
Abstract
Background and Objective
Cytochrome P450 (CYP) 2C9 is the most common CYP2C enzyme and makes up approximately onethird of total CYP protein content in the liver. It metabolises more than 100 drugs. The exposure of drugs mainly eliminated by CYP2C9 may be dramatically modified by drug–drug interactions (DDIs) and genetic variations. The objective of this study was to develop a modelling approach to predict the impact of genetic polymorphisms and DDIs on drug exposure in drugs metabolised by CYP2C9. We then developed dosing recommendations based on genotypes and compared them to current Epar/Vidal dosing guidelines.
Methods
We created two models. The genetic model was designed to predict the impact of CYP2C9 polymorphisms on drug exposure. It links the area under the concentration–time curve (AUC) ratio (mutant to wild-type patients) to two parameters: the fractional contribution of CYP2C9 to oral clearance in vivo (i.e. CR or contribution ratio), and the fractional activity of the allele combination with respect to the homozygous wild type (i.e. FA or fraction of activity). Data were available for 77 couples (substrate, genotype). We used a three-step approach: (1) initial estimates of CRs and FAs were calculated using a first bibliographic dataset; (2) external validation of these estimates was then performed through the comparison between the AUC ratios predicted by the model and the observed values, using a second published dataset; and (3) refined estimates of CRs and FAs were obtained using Bayesian orthogonal regression involving the whole dataset and initial estimates of CRs and FAs. Posterior distributions of AUC ratios, CRs and FAs were estimated using Monte-Carlo Markov chain simulation. The drug interaction model was designed to predict the impact of DDIs on drug exposure. It links the AUC ratio (ratio of drug given in combination to drug given alone) to several parameters: the CR, the inhibition ratio (IR) of an inhibitor, and the increase in clearance (IC) due to an inducer. Data were available for 80 DDIs. IRs and ICs were calculated using the interaction model and an external validation was performed. Doses adjustments were calculated in order to obtain equal values for drug exposure in extensive and poor metabolisers and then compared to Epar/Vidal dosing guidelines.
Results
CRs were assessed for 26 substrates, FAs for five genotype classes including CYP2C9*2 and *3 allelic variants, IRs for 27 inhibitors and ICs for two inducers. For the genetic model, the mean prediction error of AUC ratios was −0.01, while the mean prediction absolute error was 0.36. For the drug interaction model, the mean prediction error of AUC ratios was 0.01, while the mean prediction absolute error was 0.22. Of the 26 substrates and CYP2C9*2 and *3 variants investigated, 30 couples (substrate, genotype) lead to a dose adjustment, as opposed to only ten couples identified in the Epar/Vidal recommendations.
Conclusion
These models were already used for CYP2D6. They are accurate at predicting the impact of drug interactions and genetic polymorphisms on CYP2C9 substrate exposure. This approach will contribute to the development of personalized medicine, i.e. individualized drug therapy with specific dosing recommendations based on CYP genotype or drug associations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
The field of pharmacogenetics was born after analysis of a few rare extreme reactions (phenotypes) in some humans. These phenotypes were inherited diseases, abnormal reactions to drugs or other environmental factors. ‘Polymorphism’ has different meanings in pharmacogenetics. For this study, it means the existence of two or more variants (e.g. alleles and phenotypes) in a population that occurs at a frequency greater than 1 %. Several review articles describe genetic variations related to many mechanisms relevant to drug therapy [1–3]. Effects may be on genes that code for metabolising enzymes, transporters, receptors or ion channels. These effects may be described as pharmacokinetic (affecting drug concentrations) or pharmacodynamic (affecting drug action).
Cytochrome P450 (CYP) 2C9 is the most common CYP2C enzyme [4] and makes up approximately onethird of total CYP protein content in the liver [5]. It metabolises more than 100 drugs [6], including coumarin anticoagulants, sulfonylureas and some NSAIDs. The exposure of drugs that are mainly eliminated by CYP2C9 may be dramatically modified by drug–drug interactions (DDIs) and genetic variations. Gene coding for CYP2C9 involves numerous inherited polymorphisms. More than 50 single nucleotide polymorphisms have been described in the regulatory and coding regions of the CYP2C9 gene, but only two coding variants with functional consequences are common (CYP2C9*2 and CYP2C9*3). Their allele frequencies are around 11 % (*2) and 7 % (*3) in Caucasian subjects [6]. They have significantly lower frequencies in African and Asian subjects [7].
Our aim was to undertake a quantitative prediction of the impact of CYP2C9 polymorphisms and DDIs on drug exposure. We used the modelling approach developed by Tod et al. [8, 9], combining all available data on 26 CYP2C9 substrates, five genotypes, 27 inhibitors and two inducers. This approach describes the effect of mutations, inhibitors and inducers using a set of characteristic parameters to predict drug exposure in undocumented situations, after these parameters are estimated. Dosing recommendations based on genotype were written and compared to current Epar/Vidal dosing guidelines [10, 11].
