, and caffeine has been extensively used as a probe drug for assessing CYP1A2 activity in humans [3]. Other important substrate drugs notably include theophylline, melatonin and clozapine. CYP1A2 substrate drugs may undergo inhibition or induction via this enzyme pathway. Because some CYP1A2 inhibitors (e.g. quinolone antibiotics) and inducers (e.g. tobacco smoking) are widely used agents, drug interactions mediated by CYP1A2 are a relevant issue in clinical practice. However, only a small proportion of all possible interactions have been studied so far. Thus, prediction of CYP1A2-mediated drug interactions is desirable.
Ohno et al. [4] developed a method to forecast the magnitude of CYP3A4-mediated metabolic interactions on the basis of in vivo data. It has been applied to both CYP3A4 inhibition and induction [4, 5]. Since then, our group has refined and applied the approach to interactions mediated by other CYP isoforms, including CYP2D6, CYP2C9 and CYP2C19 [6–8]. The goal of this study was to extend this method to interactions caused by CYP1A2 inhibitors and inducers.
2 Methods
We derived the frameworks from Ohno’s group and from ours to model CYP1A2-mediated drug interactions [4–6]. Details on the physiological rationale and demonstration of the equations presented below are available in those publications.
The general metric used to quantify drug interactions is an area under the concentration–time curve (AUC) ratio. For drug interactions by inhibition, it is the ratio of the altered substrate drug AUC* to the reference AUC measured in patients without interaction, as shown in Eq. 1 [4, 6]:
where AUC is the reference AUC of the substrate drug when administered alone; AUC* is the AUC of the substrate drug when co-administered with a CYP1A2 inhibitor; CRCYP1A2 is the contribution ratio (i.e. the fraction of the substrate drug’s apparent oral clearance due to metabolism by CYP1A2); and IRCYP1A2 is the inhibition ratio for the inhibitor. IR reflects the CYP1A2 inhibition potency of the inhibitor drug. Both CRCYP1A2 and IRCYP1A2 values may range from 0 to 1. As a result, the AUC ratio for inhibition is a real positive number with a minimum value of 1 (no inhibition effect).
For drug interactions by induction, the AUC ratio is as shown in Eq. 2 [4]:
where AUC is the reference AUC of the substrate drug when it is administered alone; AUC* is the AUC of the substrate drug when it is co-administered with a CYP1A2 inducer; CRCYP1A2 is the contribution ratio (see Eq. 1); and ICCYP1A2 is the induction potency of the inducer (IC stands for ‘increase in drug clearance’). The ICCYP1A2 of an inducer is a real positive number. In the case of drug induction, the AUC ratio may range from 0 to 1; 1 indicates no induction effect.
The main objectives of this study were to estimate the parameter values of Eqs. 1 and 2 (i.e. CRCYP1A2, IRCYP1A2 and ICCYP1A2 values) for identified CYP1A2 substrates and interactors, and then to derive quantitative predictions for all possible interactions involving those drugs. A three-step approach was carried out. First, initial estimates of CRCYP1A2, IRCYP1A2 and ICCYP1A2 values were obtained from published drug interaction studies. Second, an external validation of Eqs. 1 and 2 was performed using published data not used in the first step. Third, model-based final estimates of CRCYP1A2, IRCYP1A2 and ICCYP1A2, as well as all possible AUC ratio values, were obtained by using a Bayesian regression method, and the predictive performance of the approach was assessed using all available data.
2.1 Step 1: Initial Estimation of Model Parameters
A literature search was performed to retrieve data on published drug interactions mediated by CYP1A2. The references were extracted from PubMed and Google Scholar. The main keywords were ‘pharmacokinetics’, ‘drug interactions’, ‘cytochrome P450 CYP1A2’ and the names of individual drugs. Some data were also retrieved from the summaries of product characteristics of individual drugs, provided by the European Medicines Agency (EMA). Articles published up to December 2014 were included in the analysis. As the model is valid only for drugs administered orally, data obtained for drugs administered via other routes were excluded. Only published data from humans, with oral AUC or oral clearance values, were included in the analysis. Studies providing only trough concentrations or single concentrations measured at another time point were excluded. When only clearance values were provided in the publications, the reciprocal of the clearance ratio (i.e. CL*/CL, where CL* and CL are the drug oral clearances with and without drug interactions, respectively) was used to calculate the AUC ratio (AUC*/AUC). Because CYP1A2 metabolism may be influenced by tobacco smoking, drug–drug interaction data obtained only in subgroups of smokers were excluded, except those used to quantify the effect of tobacco smoking as an inducer agent.
The values of CRCYP1A2, IRCYP1A2 and ICCYP1A2 were calculated from a subset of published AUC ratios, by rearranging Eqs. 1 and 2 as Eqs. 3, 4 and 5, as follows:
An initial CRCYP1A2 value was necessary to calculate the initial estimates of the IRCYP1A2 values. The value of 0.95 reported for the proportion of caffeine clearance mediated by CYP1A2 by Carrillo and Benitez [9] was used. As caffeine is a CYP1A2 probe drug used in many drug–drug interaction studies, this initial value allowed us to estimate IRCYP1A2 and ICCYP1A2 parameters for a number of other drugs.
When several published AUC ratios were available for a given substrate drug, data from studies with multiple doses of the inhibitor were preferred to single-dose studies, as these conditions better reflected the full magnitude of the interaction. When studies had similar designs, the median AUC ratio was selected for initial parameter estimation. All data that had not been used during the first step were considered in the external validation step.
2.2 Step 2: External Validation
The AUC ratios predicted by the initial estimates of the CRCYP1A2, IRCYP1A2 and ICCYP1A2 values in Eqs. 1 and 2 for inhibition and induction, respectively, were compared with AUC ratios observed in a second set of published data.
Predicted AUC ratios were plotted versus observed AUC ratios. Predicted values in the range of 50–200 % of the observed ratio were considered acceptable, in accordance with previous works [4, 5]. The bias and imprecision of the predicted AUC ratios were calculated as the mean prediction errors (i.e. the predicted minus the observed AUC ratios) and the mean absolute prediction errors, respectively.
