
Abstract
Cutaneous lupus erythematosus (CLE) includes a broad range of dermatologic manifestations, which may or may not be associated with systemic disease. Recent studies in this area continue to shape our understanding of this disease and treatment options. Epidemiologic studies have found an incidence of CLE of 4.30 per 100,000, which approaches similar analysis for systemic lupus erythematosus (SLE). Although there have been extensive efforts to define SLE, the classification of CLE and its subgroups remains a challenge. Currently, diagnosis relies on clinical and laboratory findings as well as skin histology. The Cutaneous Lupus Area and Severity Index™ (CLASI™) is a validated measure of disease activity and damage. CLE pathogenesis is multifactorial and includes genetic contributions as well as effects of ultraviolet (UV) light. Immune dysregulation and aberrant cell signaling pathways through cytokine cascades are also implicated. Patient education and avoidance of triggers are key to disease prevention. Antimalarials and topical steroids continue to be the standard of care; however, immunosuppressants, thalidomide analogs and monoclonal antibodies are possible systemic therapies for the treatment of recalcitrant disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A Delphi project to define cutaneous lupus erythematosus is underway. |
Pathogenesis is multifactorial, with key contributions from genetics, environmental triggers and immune dysregulation. |
Certain monoclonal antibodies and thalidomide analogs are promising new therapies. |
1 Introduction
Lupus erythematosus (LE) is an autoimmune disease with a variety of clinical manifestations ranging from multi-organ system involvement [systemic lupus erythematosus (SLE)] to limited cutaneous disease. Dermatologic findings are often an early sign of LE and are a manifestation of the disease in a majority of patients. While LE pathogenesis is not fully understood, it is a multifactorial disorder with genetic and environmental contributions, leading to immune activation. LE associated skin lesions show varied expression, leading to the broad classification as LE specific and LE nonspecific [1]. The latter include vascular changes such as livedo reticularis, periungual telangiectasias, Raynaud’s phenomenon, erythema multiforme, and calcinosis cutis, findings usually associated with SLE but also seen with other disorders. LE-specific lesions refer to the subtypes of cutaneous lupus erythematosus (CLE) that are grouped on the basis of histology, duration of lesions, laboratory abnormalities, and clinical findings. These are grouped into acute cutaneous LE (ACLE), subacute cutaneous LE (SCLE) (Fig. 1), and chronic cutaneous LE (CCLE), which includes discoid LE (DLE) (Fig. 2) and LE profundus (LEP), chilblain LE (CHLE), and LE tumidus (LET).

Subacute cutaneous lupus erythematous lesions with papulosquamous plaques on the upper back, some of which have associated white scale. Note the photodistribution