2 Methods
2.1 Impact of CYP2C9 Genetic Polymorphisms on Drug Exposure: The Genetic Model
A genetic model was designed to perform the quantitative prediction of the impact of CYP2C9 polymorphisms on drug exposure and is based on a model used to study CYP2D6 [8]. It relies mostly on in vivo data to avoid extrapolation issues and uses two characteristic parameters: one for the drug (i.e. CR or contribution ratio) and one for the genotype (i.e. FA or fraction of activity). CR2C9 is the fraction of oral clearance of the drug metabolised by CYP2C9. FA2C9 is the fraction of activity of CYP2C9 resulting from the combination of mutated alleles, relative to the activity of the reference genotype. The metric of interest is the ratio between the area under the drug concentration–time curve (AUC) in patients with mutant genotype and the AUC in patients with wild-type genotype.
Using the equation proposed by Gibbs et al. [12], the ratio of AUCXM (the AUC of the drug in patients with mutated CYP2C9 alleles: XM may refer to poor metabolisers [PMs], intermediate metabolisers [IMs] or ultra-rapid metabolisers [UMs]) to AUCEM (the AUC of the drug administered alone at the same dose in patients with wild-type genotype [extensive metabolisers; EMs]) was modified as follows (Eq. 1):
The reference value of FA in EMs who are homozygote wild-type is equal to 1. FA is <1 in IMs and PMs and >1 in UMs.
FA is assumed to be the same for all CYP2C9 substrates and only depends on the allele combination. Each allele is assumed to contribute to the overall activity independently of other alleles. Therefore, if there are m categories of alleles and n i alleles in each category (Eq. 2):
where FAi is the fraction of activity of the mutated CYP allele relative to the wild-type CYP allele.
Two reviewers performed a bibliographic search on MEDLINE from January 1966 to December 2011 to identify all available datasets of drug concentrations in humans with CYP2C9 polymorphisms. Keywords used were “pharmacogenetics”, “genotype”, “cytochrome P450”, “2C9”, “pharmacokinetics”, “pharmacodynamics”, “polymorphism”, “allele” and “metabolism”. No language restrictions were applied. In MEDLINE, we used a species filter to limit the search to “humans”. Major reviews on pharmacogenetics and drug-metabolising enzymes were also systematically searched in MEDLINE. Cross-references and quoted papers were checked in order to update the primary reference list.
A three-step approach was used to estimate CRs and FAs. Initial CRs were estimated with a set of learning data. Then, a second set of data was used for the external validation. The combined set of data was used to refine CR and FA estimates.
The first step was to allocate clinical studies by estimating CRs using pharmacokinetic data from patients with either CYP2C9*3/*3 or *1/*3 genotypes. If several studies were available for these genotypes, selection criteria of studies included the availability of results for *3/*3 genotype carrier subjects and the number of patients for each genotype group.
The second step was the external validation of initial estimates. This was performed using studies that were not included in the first set of data. We compared the AUC ratios predicted by Eq. 1 to the observed values. Visual examination of the ‘predicted versus observed AUC ratio’ plot was used for external validation. Predicted values that were 50–200 % of observed ratios were considered correct. This limit is frequently used as a criterion for model evaluation in physiologically based pharmacokinetic literature. There is no statistical reason to use this limit, but it is considered relevant from a clinical point of view. A prediction error up to a factor of 2 has no important consequences in most instances. The whole set of initial values (CRs and FAs) was considered valid if 90 % of the predicted AUC ratios were considered correct in the second step.
The third step was to improve estimation of CRs and FAs obtained after orthogonal regression, using all data (initial estimates of CRs and FAs from step 1 and observed AUC ratios used in the first two datasets). The posterior distributions of AUC ratios, CRs and FAs were estimated using Monte-Carlo Markov chain simulation with WinBugs 1.4 software (MRC Biostatistics Unit, Cambridge, UK) (for more information, see Tod et al. [8]). In this regression, we assumed the observed and predicted AUC ratios followed normal distributions centred at the predicted AUC ratios with a standard deviation of 5. Convergence was assessed by checking the stability of posterior distributions. Goodness of fit was assessed by visual examination of residual scatterplots and posterior distributions. The bias of AUC ratio prediction was evaluated by the mean prediction error. The prediction error is defined as the predicted value minus the observed value. The imprecision of the prediction was assessed by the mean absolute prediction error.
2.2 Impact of CYP2C9-Mediated Drug Interactions on Drug Exposure: The Drug Interaction Model
We carried out a bibliographic search to identify the inhibitors and inducers of CYP2C9. The same search and selection strategy was used as for the genetic model. If genetic status was reported, only data on EMs were considered. If genetic status was not reported, patients were considered as EMs (cf. low prevalence of mutations).
In previous papers [9, 13, 14], AUC* corresponded to data obtained after co-administration of an inhibitor or inducer and AUC to data obtained after administration of the victim drug taken alone at the same dose. The ratio between AUC* and AUC depends on the contribution ratio (CR2C9) defined above and the inhibition ratio (IR2C9) or the apparent increase in clearance (IC2C9) due to the interacting drug.