2.3 Step 3: Final Estimation of Model Parameters and AUC Ratios
Bayesian orthogonal regression implemented in Winbugs software (version 1.4.3) was performed to obtain final estimates of the CRCYP1A2, IRCYP1A2 and ICCYP1A2 values, as well as the AUC ratios [10]. This approach was carried out to take into account the inherent variability in those quantities that is not adequately addressed when only average estimates from published data are used. The Winbugs software provided Bayesian posterior distribution and confidence limits for all of those quantities, based on the model, data and prior distribution that were imported into the software. This approach has been described in detail in previous publications by our group [6, 7].
Briefly, for CYP1A2 inhibition, for each substrate i and inhibitor j, the predicted AUC ratio was coded in Winbugs as shown in Eq. 6 (the subscript CYP1A2 is omitted for ease of reading):
where pred ij and AUCratio ij are the predicted and observed AUC ratios for each (CR i , IR j ) pair, respectively; and CRZ i and IRZ j are the refined (Bayesian posterior) estimates of the contribution ratio and inhibition ratio, respectively. The parameter tauAUC is the precision (i.e. the reciprocal of the variance) of the AUC distribution. We assumed normal distribution for AUC ratios and logistic distribution for CR and IR values. The mean of each distribution was set to the initial estimates found in step 1. We assumed that the precision of these distributions, denoted tau, followed a gamma distribution as follows: tauCR ~ G(4, 1), tauIR ~ G(4, 1) and tauAUC ~ G(0.1, 2.5) for CR, IR and AUC distributions, respectively. These gamma distributions were set so that the expected standard deviations of CR and IR on the logit scale and of the AUC ratio were 0.5, 0.5 and 5, respectively. In Bayesian statistics, the gamma distribution G(α, β) is commonly used as a prior for the precision (i.e. the reciprocal of the variance) of a normal distribution [11].
For CYP1A2 induction, a similar Bayesian regression approach was carried out, except that only ICCYP1A2 values and AUC ratios were estimated. CRCYP1A2 values were fixed at their point estimates obtained from data on CYP1A2 inhibition (CRZ i ). For each substrate i and inducer k, the regression equation was as shown in Eq. 7:
where pred ik and AUCratio ik are the predicted and observed AUC ratios for each (CR i , IC k ) pair, respectively; CRZ i is the final point estimate of the contribution ratio; and ICZ k is the refined (Bayesian posterior) estimate of the parameter ICCYP1A2. Because ICCYP1A2 is a real positive number, we assumed log-normal distribution for this parameter. The precision tauAUC was set as for inhibition (tauAUC ~ G(0.1, 2.5)).
The posterior distributions of the AUC ratios, CRCYP1A2, IRCYP1A2, and ICCYP1A2 were obtained by Monte Carlo Markov chain simulation within Winbugs. Convergence was assessed by examining trace and history plots of the parameter sample values versus the number of iterations to check the stability of the posterior distributions. The precision of the posterior estimates was assessed using the Monte Carlo error provided by Winbugs. A Monte Carlo error on a parameter estimate of less than 5 % of the sample standard deviation was considered acceptable [10].
The means of the posterior distributions were used as point estimates of the CRCYP1A2, IRCYP1A2, ICCYP1A2 and AUC ratios. A 90 % confidence interval was calculated for each quantity as the interval between the 5th and the 95th percentiles of the posterior distribution of each variable.
In order to assess the predictive performance of the final model parameter estimates, we examined the plot of predicted (mean point estimates) versus observed AUC ratios and calculated the bias and imprecision of the AUC ratio predictions, as was done in the external validation step.
In the end, the final estimates of CRCYP1A2, IRCYP1A2 and ICCYP1A2 were used to derive AUC ratios for all possible substrate–inhibitor and substrate–inducer pairs, using Eqs. 1 and 2, respectively. This provided insights into the potential magnitude of CYP1A2-mediated drug interactions for unpublished and rare cases of interactions.
3 Results
3.1 Initial Estimation
A total of 100 AUC ratios reported in 78 studies were retrieved from the literature search. Initial values of CRCYP1A2, IRCYP1A2 and ICCYP1A2 were calculated for 19 CYP1A2 substrate drugs, 26 inhibitors and seven inducers, respectively [9, 12–57]. Those initial estimates, as well as the corresponding references, are given in Tables 1, 2 and 3, respectively. Many studies used oral caffeine or theophylline as CYP1A2 probe drugs to assess potential inhibitors and inducers. Four drugs were identified as highly sensitive CYP1A2 substrates, with an estimated 100 % of oral clearance being mediated by CYP1A2: agomelatine, melatonin, tacrine and tizanidine. Actually, for those drugs, the initial estimates of CRCYP1A2 derived from published data were greater than 1, which is not compatible with the model theory, so the value was rounded down to 1. A number of quinolone antibiotics were identified as CYP1A2 inhibitors, with ciprofloxacin and enoxacin appearing to be the most potent. Inducers notably included carbamazepine and rifampicin, which are inducers of other CYP isoforms, and tobacco smoking.
3.2 External Validation
Fifty AUC ratios not used in the initial step were available for external validation [20, 26, 31, 36, 42, 49, 56, 58–86].
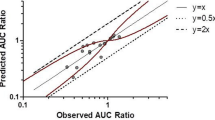
The plot of predicted versus observed AUC ratios is shown in Fig. 1. Seven predicted AUC ratios from six different studies were outside the 50–200 % range of the observed ratios. Among those, four ratios were overpredicted (erlotinib–tobacco smoking and theophylline–rofecoxib [three AUC ratios from the same study]) and three ratios were underpredicted (tizanidine–ciprofloxacin, melatonin–ethinylestradiol and melatonin–fluvoxamine). There was no obvious reason for these discrepancies. However, the model provided acceptable predictive performance at that step, with bias of −0.22 and imprecision of 0.93 (37 %).

Predicted versus observed area under the concentration–time curve (AUC) ratios for cytochrome P450 (CYP) 1A2-mediated drug interactions in the external validation step
3.3 Final Estimation
The final mean point estimates of the CRCYP1A2, IRCYP1A2 and ICCYP1A2 values provided by Winbugs, along with their 90 % confidence intervals, are given in Tables 4, 5 and 6, respectively. The accuracy of the parameter estimates was good, with Monte Carlo errors of less than 2 %, except for two CRCYP1A2 estimates (agomelatine 5.8 %, tizanidine 4.5 %) and one IRCYP1A2 estimate (fluvoxamine 6 %), which were very close to the asymptotic value of 1.