For an inhibitor (Eq. 3):
For an inducer (Eq. 4):
In these equations, IR2C9 is a metric of inhibitor potency, ranges from 0 to 1, depends on the dose of the inhibitor and is related to its inhibition constant Ki; and IC2C9 is a metric of inducer potency, depends on the dose of the inducer and ranges from 0 to a maximal value that may be greater than 1. Using the AUC ratio measured in a clinical DDI study, and the CR2C9 of the victim drug determined from a pharmacogenetic study, the IR or the IC may be calculated by solving Eqs. 3 and 4. Alternatively, if the AUC ratio and the IR of the inhibitor are known, the CR may be calculated (the ‘interaction method’).
Sometimes, DDIs on CYP2C9 also involve displacement from drug binding to plasma proteins. This approach is valid as long as the AUC ratio of total concentrations (AUC*/AUC) in Eqs. 3 and 4 is replaced by the AUC ratio of unbound concentrations (fAUC*/fAUC).
For an inhibitor (Eq. 5):
For an inducer (Eq. 6):
IRs or ICs were calculated by solving Eqs. 3, 4, 5 or 6. Then, an external validation was carried out like the one used for the genetic model.
2.3 Impact of Genetic Polymorphisms and Drug Interactions on Dosing Recommendations
We assumed that a substrate with a CR2C9 greater than 0.5 may require a dose adjustment. A genetic polymorphism or a DDI leading to an AUC ratio greater than 2 could also require a dose adjustment.
Dose adjustment was calculated in order to obtain equal values for drug exposure in EMs and PMs (Eq. 7):
Dose adjustments in PMs were then compared to Epar/Vidal dosing guidelines [10, 11] in patients with CYP2C9 polymorphisms for the 20 substrates with a CR2C9 greater than 0.5.
3 Results
3.1 Model Predictions for CYP2C9 Polymorphisms
The bibliographic search identified 32 relevant articles. Twenty-one of these articles were used for the initial estimates of CRs (first step) and 11 for external validation (second step). For the re-estimations of parameters (third step), all 32 articles were used. The description of each study is given in Appendices 1 and 2 of the Online Resource (drug, dose value, type of study, number of subjects, healthy volunteers or patients, and sampling schedule).
3.2 Initial Estimates for Contribution Ratios (CRs) and Fractions of Activity (FAs)
Initial FA values were estimated from S-warfarin which was assumed to be completely metabolised by CYP2C9 (i.e. CR = 1) [15]. The initial FA values for CYP2C9 genotypes *1/*3, *3/*3, *1/*2, *2/*2 and *2/*3 were 0.55, 0.1, 0.84, 0.68 and 0.39, respectively.
Initial CR values were estimated for 26 substrates (19 drugs and 7 enantiomers). CRs were estimated for 23 of them with pharmacogenetic/pharmacokinetic studies. For indomethacin, naproxen, and tenoxicam, CRs were estimated based on clinical pharmacokinetic data (the fraction of dose recovered as metabolites) and in vitro data. Most pharmacogenetic studies were done in healthy volunteers. The number of subjects included in studies varied between 5 and 108 (for more details, please refer to Appendix 1 of the Online Resource).
3.3 External Validation of Initial Estimates
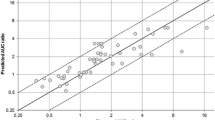
Eleven pharmacogenetic/pharmacokinetic studies were used for external validation. Six were steady-state studies, one was a multiple-dose pharmacokinetic study, and four were single-dose pharmacokinetic studies. Twenty-four AUC ratios were available for external validation. The number of PMs and IMs varied between 1 and 9 (please see Appendix 2 of the Online Resource). The agreement between predicted and observed AUC ratio is shown in Fig. 1. Two points are outside the acceptable range of prediction. One outlier is underestimated and corresponds to losartan for genotype CYP2C9*1/*2 [16]. The other one is overestimated and corresponds to S-ibuprofen for genotype *2/*3. However, the acceptance criterion for CR and FA initial values was fulfilled (22/24 = 92% of acceptable predictions). The mean prediction error of the AUC ratio was −0.01 and the mean prediction absolute error was 0.36.

Predicted versus observed AUC ratios for the external validation set of the pharmacogenetic part. The solid line is the identity line (y = x). The upper dotted line represents (y = 2x) and the lower dotted line represents (y = 0.5x). AUC area under the concentration–time curve
3.4 Final Estimates for CRs and FAs
The final estimates of CRs and FAs are shown in Tables 1 and 2. The agreement between predicted and observed AUC ratio with final estimates of CR2C9 and FA2C9 is shown in Fig. 2. The mean prediction error of the AUC ratio was −0.05 and the mean prediction absolute error was 0.25.

Predicted versus observed AUC ratios with final estimates of CR2C9 and FA2C9. The solid line is the identity line (y = x). The upper dotted line represents (y = 2x) and the lower dotted line represents (y = 0.5x). AUC area under the concentration–time curve, CR 2C9 fractional contribution of CYP2C9 to oral clearance in vivo, CYP cytochrome P450, FA 2C9 fraction of activity of CYP2C9
We found that the relative activity of CYP2C9*2*2 and *3*3 compared with *1*1 are 70 and 13 %, respectively. This is fully consistent with the meta-analysis carried out by Dickinson et al. [17], who reported 70 and 10 %.
3.5 Model Predictions for CYP2C9-Mediated Drug–Drug Interactions
The bibliographic search identified 59 relevant articles. Descriptions of each study are shown in Appendix 3 of the Online Resource (drug, dose value, type of study, number of subjects, healthy volunteers or patients, and sampling schedule). We estimated the CR2C9 for four additional drugs (cyclophosphamide: 0.35; meloxicam: 0.59; rosuvastatin: 0.26; and sulfamethoxazole: 0.45) using the interaction method. We calculated the IR2C9 for 27 inhibitors and the IC2C9 for two inducers. IR2C9 values are shown in Table 3. Five inhibitors were actually non-inhibitors (clopidogrel, glibenclamide [glyburide], ibuprofen, losartan and tolbutamide). The IC2C9 values of rifampicin (rifampin) 600 mg/day and bosentan 1,000 mg/day were 1.22 and 0.41, respectively.
External validation was based on 46 AUC ratios. The relationship between the predicted and observed AUC ratio is shown in Fig. 3. The mean prediction error of the AUC ratios was −0.01 and the mean absolute prediction error was 0.22. Two AUC ratios (S-warfarin-bucolome and phenytoin-valproate interactions) were predicted using unbound concentrations (Eq. 5). All predicted ratios were 50–200 % of observed values.

Predicted versus observed AUC ratios for the external validation set (drug interaction model). The solid line is the identity line (y = x). The upper dotted line represents (y = 2x) and the lower dotted line represents (y = 0.5x). Ratios lower than 1 are inducers. Ratios greater than 1 are inhibitors. AUC area under the concentration–time curve
3.6 Drug Dosage Adjustment
Table 4 summarizes drug dosage adjustments predicted by our models, compared with Epar/Vidal dosing guidelines. Twenty out of 26 substrates included in our models had a CR value greater or equal to 0.5 (Table 1). Only CYP2C9*2/*3 and *3/*3 genotypes exhibit an AUC higher than twice the AUC of patients with wild-type genotype. With our models, 33 couples (substrate, genotype) may lead to a dose adjustment, as opposed to only ten couples found in Epar/Vidal dosing guidelines [10, 11].
4 Discussion
DDIs and genetic polymorphisms may increase or reduce drug clearance, making it necessary to alter the dose to keep drug exposure in the normal range. The consequences of DDIs and genetic polymorphisms are the same, as long as the predicted AUC ratios (AUCXM/AUCEM and AUC*/AUC) are the same.
The drug interaction model (Eqs. 3 and 4) predicts AUC ratios for about 700 drug combinations, only 10 % of which have been evaluated in clinical studies. Strong interactions were observed for victim drugs with CRs greater than 0.8 (12 drugs), combined with rifampicin or inhibitors with an IR greater than 0.8 (bucolome, fluoxetine and sulfaphenazole).
4.1 Implications on Some CYP2C9 Substrates
This part is focused on the impact of genetic polymorphism. Standard drug doses achieve normal concentrations and effects in homozygous EMs, but may be toxic in PMs (and in heterozygous EMs or IMs) and ineffective in UMs. Genetic polymorphisms may be clinically relevant if the signal of their effect is greater than the noise from all other sources of inter-individual variability and their therapeutic index is narrow.
The pharmacogenetics of blood concentrations, assuming that blood concentrations mirror the concentrations at target sites, is accepted by regulatory authorities. It is possible to find equivalent doses for different genotypes that modify drug kinetics. However, differences in drug effects between genotype groups are almost always smaller than pharmacokinetic differences. Minor dose adjustments are not needed. For some drugs, plasma concentrations correlate poorly with clinical efficacy and empirical dose-finding rather than drug concentrations is used to avoid adverse drug effects.
4.2 Vitamin K Antagonists
The therapeutic use of vitamin K antagonists is difficult because of a narrow therapeutic index: an international normalized ratio (INR) target between 2.0 and 3.0 is recommended for most indications [18, 19]. The dose-response relationship is not easy to predict, causing frequent bleeding complications or insufficient anticoagulation. CYP2C9 has clearly been established as the predominant catalyst for the metabolism of its more potent S-enantiomer [19]. Vitamin K epoxide reductase complex, subunit 1 (VKORC1) genetic polymorphisms are also an important predictor of acenocoumarol pharmacokinetic–pharmacodynamic responses at the initiation of therapy [20].
For S-acenocoumarol and S-warfarin, our model suggests a decrease of the standard dose by 85 % for homozygous CYP2C9*3 patients and 65 % for CYP2C9*2/*3 patients (Table 4). However, Epar/Vidal specifies that the genetic variability related to CYP2C9 represents roughly 14 % of the inter-subject response variability and no drug dosing adjustment is recommended in addition to INR-based dosages and follow-up. In our opinion, a dose adjustment for S-acenocoumarol and S-warfarin could be beneficial for PM patients at the initiation of therapy in relation to pharmacokinetic variability induced by the CYP2C9 genotype. However, early INR values are routinely determined in clinical practice and dosing is almost continuously adjusted in the early phase of treatment.