Melatonin and the melatonin receptor agonist agomelatine were found to be very sensitive CYP1A2 substrates, as was tizanidine, with CRCYP1A2 values close to the maximum value of 1. Among currently marketed drugs, the most potent CYP1A2 inhibitors were fluvoxamine, rofecoxib, enoxacin and ciprofloxacin, with IRCYP1A2 values greater than 0.90. The inducers with the highest inducing potency were carbamazepine, rifampicin and tobacco smoking, but their predicted effects remained moderate, with mean predicted AUC ratios not less than 0.4, even for the most sensitive substrate (agomelatine).
The relationship between the observed AUC ratios and the final predictions is plotted in Fig. 2. Only three outliers were observed. Those corresponded to slight overpredictions for caffeine–ciprofloxacin (in two studies) and erlotinib–tobacco smoking interactions. The mean prediction error of the AUC ratio was 0.11, and the mean absolute error was 0.43 (21.9 %).

Predicted versus observed area under the concentration–time curve (AUC) ratios for cytochrome P450 (CYP) 1A2-mediated drug interactions, with final estimates of the contribution ratio (CR), inhibition ratio (IR) and increase in drug clearance (IC) values
3.4 Prediction of Unpublished Drug Interactions
The AUC ratios corresponding to interactions between all possible substrate–inhibitor and substrate–inducer pairs were computed, along with their confidence intervals. This yielded a total of 492 predictions for CYP1A2 inhibition and 133 interactions for induction.
As an example, Fig. 3 shows the model-based predicted AUC ratios for the interactions of three CYP1A2 substrates (erlotinib, theophylline and tizanidine) with two inducers (rifampicin and tobacco smoking) and three inhibitors (ethinylestradiol, ciprofloxacin and fluvoxamine).

Predicted area under the concentration–time curve (AUC) ratios for the interactions of three cytochrome P450 (CYP) 1A2 substrate drugs with two inducers and three inhibitors. Each bar represents the mean point estimate of the AUC ratio. Each vertical black line represents the lower (5th percentile) or upper (95th percentile) confidence limit of the AUC ratio for the inducer or inhibitor, respectively
4 Discussion
A few years ago, Ohno et al. [4, 5] proposed a quantitative approach for predicting drug–drug interactions mediated by CYP3A4 on the basis of in vivo data. Since then, our group has extended this approach to other major drug-metabolizing CYPs, including CYP2D6, CYP2C9 and CYP2C19 [6–8]. In those applications, the method has been refined by incorporating Bayesian estimation of the model parameters. Importantly, it has been shown that the model can be modified to predict the influence of cytochrome genetic polymorphisms on substrate drug exposure and drug interactions [87, 88].
In this study, the method was applied to predict drug interactions caused by inhibitors and inducers of CYP1A2. In accordance with the results from our previous applications, this approach showed good predictive performance, as the mean AUC ratios for the CYP1A2 substrates were predicted with limited bias and acceptable precision. This approach has permitted quantification of the sensitivity of 19 substrate drugs, as quantified by their CRCYP1A2 values, and the inhibition potency (IRCYP1A2) and induction potency (ICCYP1A2) values of 26 inhibitors and seven inducers, respectively. Finally, predicted AUC ratios corresponding to a total of 625 potential interactions were predicted, including a number of unpublished cases.
Co-administration of sensitive CYP1A2 substrate drugs with potent inhibitors may result in large increases in the substrate drug exposure. For example, an AUC ratio greater than 5 is predicted to occur for substrates such as agomelatine, melatonin or tizanidine if they are co-administered with inhibitors such as fluvoxamine, rofecoxib or ciprofloxacin.
This study also provides insights into the drug–drug interaction profile of recently marketed drugs such as erlotinib, pomalidomide and vemurafenib.
The induction potency of a non-pharmaceutical, widely used product—tobacco smoking—was also quantified in this study. Tobacco smoking, with a consumption of 10–20 cigarettes per day, appears to be a weak to moderate inducer of CYP1A2, depending on the CRCYP1A2 of the substrate drug. The mean predicted decrease in drug exposure should not exceed 42 %, even for the most sensitive substrates (agomelatine and tizanidine). However, the interactions may be clinically significant for drugs with a narrow therapeutic margin, such as theophylline, for which a 29 % mean decrease in the AUC is predicted.
Such quantitative prediction represents progress in comparison with the classical approach used in clinical practice for managing drug–drug interactions, which is mostly qualitative and is limited to the available data from the Summaries of Product Characteristics or research publications.
Other approaches for quantitative prediction of drug–drug interactions notably include the method based on in vitro enzyme kinetic parameters (i.e. [I], the inhibitor concentration available to the enzyme; and Ki, the inhibition constant of the inhibitor) and physiologically based pharmacokinetic modelling [89–91]. Those two approaches are more complex than the modelling approach based on in vivo data used in our study, which basically requires only two characteristic parameters: the substrate CR and the inhibitor IR or inducer IC.
The application of the in vitro data-based and physiologically based pharmacokinetic (PBPK) methods to CYP1A2-mediated drug interactions has received little attention so far. Thus, it is hardly possible to compare the predictive ability of our approach based on in vivo data with that of other methods. Using in vitro data and various models for hepatic clearance, Obach et al. predicted theophylline AUC increases of 1.03–1.39 and 2.74–4.95 in cases of co-administration with enoxacin and fluvoxamine, respectively. Those estimates are grossly consistent with our model-based mean predictions of 2.12 and 2.31 and with literature values of 1.63 and 3.85 for enoxacin and 3.33 for fluvoxamine [49, 60, 79]. Further research is necessary to compare the various prediction methods for drug interactions. It has been shown that prediction of drug interactions from in vitro data can be tricky, as it strongly depends on experimental conditions and model parameterization [92]. For example, tolfenamic acid was found to be a potent inhibitor of CYP1A2 in vitro when studied without albumin, while no significant interaction was observed in vivo. This overestimation of the inhibition potency was due to the high protein binding of tolfenamic acid [93]. By contrast, our prediction approach is straightforward, as it relies only on in vivo data.