Lacut et al. [21] showed that VKORC1 genotypes strongly affect the anticoagulation effects of fluindione. However, the influence of CYP2C9, CYP4F2 and EPHX1 genotypes on fluindione seems weaker than on S-acenocoumarol and S-warfarin. According to our models, the standard dose should be reduced by 70 % for CYP2C9*3/*3, but there are no specific dosing guidelines for these drugs in Epar/Vidal (Table 4).
4.3 NSAIDs
Dosing guidelines are not well-established for NSAIDs, even though they may cause frequent adverse events, such as hepatotoxicity, gastrointestinal and nephrotoxicity, especially with chronic pain [22]. Our study included six NSAIDs: celecoxib, flurbiprofen, ibuprofen, lornoxicam, naproxen and piroxicam. All but naproxen had a CR2C9 value greater than 0.5. We did not find any Epar/Vidal dosing guidelines for four of them (ibuprofen, flurbiprofen, lornoxicam and naproxen). Piroxicam was recommended to be administered with caution for patients with the CYP2C9*3 allele. Epar and Vidal recommend a onethird decrease in the standard dose of celecoxib in PMs.
Ibuprofen is one of the most frequently used NSAIDs. Most preparations contain the racemic mixture of R- and S-ibuprofen [23]. Data from biochemical studies indicate that CYP2C9 may be the major S-ibuprofen hydroxylase, whereas R-ibuprofen appears to be metabolised by CYP2C8 [23]. Our models predict a 2.7-fold increase in AUC for S-ibuprofen in CYP2C9*3/*3 patients, which should lead to a recommended dose reduction of 60 %.
Only one clinical study has assessed the impact of CYP2C9 polymorphisms on the pharmacokinetics of flurbiprofen but no homozygous PMs (i.e. *2/*3, *3/*3 or *2/*2 genotypes) were included [24]. With our model, the standard dose should be reduced by 80 and 60 % for the CYP2C9*3/*3 and CYP2C9*2/*3 genotypes, respectively.
For lornoxicam, our results predicted a sevenfold increase in AUC for CYP2C9*3/*3 genotype patients compared with *1/*1 genotype patients, suggesting a corresponding recommendation of dose reduction.
Piroxicam is the only NSAID metabolised by CYP2C9 for which dosing caution is required because of a high risk of dose-related adverse events in PMs. Our results are in line with these guidelines and suggest a dose reduction of 75 and 55 % for the CYP2C9*3/*3 and *2/*3 genotypes, respectively.
For celecoxib, four clinical studies assessed the impact of CYP2C9 polymorphisms on pharmacokinetic parameters [25–27]. However, there was high variability between studies. The AUC ratio for CYP2C9 homozygous *3 allele patients out of wild-type patients varied between 0.91 and 7.78. This may be because of the low number of patients with this genotype included in the studies (a maximum of three patients with the *3/*3 genotype). Our model suggests decreasing the standard dose by 85 % for homozygous CYP2C9*3 patients and 60 % for CYP2C9*2/*3 patients and this is in line with Epar/Vidal dosing guidelines.
4.4 Oral Antihyperglycaemic Agents
Oral antihyperglycaemic drugs are prescribed for type 2 diabetes mellitus. Although there is no specific therapeutic concentration range, severe adverse effects can occur if concentrations vary too much [28]. We only found one recommendation for nateglinide when administered to PMs.
According to three clinical studies [29–31], exposure to glibenclamide (AUC) varies between 1.4- and 2.6-fold for heterozygous patients with the CYP2C9*3 allele, compared with homozygous patients with the wild-type allele. However, it does not seem to affect the pharmacokinetics of glibenclamide in two studies. Our model suggests being cautious with homozygous *3/*3 patients, in whom the AUC is twofold greater than in wild-type patients.
For nateglinide, data indicate that this substrate is predominantly metabolised by CYP2C9, and by CYP3A4 to a smaller extent. In an interaction trial with sulfinpyrazone, a CYP2C9 inhibitor, a modest increase in nateglinide AUC (~28 %) was observed in healthy volunteers, with no changes in the mean maximum plasma drug concentration and elimination half-life. A more prolonged effect and a potential risk of hypoglycaemia cannot be excluded in patients when nateglinide is co-administered with CYP2C9 inhibitors. Particular caution is recommended in Epar/Vidal dosing guidelines only when nateglinide is co-administered with more potent CYP2C9 inhibitors, e.g. fluconazole or gemfibrozil, or in patients who are known to be PMs for CYP2C9. With our model, no dose adjustment was required.
Tolbutamide is not often prescribed because of a possible increased risk of cardiovascular mortality and adverse effects. Kirchheiner et al. [32] showed a ratio of AUCPM to AUCEM of more than sixfold in homozygous CYP2C9*3 patients. Our results suggest a dose reduction of 80 and 55 % for patients with *3/*3 and *2/*3, respectively, but no specific dosing guidelines are proposed in Epar/Vidal.
4.5 Sartans
The most common drug reactions of sartans are headaches, upper respiratory tract infections, diarrhoea, fatigue and cough [33, 34]. For irbesartan, clinical studies show no relationship between the dosage and overall frequency of adverse reactions. The only clinical study we found in the pharmacogenic literature on irbesartan only included IMs. Our model estimated the CR value at 0.84 for irbesartan, which resulted in a dose reduction of 75 and 55 % for CYP2C9*3/*3 and *2/*3 genotypes, respectively.