Regarding the estimates of substrate drug sensibility (CRCYP1A2) and induction/inhibition potency, our results compare quite well with the categorical classification provided by the US Food and Drug Administration’s (FDA’s) 2012 guidelines on drug interaction studies [94]. For example, those guidelines classify ciprofloxacin, enoxacin and fluvoxamine as strong inhibitors, and each has an estimated IRCYP1A2 value greater than 0.90. Methoxsalen, mexiletine, oral contraceptives, phenylpropanolamine, thiabendazole and vemurafenib are cited as moderate inhibitors in the guidelines, which is in accordance with our estimates of the IRCYP1A2 values, which range from 0.37 to 0.80 for these agents. Caffeine, cimetidine, daidzein, norfloxacin and propafenone are classified as weak inhibitors; this is also in agreement with their estimated IRCYP1A2 values, not exceeding 0.45.
According to the guidelines, there is no strong CYP1A2 inducer, and sensitive substrates include caffeine, duloxetine, melatonin, ramelteon, tacrine and tizanidine. Our findings appear consistent with all of the above.
This study had several limitations that should be considered. First, some drugs known as sensitive CYP1A2 substrates on the basis of experimental data, such as triamterene or riluzole, were not included in this analysis, because of the lack of in vivo data [95]. It is likely that significant CYP1A2-mediated drug interactions may occur with those drugs, but we were not able to quantify those.
Other limitations were related to the model that was used and have been described in detail elsewhere [4, 6]. Briefly, the model is valid only for orally administered drugs with linear pharmacokinetics. Also, while the inhibition/induction potency is thought to increase with the inhibitor/inducer dose [4], this feature was not included in the results. Studies performed with various doses of the inhibitors or inducers were sparse, and some showed unexpected results. For example, the observed AUC ratios for caffeine when it was co-administered with ciprofloxacin were 1.57, 2.27, 1.59 and 2.44 for ciprofloxacin daily doses of 500, 1000, 1500 and 1500 mg, respectively. For this drug, those results do not suggest a clear dose–response relationship in the inhibition potency, at least over this range of doses. As a result, single IRCYP1A2 and ICCYP1A2 values are reported for each agent and are assumed to hold true over the range of doses used in published studies.
While the dose-dependent effect of the interactor drug is a general feature of drug interaction theory, the ability to observe it probably depends on each drug’s interaction properties, as well as on the study design and the range of doses used in clinical practice. In a crossover study, Jeppesen et al. [96] showed that the dose-dependent inhibitory effects of several serotonin selective reuptake inhibitors on CYP2D6 activity, as measured by the sparteine metabolic ratio, greatly varied between agents. While the CYP2D6 inhibition potency of paroxetine clearly increased with the dose, that of fluoxetine showed little or no dose dependency for doses ranging from 20 to 80 mg [96].
Another clear limitation is the paucity of data reported for some agents. Some reports, especially the Summaries of Product Characteristics, do not always specify the inhibitor/inducer dose used to obtain the data, the number of subjects involved and the variability of the results.
For some substrates (e.g. clozapine), one can notice that the 90 % confidence interval of the CR is quite large. This is due to the Bayesian nature of the estimation method used to provide these confidence limits. Bayesian estimation depends on the prior information and on the data. As we set moderately informative priors for parameters and AUC ratio distributions, the variance of the posterior distribution remained quite large when only a few observed data were available. Thus, we recommend considering the mean point estimates of the parameters and AUC ratios as the most reasonable estimates of drug interaction potential, while the confidence intervals are of interest to assess the uncertainty around those predictions.
In addition, the model—at least in its basic form used here—cannot deal with multiple, complex drug interaction patterns and can accommodate only one cytochrome pathway at a time. An interesting example of an interaction involving multiple enzyme pathways is the melatonin receptor agonist ramelteon. Ramelteon is a substrate of CYP1A2 but also of the CYP2C19 and CYP3A4 isoenzymes [97]. Considerable changes in ramelteon exposure have been described when it is co-administered with fluvoxamine and rifampicin, with mean AUC ratios of 190 and 0.2, respectively [77]. However, prediction of the magnitudes of those interactions was not possible with the basic model, because both fluvoxamine and rifampicin alter the activity of the three CYP isoforms responsible for the metabolism of ramelteon. In order to examine this case more deeply, we used the general model for multiple CYP-mediated drug interactions published by our group [88]. The results are detailed in the “Appendix” and confirm that the general model can accommodate such complex cases quite satisfactorily.
It should be noted that human CYP1A2 metabolism, and thus drug–drug interaction data from individuals, may be influenced by a number of other determinants, including the CYP1A2 genotype, smoking, use of oestrogen-containing contraceptives and caffeine intake [98].
More than 20 variant alleles of the CYP1A2 gene have been identified. However, in contrast to other drug-metabolizing CYPs, such as CYP2D6 or CYP2C19, the mutation frequencies appear to be very low, and only a few variants associated with phenotypic variability or inducibility in CYP1A2 gene expression have been identified so far [99]. Although the influence of the genotype on drug–drug interactions can be theoretically handled by the approach [7], the available data on drug interactions in patients with CYP1A2 mutations were much too sparse to be included in this study. Thus, it was assumed that all subjects were CYP1A2 extensive metabolizers, and it may not be possible to extrapolate the results to populations of patients with mutated CYP1A2 genotypes. Modelling of the influence of the CYP1A2 genotype on drug exposure is an area for further research.
As most drug–drug interaction studies are traditionally performed in healthy volunteers, little is known about the potential effects of medical conditions, such as hepatic or renal impairment, on the magnitude of CYP1A2-mediated drug–drug interactions, which is also a limitation of this work based on such data.
Regarding other influencing factors, caffeine and ethinylestradiol-containing oral contraceptives were found to be modest CYP1A2 inhibitors, while tobacco smoking was identified as a weak inducer, as shown by their respective IRCYP1A2 and ICCYP1A2 values. Thus, concomitant use of these agents may modify the clearance of other CYP1A2 substrate drugs and hence the magnitude of CYP1A2-mediated interactions. Unfortunately, most drug–drug interaction studies have not provided details on the use of these products, except for specific studies that quantified their effect. As a result, co-intake of these products cannot be ruled out, and this may have influenced the original data and our model-based parameter estimates for other drugs. However, we believe that this should not undermine our results, because their effects appear to be weak, and because our final Bayesian estimation provided average parameter and AUC values from all pooled data. Also, these estimates reflect ‘real-life’ condition of drug use.