4.6 Anticonvulsant Drugs
Phenytoin exhibits a non-linear relationship between dose and serum concentrations, and the therapeutic index is narrow, with a range usually between 5 and 25 μg/mL. Of the orally administrated dose of phenytoin, 70–90 % is oxidized by CYP2C9, and a minor amount by CYP2C19, leading to S-5-(4p-hydroxyphenyl)-5-phenylhydantoin in humans. The relative contribution of CYPC219 to phenytoin metabolism increases as phenytoin concentrations increase, due to saturation of CYP2C9 [35]. We found no Vidal/Epar guidelines on dose adjustment for phenytoin, whereas the AUC ratio of CYP2C9*3*3 patients compared to *1*1 patients is about threefold. Taking into account the narrow therapeutic index of phenytoin, we think that this increase may be clinically significant. This is in agreement with a retrospective analysis of 269 patients with epilepsy. The maximal dose of phenytoin was stratified by the CYP2C9 genotype [36]. Carriers of one or two defective CYP2C9 alleles (i.e. CYP2C9 heterozygous EMs or PMs) apparently required a 13 and 30 % lower dose, respectively [35]. Other studies also indicated that genetic polymorphisms of CYP2C9 and CYP2C19 are closely related to the dose required for the control of seizures in patients [36–40].
Phenobarbital was included in our study, because several pathways (including CYP2C9) are responsible for various proportions of total phenobarbital elimination. Although phenobarbital p-hydroxylation co-segregates with CYP2C19 activity [41], our results show that CYP2C9 is a major pathway for phenobarbital metabolism. This is another reason to monitor the phenobarbital concentration in plasma. However, the use of this drug in developed countries is declining in favour of alternative drugs with improved safety.
4.7 Cholesterol-Lowering Drugs
Kirchheiner et al. [42] studied the impact of CYP2C9*2 and *3 genotypes on the pharmacokinetics and pharmacodynamics of fluvastatin in healthy volunteers. The pharmacokinetics of both enantiomers showed statistically significant differences according to the number of *3 alleles and no effect on *2 was observed. No correlation with the CYP2C9 genotype was found with either pharmacokinetics or cholesterol-lowering effects. However, Sabia et al. [43] reported that high fluvastatin concentrations may be related to adverse drug effects such as diarrhoea, headaches and clinically relevant elevations in serum transaminase concentrations. Our results suggest decreasing the dose because we observed a 2.6-fold increase in AUC for the 3R-5S active enantiomer in patients with the CYP2C9*3/*3 genotype.
4.8 Limits of the Proposed Modelling Approach
Our models rely on several assumptions. Firstly, the FA of a variant (relative to wild-type) is assumed to be the same with respect to all substrates. This strong assumption has been questioned after results from in vitro experiments [44]. Secondly, the method may only be used for drugs that have linear kinetics. The predictions would be biased if the method is used for drugs exhibiting non-linear kinetics at the usual dosing regimen. The large variability of AUC ratio predictions in XMs and EMs for phenytoin (95 % CI 1.7–4.9) may be explained by its non-linear kinetics. The clinical interpretation of an AUC ratio may also be difficult with respect to drugs that have enantiomers and/or active metabolites. The quantitative prediction of the impact of an almost complete loss of CYP2C9 activity on a substrate that is almost exclusively cleared by CYP2C9 metabolism would probably be imprecise because a small variation in CR and/or FA values may result in a large variation in AUC ratios.
Other limitations are specific to drug polymorphisms. Firstly, the analyses were not conducted for other important variants of CYP2C9, such as *5, *6, *8 or *11, because of a paucity of data. These CYP2C9 alleles were only detected in African populations, such as CYP2C9*5, CYP2C9*6, CYP2C9*8 and CYP2C9*11 (allele frequencies in African populations of 1.8, <1, 8 and 2.7 %, respectively) [45–47]. A single African subject with epilepsy was identified as carrying a homozygous deletion of the CYP2C9 gene (CYP2C9*6), which means it is possible to live without this gene [48]. The allele frequency of this variant was 0.6 % in the African population studied, and this allele was not found in Asian or Caucasian populations. It remains to be seen whether some of the numerous non-coding CYP2C9 polymorphisms can contribute to the genotypic prediction of the CYP2C9 metabolic phenotype. Our approach may be extended to these variants once sufficient data become available. Secondly, the impact of genetic polymorphisms may interact with other factors such as race, which is not accounted for in the present models and may result in some bias.
5 Conclusion
As with many complex and new technologies, there are problems and delays in the transfer of scientific pharmacogenetic knowledge from bench to bedside. Specific translational research has to be supported. This modelling approach can only suggest some recommendations in terms of dosing adjustment in patients with specific CYP2C9 genotypes or co-medications. However, their clinical usefulness in terms of efficacy/toxicity clearly needs to be further quantified and validated. The interest of CYP2C9 genotyping before drug prescription should be assessed in prospective randomized clinical trials, in which one group of patients is treated with consideration for genotype information (and subsequent dosing adjustment) and compared with another group, in which physicians do not have genotype information and conventional empirical therapy is used. Our models may help design such studies and select study drugs.
References
Weinshilboum R. Richard Weinshilboum: pharmacogenetics: the future is here! Mol Interv. 2003;3(3):118–22.
Meyer UA. Pharmacogenetics and adverse drug reactions. Lancet. 2000;356(9242):1667–71.
Wolf CR, Smith G, Smith RL. Science, medicine, and the future: pharmacogenetics. BMJ. 2000;320(7240):987–90.
Lapple F, et al. Differential expression and function of CYP2C isoforms in human intestine and liver. Pharmacogenetics. 2003;13(9):565–75.
Lasker JM, et al. Characterization of CYP2C19 and CYP2C9 from human liver: respective roles in microsomal tolbutamide, S-mephenytoin, and omeprazole hydroxylations. Arch Biochem Biophys. 1998;353(1):16–28.
Kirchheiner J, Brockmoller J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther. 2005;77(1):1–16.
Xie HG, et al. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv Drug Deliv Rev. 2002;54(10):1257–70.
Tod M, Goutelle S, Gagnieu MC. Genotype-based quantitative prediction of drug exposure for drugs metabolized by CYP2D6. Clin Pharmacol Ther. 2011;90(4):582–7.
Tod M, Goutelle S, Clavel-Grabit F, Nicolas G, Charpiat B. Quantitative prediction of cytochrome P450 (CYP) 2D6-mediated drug interactions. Clin Pharmacokinet. 2011;50(8):519–30.
European public assessment reports. 2012. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124. Accessed 1 Jun 2012.
VIDAL. Fiches médicaments. 2012. http://www.vidalpro.net/. Accessed 1 Jun 2012.
Gibbs JP, Hyland R, Youdim K. Minimizing polymorphic metabolism in drug discovery: evaluation of the utility of in vitro methods for predicting pharmacokinetic consequences associated with CYP2D6 metabolism. Drug Metab Dispos. 2006;34(9):1516–22.
Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–96.
Ohno Y, Hisaka A, Ueno M, Suzuki H. General framework for the prediction of oral drug interactions caused by CYP3A4 induction from in vivo information. Clin Pharmacokinet. 2008;47(10):669–80.
Takahashi H, Echizen H. Pharmacogenetics of CYP2C9 and interindividual variability in anticoagulant response to warfarin. Pharmacogenomics J. 2003;3(4):202–14.
Fischer TL, et al. Evaluation of potential losartan-phenytoin drug interactions in healthy volunteers. Clin Pharmacol Ther. 2002;72(3):238–46.
Dickinson GL, et al. The use of mechanistic DM-PK-PD modelling to assess the power of pharmacogenetic studies—CYP2C9 and warfarin as an example. Br J Clin Pharmacol. 2007;64(1):14–26.
Ansell J, et al. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):204S–33S.
Ufer M. Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin Pharmacokinet. 2005;44(12):1227–46.
Verstuyft C, Delavenne X, Rousseau A, Robert A, Tod M, Diquet B, et al. A pharmacokinetic-pharmacodynamic model for predicting the impact of CYP2C9 and VKORC1 polymorphisms on fluindione and acenocoumarol during induction therapy. Clin Pharmacokinet. 2012;51(1):41–53.
Lacut K, Ayme-Dietrich E, Gourhant L, Poulhazan E, Andro M, Becquemont L, et al. Impact of genetic factors (VKORC1, CYP2C9, CYP4F2 and EPHX1) on the anticoagulation response to fluindione. Br J Clin Pharmacol. 2012;73(3):428–36.
Herndon CM, et al. Management of chronic nonmalignant pain with nonsteroidal antiinflammatory drugs: joint opinion statement of the Ambulatory Care, Cardiology, and Pain and Palliative Care Practice and Research Networks of the American College of Clinical Pharmacy. Pharmacotherapy. 2008;28(6):788–805.
Kirchheiner J, et al. Enantiospecific effects of cytochrome P450 2C9 amino acid variants on ibuprofen pharmacokinetics and on the inhibition of cyclooxygenases 1 and 2. Clin Pharmacol Ther. 2002;72(1):62–75.
Lee CR, et al. Differences in flurbiprofen pharmacokinetics between CYP2C9*1/*1, *1/*2, and *1/*3 genotypes. Eur J Clin Pharmacol. 2003;58(12):791–4.
Tang C, et al. In-vitro metabolism of celecoxib, a cyclooxygenase-2 inhibitor, by allelic variant forms of human liver microsomal cytochrome P450 2C9: correlation with CYP2C9 genotype and in-vivo pharmacokinetics. Pharmacogenetics. 2001;11(3):223–35.
Sandberg M, et al. Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br J Clin Pharmacol. 2002;54(4):423–9.
Brenner SS, Herrlinger C, Dilger K, Mürdter TE, Hofmann U, Marx C, et al. Influence of age and cytochrome P450 2C9 genotype on the steady-state disposition of diclofenac and celecoxib. Clin Pharmacokinet. 2003;42(3):283–92.