This work may have important implications for clinical practice. Indeed, the predicted AUC ratios may be used as correction factors to adjust the maintenance doses of the substrate drugs in case of a potential interaction, when it seems clinically relevant to do so. However, one should be careful in the interpretation of the AUC ratio, taking into account the entire clinical pharmacokinetics and pharmacodynamics, especially when prodrugs or active metabolites are involved in the drug response. It should be noted that this approach provides only an average prediction of the alteration in CYP1A2 substrate drug exposure, and so the clinical alteration may vary between individuals. In addition, as drug–drug interactions depend on the pharmacokinetics of the substrate and interactor drugs, which may exhibit intra-individual variability, the quantitative effect of drug–drug interactions is likely to vary within individuals during prolonged co-therapy.
5 Conclusion
The framework introduced by Ohno for CYP-mediated drug interactions has been extended successfully to CYP1A2. The method displayed good predictive performance in predicting the magnitude of the interactions caused by CYP1A2 inhibitors and inducers. It provided quantitative predictions of the effects of 26 inhibitors and seven inducers on the exposure to 19 oral CYP1A2 substrate drugs. This analysis has provided insights into the interaction profiles of drugs that have been poorly studied so far and can help to manage significant interactions in clinical practice. Quantitative predictions corresponding to 625 possible interactions are available via a web tool (http://www.ddi-predictor.org) [100].
References
Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–23.
Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:825–33.
Rasmussen BB, Brøsen K. Theophylline has no advantages over caffeine as a putative model drug for assessing CYPIA2 activity in humans. Br J Clin Pharmacol. 1997;43:253–8.
Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46:681–96.
Ohno Y, Hisaka A, Ueno M, Suzuki H. General framework for the prediction of oral drug interactions caused by CYP3A4 induction from in vivo information. Clin Pharmacokinet. 2008;47:669–80.
Tod M, Goutelle S, Clavel-Grabit F, Nicolas G, Charpiat B. Quantitative prediction of cytochrome P450 (CYP) 2D6-mediated drug interactions. Clin Pharmacokinet. 2011;50:519–30.
Goutelle S, Bourguignon L, Bleyzac N, Berry J, Clavel-Grabit F, Tod M. In vivo quantitative prediction of the effect of gene polymorphisms and drug interactions on drug exposure for CYP2C19 substrates. AAPS J. 2013;15:415–26.
Castellan A-C, Tod M, Gueyffier F, Audars M, Cambriels F, Kassaï B, et al. Quantitative prediction of the impact of drug interactions and genetic polymorphisms on cytochrome P450 2C9 substrate exposure. Clin Pharmacokinet. 2013;52:199–209.
Carrillo JA, Benitez J. Clinically significant pharmacokinetic interactions between dietary caffeine and medications. Clin Pharmacokinet. 2000;39:127–53.
Spiegelhalter D, Thomas A, Best N, Lunn D. Winbugs 1.4.3 user manual. Cambridge: MRC Biostatistics Unit; 2007.
Congdon P. Bayesian statistical modelling. Chichester: Wiley; 2001.
Becquemont L, Ragueneau I, Le Bot MA, Riche C, Funck-Brentano C, Jaillon P. Influence of the CYP1A2 inhibitor fluvoxamine on tacrine pharmacokinetics in humans. Clin Pharmacol Ther. 1997;61:619–27.
Buchan P, Wade A, Ward C, Oliver SD, Stewart AJ, Freestone S. Frovatriptan: a review of drug–drug interactions. Headache. 2002;42(Suppl 2):S63–73.
Granfors MT, Backman JT, Neuvonen M, Ahonen J, Neuvonen PJ. Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction. Clin Pharmacol Ther. 2004;75:331–41.
Hägg S, Spigset O, Mjörndal T, Dahlqvist R. Effect of caffeine on clozapine pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2000;49:59–63.
Härtter S, Nordmark A, Rose D-M, Bertilsson L, Tybring G, Laine K. Effects of caffeine intake on the pharmacokinetics of melatonin, a probe drug for CYP1A2 activity. Br J Clin Pharmacol. 2003;56:679–82.
Kaye CM, Nicholls B. Clinical pharmacokinetics of ropinirole. Clin Pharmacokinet. 2000;39:243–54.
Klein A, Sami M, Selinger K. Mexiletine kinetics in healthy subjects taking cimetidine. Clin Pharmacol Ther. 1985;37:669–73.
Kletzl H, Zwanziger E, Kirkpatrick C, Luedin E. Effect of ciprofloxacin on the systemic exposure to erlotinib [abstract]. J Clin Oncol. 2008;26(Suppl):abstract no. 19047.
Kunii T, Fukasawa T, Yasui-Furukori N, Aoshima T, Suzuki A, Tateishi T, et al. Interaction study between enoxacin and fluvoxamine. Ther Drug Monit. 2005;27:349–53.
Lahu G, Nassr N, Herzog R, Elmlinger M, Ruth P, Hinder M, et al. Effect of steady-state enoxacin on single-dose pharmacokinetics of roflumilast and roflumilast N-oxide. J Clin Pharmacol. 2011;51:586–93.
Lobo ED, Bergstrom RF, Reddy S, Quinlan T, Chappell J, Hong Q, et al. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin Pharmacokinet. 2008;47:191–202.
Mirghani RA, Hellgren U, Westerberg PA, Ericsson O, Bertilsson L, Gustafsson LL. The roles of cytochrome P450 3A4 and 1A2 in the 3-hydroxylation of quinine in vivo. Clin Pharmacol Ther. 1999;66:454–60.
Orlando R, Padrini R, Perazzi M, De Martin S, Piccoli P, Palatini P. Liver dysfunction markedly decreases the inhibition of cytochrome P450 1A2-mediated theophylline metabolism by fluvoxamine. Clin Pharmacol Ther. 2006;79:489–99.
European Medicines Agency. Valdoxan: agomelatine. London: European Medicines Agency; 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000915/human_med_001123.jsp&mid=WC0b01ac058001d124. Accessed 17 Feb 2015.
European Medicines Agency. Zyprexa: olanzapine. London: European Medicines Agency; 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000115/human_med_001189.jsp&mid=WC0b01ac058001d124. Accessed 17 Feb 2015.
European Medicines Agency. Sycrest: asenapine. London: European Medicines Agency; 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001177/human_med_001379.jsp&mid=WC0b01ac058001d124. Accessed 17 Feb 2015.
European Medicines Agency. Imnovid (previously pomalidomide Celgene): pomalidomide. London: European Medicines Agency; 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002682/human_med_001669.jsp&mid=WC0b01ac058001d124. Accessed 17 Feb 2015.
European Medicines Agency. Azilect: rasagiline. London: European Medicines Agency; 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000574/human_med_000667.jsp&mid=WC0b01ac058001d124. Accessed 17 Feb 2015.
Backman JT, Karjalainen MJ, Neuvonen M, Laitila J, Neuvonen PJ. Rofecoxib is a potent inhibitor of cytochrome P450 1A2: studies with tizanidine and caffeine in healthy subjects. Br J Clin Pharmacol. 2006;62:345–57.
Balogh A, Klinger G, Henschel L, Börner A, Vollanth R, Kuhnz W. Influence of ethinylestradiol-containing combination oral contraceptives with gestodene or levonorgestrel on caffeine elimination. Eur J Clin Pharmacol. 1995;48:161–6.
Bapiro TE, Sayi J, Hasler JA, Jande M, Rimoy G, Masselle A, et al. Artemisinin and thiabendazole are potent inhibitors of cytochrome P450 1A2 (CYP1A2) activity in humans. Eur J Clin Pharmacol. 2005;61:755–61.
Bendriss EK, Bechtel Y, Bendriss A, Humbert PH, Paintaud G, Magnette J, et al. Inhibition of caffeine metabolism by 5-methoxypsoralen in patients with psoriasis. Br J Clin Pharmacol. 1996;41:421–4.
Brazier JL, Descotes J, Lery N, Ollagnier M, Evreux JC. Inhibition by idrocilamide of the disposition of caffeine. Eur J Clin Pharmacol. 1980;17:37–43.
Carbó M, Segura J, De la Torre R, Badenas JM, Camí J. Effect of quinolones on caffeine disposition. Clin Pharmacol Ther. 1989;45:234–40.
Christensen M, Tybring G, Mihara K, Yasui-Furokori N, Carrillo JA, Ramos SI, et al. Low daily 10-mg and 20-mg doses of fluvoxamine inhibit the metabolism of both caffeine (cytochrome P4501A2) and omeprazole (cytochrome P4502C19). Clin Pharmacol Ther. 2002;71:141–52.
Efthymiopoulos C, Bramer SL, Maroli A, Blum B. Theophylline and warfarin interaction studies with grepafloxacin. Clin Pharmacokinet. 1997;33(Suppl 1):39–46.
Jonkman JH, Sollie FA, Sauter R, Steinijans VW. The influence of caffeine on the steady-state pharmacokinetics of theophylline. Clin Pharmacol Ther. 1991;49:248–55.
Kinzig-Schippers M, Fuhr U, Zaigler M, Dammeyer J, Rüsing G, Labedzki A, et al. Interaction of pefloxacin and enoxacin with the human cytochrome P450 enzyme CYP1A2. Clin Pharmacol Ther. 1999;65:262–74.
Lake CR, Rosenberg D, Quirk R. Phenylpropanolamine and caffeine use among diet center clients. Int J Obes. 1990;14:575–82.
Liu L, Pan X, Liu H, Liu X, Yang H, Xie L, et al. Modulation of pharmacokinetics of theophylline by antofloxacin, a novel 8-amino-fluoroquinolone, in humans. Acta Pharmacol Sin. 2011;32:1285–93.
May DC, Jarboe CH, VanBakel AB, Williams WM. Effects of cimetidine on caffeine disposition in smokers and nonsmokers. Clin Pharmacol Ther. 1982;31:656–61.
Mays DC, Camisa C, Cheney P, Pacula CM, Nawoot S, Gerber N. Methoxsalen is a potent inhibitor of the metabolism of caffeine in humans. Clin Pharmacol Ther. 1987;42:621–6.
Michaud V, Mouksassi MS, Labbé L, Bélanger P-M, Ferron LA, Gilbert M, et al. Inhibitory effects of propafenone on the pharmacokinetics of caffeine in humans. Ther Drug Monit. 2006;28:779–83.
Momo K, Homma M, Osaka Y, Inomata S, Tanaka M, Kohda Y. Effects of mexiletine, a CYP1A2 inhibitor, on tizanidine pharmacokinetics and pharmacodynamics. J Clin Pharmacol. 2010;50:331–7.
Nicolau DP, Nightingale CH, Tessier PR, Fu Q, Xuan DW, Esguerra EM, et al. The effect of fleroxacin and ciprofloxacin on the pharmacokinetics of multiple dose caffeine. Drugs. 1995;49(Suppl 2):357–9.
Peng W-X, Li H-D, Zhou H-H. Effect of daidzein on CYP1A2 activity and pharmacokinetics of theophylline in healthy volunteers. Eur J Clin Pharmacol. 2003;59:237–41.
Sofowora GG, Choo EF, Mayo G, Shyr Y, Wilkinson GR. In vivo inhibition of human CYP1A2 activity by oltipraz. Cancer Chemother Pharmacol. 2001;47:505–10.
Takagi K, Hasegawa T, Ogura Y, Suzuki R, Yamaki K, Watanabe T, et al. Comparative studies on interaction between theophylline and quinolones. J Asthma. 1988;25:63–71.
Trépanier EF, Nafziger AN, Amsden GW. Effect of terbinafine on theophylline pharmacokinetics in healthy volunteers. Antimicrob Agents Chemother. 1998;42:695–7.
European Medicines Agency. Zelboraf: vemurafenib. London: European Medicines Agency; 2015. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002409/human_med_001544.jsp&mid=WC0b01ac058001d124. Accessed 17 Feb 2015.
Backman JT, Granfors MT, Neuvonen PJ. Rifampicin is only a weak inducer of CYP1A2-mediated presystemic and systemic metabolism: studies with tizanidine and caffeine. Eur J Clin Pharmacol. 2006;62:451–61.