Becker ML, et al. Cytochrome P450 2C9 *2 and *3 polymorphisms and the dose and effect of sulfonylurea in type II diabetes mellitus. Clin Pharmacol Ther. 2008;83(2):288–92.
Kirchheiner J, et al. Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther. 2002;71(4):286–96.
Niemi M, et al. Glyburide and glimepiride pharmacokinetics in subjects with different CYP2C9 genotypes. Clin Pharmacol Ther. 2002;72(3):326–32.
Yin OQ, Tomlinson B, Chow MS. CYP2C9, but not CYP2C19, polymorphisms affect the pharmacokinetics and pharmacodynamics of glyburide in Chinese subjects. Clin Pharmacol Ther. 2005;78(4):370–7.
Kirchheiner J, Bauer S, Meineke I, Rohde W, Prang V, Meisel C, et al. Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics. 2002;12(2):101–9.
Bourrie M, et al. Role of cytochrome P-4502C9 in irbesartan oxidation by human liver microsomes. Drug Metab Dispos. 1999;27(2):288–96.
Simon TA, et al. Safety of irbesartan in the treatment of mild to moderate systemic hypertension. Am J Cardiol. 1998;82(2):179–82.
Saruwatari J, Ishitsu T, Nakagawa K. Update on the genetic polymorphisms of drug-metabolizing enzymes in antiepileptic drug therapy. Pharmaceuticals. 2010;3(8):2709–32.
Tate SK, et al. Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc Natl Acad Sci USA. 2005;102(15):5507–12.
Mamiya K, et al. The effects of genetic polymorphisms of CYP2C9 and CYP2C19 on phenytoin metabolism in Japanese adult patients with epilepsy: studies in stereoselective hydroxylation and population pharmacokinetics. Epilepsia. 1998;39(12):1317–23.
Hung CC, et al. Dosage recommendation of phenytoin for patients with epilepsy with different CYP2C9/CYP2C19 polymorphisms. Ther Drug Monit. 2004;26(5):534–40.
Odani A, et al. Genetic polymorphism of the CYP2C subfamily and its effect on the pharmacokinetics of phenytoin in Japanese patients with epilepsy. Clin Pharmacol Ther. 1997;62(3):287–92.
van der Weide J, et al. The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics. 2001;11(4):287–91.
Hadama A, et al. P-hydroxylation of phenobarbital: relationship to (S)-mephenytoin hydroxylation (CYP2C19) polymorphism. Ther Drug Monit. 2001;23(2):115–8.
Kirchheiner J, Kudlicz D, Meisel C, Bauer S, Meineke I, Roots I, et al. Influence of CYP2C9 polymorphisms on the pharmacokinetics and cholesterol-lowering activity of (−)-3S,5R-fluvastatin and (+)-3R,5S-fluvastatin in healthy volunteers. Clin Pharmacol Ther. 2003;74(2):186–94.
Sabia H, et al. Safety, tolerability, and pharmacokinetics of an extended-release formulation of fluvastatin administered once daily to patients with primary hypercholesterolemia. J Cardiovasc Pharmacol. 2001;37(5):502–11.
Shen H, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos. 2007;35(8):1292–300.
Dickmann LJ, et al. Identification and functional characterization of a new CYP2C9 variant (CYP2C9*5) expressed among African Americans. Mol Pharmacol. 2001;60(2):382–7.
Yasar U, et al. Analysis of CYP2C9*5 in Caucasian, Oriental and black-African populations. Eur J Clin Pharmacol. 2002;58(8):555–8.
Allabi AC, et al. Functional impact of CYP2C95, CYP2C96, CYP2C98, and CYP2C911 in vivo among black Africans. Clin Pharmacol Ther. 2004;76(2):113–8.
Kidd RS, et al. Identification of a null allele of CYP2C9 in an African-American exhibiting toxicity to phenytoin. Pharmacogenetics. 2001;11(9):803–8.
Kirchheiner J, et al. Effects of CYP2C9 polymorphisms on the pharmacokinetics of R- and S-phenprocoumon in healthy volunteers. Pharmacogenetics. 2004;14(1):19–26.
Loebstein R, et al. Interindividual variability in sensitivity to warfarin: nature or nurture? Clin Pharmacol Ther. 2001;70(2):159–64.
Acknowledgments
This study was partially funded by the BIMBO project (ANR SYSCOMM 2008-002). The authors have no conflicts of interest that are directly relevant to the content of this study. The authors would like to thank Kent Neal (supported by the French Cochrane Center) for proofreading the manuscript.
Members of the Genophar II Working Group
Nathalie Bleyzac, Laurent Bourguignon, Bruno Charpiat, Sylvain Goutelle, Emilie Hénin and Marie-Claude Gagnieu.
Author information
Authors and Affiliations
Consortia
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Castellan, AC., Tod, M., Gueyffier, F. et al. Quantitative Prediction of the Impact of Drug Interactions and Genetic Polymorphisms on Cytochrome P450 2C9 Substrate Exposure. Clin Pharmacokinet 52, 199–209 (2013). https://doi.org/10.1007/s40262-013-0031-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0031-3