Backman JT, Schröder MT, Neuvonen PJ. Effects of gender and moderate smoking on the pharmacokinetics and effects of the CYP1A2 substrate tizanidine. Eur J Clin Pharmacol. 2008;64:17–24.
Darwish M, Kirby M, Robertson P, Hellriegel ET. Interaction profile of armodafinil with medications metabolized by cytochrome P450 enzymes 1A2, 3A4 and 2C19 in healthy subjects. Clin Pharmacokinet. 2008;47:61–74.
Dilger K, Zheng Z, Klotz U. Lack of drug interaction between omeprazole, lansoprazole, pantoprazole and theophylline. Br J Clin Pharmacol. 1999;48:438–44.
Gillum JG, Sesler JM, Bruzzese VL, Israel DS, Polk RE. Induction of theophylline clearance by rifampin and rifabutin in healthy male volunteers. Antimicrob Agents Chemother. 1996;40:1866–9.
Lucas RA, Gilfillan DJ, Bergstrom RF. A pharmacokinetic interaction between carbamazepine and olanzapine: observations on possible mechanism. Eur J Clin Pharmacol. 1998;54:639–43.
Bachmann K, White D, Jauregui L, Schwartz JI, Agrawal NGB, Mazenko R, et al. An evaluation of the dose-dependent inhibition of CYP1A2 by rofecoxib using theophylline as a CYP1A2 probe. J Clin Pharmacol. 2003;43:1082–90.
Batty KT, Davis TM, Ilett KF, Dusci LJ, Langton SR. The effect of ciprofloxacin on theophylline pharmacokinetics in healthy subjects. Br J Clin Pharmacol. 1995;39:305–11.
Beckmann J, Elsässer W, Gundert-Remy U, Hertrampf R. Enoxacin—a potent inhibitor of theophylline metabolism. Eur J Clin Pharmacol. 1987;33:227–30.
Broughton LJ, Rogers HJ. Decreased systemic clearance of caffeine due to cimetidine. Br J Clin Pharmacol. 1981;12:155–9.
Fukasawa T, Yasui-Furukori N, Suzuki A, Ishii G, Inoue Y, Tateishi T, et al. Effects of caffeine on the kinetics of fluvoxamine and its major metabolite in plasma after a single oral dose of the drug. Ther Drug Monit. 2006;28:308–11.
Fulton B, Goa KL. Olanzapine: a review of its pharmacological properties and therapeutic efficacy in the management of schizophrenia and related psychoses. Drugs. 1997;53:281–98.
Gardner MJ, Tornatore KM, Jusko WJ, Kanarkowski R. Effects of tobacco smoking and oral contraceptive use on theophylline disposition. Br J Clin Pharmacol. 1983;16:271–80.
Granfors MT, Backman JT, Laitila J, Neuvonen PJ. Oral contraceptives containing ethinyl estradiol and gestodene markedly increase plasma concentrations and effects of tizanidine by inhibiting cytochrome P450 1A2. Clin Pharmacol Ther. 2005;78:400–11.
Granfors MT, Backman JT, Neuvonen M, Neuvonen PJ. Ciprofloxacin greatly increases concentrations and hypotensive effect of tizanidine by inhibiting its cytochrome P450 1A2-mediated presystemic metabolism. Clin Pharmacol Ther. 2004;76:598–606.
Hamilton M, Wolf JL, Rusk J, Beard SE, Clark GM, Witt K, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res. 2006;12:2166–71.
Healy DP, Polk RE, Kanawati L, Rock DT, Mooney ML. Interaction between oral ciprofloxacin and caffeine in normal volunteers. Antimicrob Agents Chemother. 1989;33:474–8.
Hurwitz A, Vacek JL, Botteron GW, Sztern MI, Hughes EM, Jayaraj A. Mexiletine effects on theophylline disposition. Clin Pharmacol Ther. 1991;50:299–307.
Jeppesen U, Loft S, Poulsen HE, Brśen K. A fluvoxamine–caffeine interaction study. Pharmacogenetics. 1996;6:213–22.
Joeres R, Klinker H, Heusler H, Epping J, Richter E. Influence of mexiletine on caffeine elimination. Pharmacol Ther. 1987;33:163–9.
Kusumoto M, Ueno K, Tanaka K, Takeda K, Mashimo K, Kameda T, et al. Lack of pharmacokinetic interaction between mexiletine and omeprazole. Ann Pharmacother. 1998;32:182–4.
Labbé L, Abolfathi Z, Robitaille NM, St-Maurice F, Gilbert M, Turgeon J. Stereoselective disposition of the antiarrhythmic agent mexiletine during the concomitant administration of caffeine. Ther Drug Monit. 1999;21:191–9.
Labbé L, Robitaille NM, Lefez C, Potvin D, Gilbert M, O’Hara G, et al. Effects of ciprofloxacin on the stereoselective disposition of mexiletine in man. Ther Drug Monit. 2004;26:492–8.
Larsen JT, Hansen LL, Spigset O, Brøsen K. Fluvoxamine is a potent inhibitor of tacrine metabolism in vivo. Eur J Clin Pharmacol. 1999;55:375–82.
Mahr G, Sörgel F, Granneman GR, Kinzig M, Muth P, Patterson K, et al. Effects of temafloxacin and ciprofloxacin on the pharmacokinetics of caffeine. Clin Pharmacokinet. 1992;22(Suppl 1):90–7.
McGechan A, Wellington K. Ramelteon. CNS Drugs. 2005;19:1057–65 (discussion 1066–7).
Pentikäinen PJ, Koivula IH, Hiltunen HA. Effect of rifampicin treatment on the kinetics of mexiletine. Eur J Clin Pharmacol. 1982;23:261–6.
Rasmussen BB, Jeppesen U, Gaist D, Brøsen K. Griseofulvin and fluvoxamine interactions with the metabolism of theophylline. Ther Drug Monit. 1997;19:56–62.
Robson RA, Begg EJ, Atkinson HC, Saunders DA, Frampton CM. Comparative effects of ciprofloxacin and lomefloxacin on the oxidative metabolism of theophylline. Br J Clin Pharmacol. 1990;29:491–3.
Rost KL, Fuhr U, Thomsen T, Zaigler M, Brockmöller J, Bohnemeier H, et al. Omeprazole weakly inhibits CYP1A2 activity in man. Int J Clin Pharmacol Ther. 1999;37:567–74.
Schwartz J, Jauregui L, Lettieri J, Bachmann K. Impact of ciprofloxacin on theophylline clearance and steady-state concentrations in serum. Antimicrob Agents Chemother. 1988;32:75–7.
Spigset O, Carleborg L, Hedenmalm K, Dahlqvist R. Effect of cigarette smoking on fluvoxamine pharmacokinetics in humans. Clin Pharmacol Ther. 1995;58:399–403.
Stille W, Harder S, Mieke S, Beer C, Shah PM, Frech K, et al. Decrease of caffeine elimination in man during co-administration of 4-quinolones. J Antimicrob Chemother. 1987;20:729–34.
Stoysich AM, Mohiuddin SM, Destache CJ, Nipper HC, Hilleman DE. Influence of mexiletine on the pharmacokinetics of theophylline in healthy volunteers. J Clin Pharmacol. 1991;31:354–7.
Von Richter O, Lahu G, Huennemeyer A, Herzog R, Zech K, Hermann R. Effect of fluvoxamine on the pharmacokinetics of roflumilast and roflumilast N-oxide. Clin Pharmacokinet. 2007;46:613–22.
Tod M, Goutelle S, Gagnieu MC. Genotype-based quantitative prediction of drug exposure for drugs metabolized by CYP2D6. Clin Pharmacol Ther. 2011;90:582–7.
Tod M, Nkoud-Mongo C, Gueyffier F. Impact of genetic polymorphism on drug–drug interactions mediated by cytochromes: a general approach. AAPS J. 2013;15:1242–52.
Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug–drug interactions from in vitro data. Br J Clin Pharmacol. 2004;57:473–86.
Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug–drug interactions. J Pharmacol Exp Ther. 2006;316:336–48.
Marsousi N, Daali Y, Rudaz S, Almond L, Humphries H, Desmeules J, et al. Prediction of metabolic interactions with oxycodone via CYP2D6 and CYP3A inhibition using a physiologically based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2014;3:e152.
Karjalainen MJ, Neuvonen PJ, Backman JT. In vitro inhibition of CYP1A2 by model inhibitors, anti-inflammatory analgesics and female sex steroids: predictability of in vivo interactions. Basic Clin Pharmacol Toxicol. 2008;103(2):157–65.
Karjalainen MJ, Neuvonen PJ, Backman JT. Tolfenamic acid is a potent CYP1A2 inhibitor in vitro but does not interact in vivo: correction for protein binding is needed for data interpretation. Eur J Clin Pharmacol. 2007;63(9):829–36.
US Food and Drug Administration. Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Rockville: US Food and Drug Administration, 2012. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. Accessed 15 Dec 2012.
Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why, how, and when? Basic Clin Pharmacol Toxicol. 2005;97:125–34.
Jeppesen U, Gram LF, Vistisen K, Loft S, Poulsen HE, Brøsen K. Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol. 1996;51(1):73–8.
Obach RS, Ryder TF. Metabolism of ramelteon in human liver microsomes and correlation with the effect of fluvoxamine on ramelteon pharmacokinetics. Drug Metab Dispos. 2010;38:1381–91.
Zhou S-F, Yang L-P, Zhou Z-W, Liu Y-H, Chan E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11:481–94.
PharmGKB. Gene: CYP1A2. Stanford: PharmGKB; 2015. https://www.pharmgkb.org/gene/PA27093#tabview=tab3&subtab=32. Accessed 29 Aug 2015.
Genophar II Working Group. DDI-predictor. Lentilly: Genophar II Working Group; 2015. http://www.ddi-predictor.org. Accessed 1 Feb 2016.
Acknowledgments
This work was presented in part at the 2015 Meeting of the French Society of Pharmacology and Therapeutics (SFPT); Caen, France; 21–23 April 2015. This study was not supported by any academic, company or sponsor funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Laurence Gabriel, Michel Tod and Sylvain Goutelle have no potential conflicts of interest that might be relevant to the content of this study.
Appendix
Appendix
This “Appendix” illustrates the application of the general model for multiple CYP-mediated drug interactions to the case of interactions with ramelteon as the victim drug.
The model for an interaction mediated by three cytochromes (denoted 1, 2 and 3) and caused by an inhibitor and an inducer is described by Eqs. 8 and 9, respectively [88]:
The estimated IRs of fluvoxamine are as follows: ICCYP1A2 = 1, ICCYP2C19 = 0.98, IRCYP3A4 = 0.3.
The estimated ICs of rifampicin are as follows: ICCYP1A2 = 1.45, ICCYP2C19 = 4.2, ICCYP3A4 = 7.7.
Ramelteon is metabolized by CYP1A2, CYP2C19 and CYP3A4, with contribution ratios estimated from in vitro data as follows [93]: CRCYP1A2 = 0.49, CRCYP2C19 = 0.42, CRCYP3A4 = 0.086 (CR set 1).
Because the CRs of ramelteon have not been estimated in vivo, one might also consider another set of CR values. For example, we may assume that the contribution of CYP3A4 might be lower or even negligible in vivo, with CRCYP1A2 = 0.57, CRCYP2C19 = 0.42, CRCYP3A4 = 0.01 (CR set 2), or CRCYP1A2 = 0.58, CRCYP2C19 = 0.42, CRCYP3A4 = 0 (CR set 3).
The predicted magnitude of the drug interactions is shown in Table 7.
As one can see, for the interaction caused by fluvoxamine, the predicted AUC ratio strongly depends on the estimated CRs for ramelteon, because fluvoxamine is a very potent inhibitor of CYP1A2 and CYP2C19, with IR values close to 1. Decreasing the CR of CYP3A4 and increasing the CR of CYP1A2/CYP2C19 result in a larger predicted AUC ratio, which better agrees with the observed ratio.
Rights and permissions
About this article

Cite this article
Gabriel, L., Tod, M. & Goutelle, S. Quantitative Prediction of Drug Interactions Caused by CYP1A2 Inhibitors and Inducers. Clin Pharmacokinet 55, 977–990 (2016). https://doi.org/10.1007/s40262-016-0371-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0371-x