Discoid lupus erythematosus lesion with erythema and hyperkeratosis with hypopigmentation and hyperpigmentation at the periphery
CLE may result in significant disfigurement and discomfort, leading to poor quality of life. Avoidance of triggers such as significant sun exposure is key to preventing symptoms. Treatment with topical steroids may be sufficient for mild disease. Oral antimalarials are the standard of care for more severe presentations. If these do not work or if the disease is more aggressive, the use of other immunomodulatory and immunosuppressant therapies, as well as biologic therapies, are possible alternatives.
2 Epidemiology
The annual incidence of SLE is approximately 1–10 per 100,000, and the prevalence of SLE is approximately 5.8–130 per 100,000 [2, 3]. These estimates vary based on the sex, race, age, and ethnicity of the population. Although SLE epidemiology has been relatively well studied in a variety of worldwide populations, there is a lack of knowledge about CLE. Within the population of Olmsted County, Minnesota, from 1965 to 2005 the incidence of CLE was 4.30 per 100,000, with a female predominance and an average age of onset of 48.5 years [4]. Subsequent studies in Sweden largely support these data [5]. In a more recent examination of the same population from 1993 to 2005, a direct epidemiologic comparison was performed between SLE and CLE and found that the incidence of SLE was similar to that of CLE (2.9 per 100,000 and 4.2 per 100,000, respectively, P = 0.10). CLE was three times more common than SLE in men [6]. However, it is important to note that the populations studied, both in Minnesota and Sweden, are predominantly white and located in the upper latitudes of the globe. The expression of SLE is influenced by race, particularly affecting African-American and Asian populations [7–9]. Given this and the known photosensitivity of CLE subtypes, further epidemiologic studies with a more racially diverse population in varied locales would be valuable.
3 CLE Subtype Classification
The American College of Rheumatology (ACR) initially established the criteria for the classification of SLE, and these were last revised in 1997. These criteria consist of 11 clinical and laboratory components, four of which must be met to diagnose SLE. The dermatology community has raised concerns about the ACR criteria over the lack of specificity and the large number of skin criteria. In particular, four criteria designate mucocutaneous involvement (malar rash, discoid lesions, photosensitivity, and oral ulcers), and these can potentially be used to diagnose SLE in patients with skin disease [10]. In 2012, the Systemic Lupus International Collaborating Clinics (SLICC) undertook a revision of the ACR criteria to address these and other concerns. Meant to be more clinically relevant, they proposed SLE classification criteria based on fulfilling at least four criteria as well, with at least one immunologic and one clinical, also making an allowance for proven lupus nephritis in the presence of antinuclear antibodies (ANAs) or anti-double-stranded DNA (anti-dsDNA) antibodies [11]. Of the clinical criteria, four are mucocutaneous; however, malar rash and photosensitivity are grouped together under the acute cutaneous criterion, and nonscarring alopecia is a new, separate category. The SLICC criteria are more sensitive than the ACR criteria but less specific, and although fewer patients were misclassified using the SLICC versus the ACR criteria, the difference between the two was not statistically significant [11]. Currently, both the ACR and SLICC criteria are used to classify SLE, and the choice is ultimately based on user preference.
While the definition of SLE has been extensively studied, systematic work to define CLE is just beginning. In 1981, Gilliam and Sontheimer posed a schema to organize LE lesions mainly on the basis of clinical appearance, grouping lesions as CCLE, SCLE, and ACLE [1]. Within these subgroups, there are general commonalties in the duration of lesions, histology, and laboratory findings. To date there is no uniform CLE definition despite several proposals by the LE community. In 2013, the 3rd International Meeting on Cutaneous Lupus Erythematosus (ICCLE) was held to develop a process to achieve consensus of uniform definitions, diagnostic criteria, and classification of CLE. Out of this meeting came the decision to employ the Delphi consensus method as a means of developing a classification scheme [12]. In a follow-up to the ICCLE, a “pre-Delphi” questionnaire evaluated by experts in the field found a consensus that there is a need for a new CLE definition and current schemes are inadequate for communication and prognostic information [13]. A formal Delphi process is currently underway, with a focus on initially characterizing DLE.
4 Diagnosis and Assessment
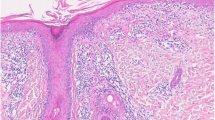
The diagnosis of CLE is based on clinical and laboratory findings as well as histologic assessment of skin biopsy specimens. Cutaneous histopathology is similar between LE subtypes. Histologic features include interface dermatitis with a mononuclear cellular infiltrate at the dermoepidermal junction (DEJ), and perivascular and periadnexal inflammation. More pronounced dermal edema and epidermal atrophy may be notice in SCLE specimens, while DLE may demonstrate more follicular plugging and inflammation that extends in the dermis (Fig. 3). All forms of LE may show a deposition of immunoglobulin (usually IgG) and complement components (usually C3) at the DEJ of lesional skin. Histopathology and immunopathology may be helpful in the diagnosis of LE but have little utility in determining clinical subtype [14].

Discoid lupus erythematosus. This scanning view shows an atrophic epidermis, vacuolar change, hyperkeratosis, follicular plugging, and a perivascular and periadnexal chronic inflammatory cell infiltrate. H & E, ×10
Since SLE may initially present with cutaneous manifestations, all patients presenting with CLE should be evaluated for systemic involvement. This includes a full history, review of systems and physical exam, as well as laboratory testing. A complete blood count, liver function tests, measurements of renal function, and urinalysis are appropriate. Serologic assessment of autoantibodies should also be performed. ANAs are not specific for SLE, but higher titers (≥1:320) are more suggestive of SLE over CLE [15]. Anti-dsDNA and anti-Smith (anti-Sm) are highly specific for SLE, but are present in about 70 and 25 % of patients, respectively. Anti-La (SSB) and anti-Ro (SSA) may also be present with SLE, although less specific [16]. The frequency of antiphospholipid antibodies is between 25 and 61 % in SLE [17] and 5.8 and 68 % in CLE [18]. Further work-up should be performed in LE patients with livedo reticularis or signs of thrombosis (i.e., stroke, recurrent miscarriage, deep venous thrombosis) to include the dilute Russell viper venom test, lupus anticoagulant-sensitive partial thromboplastin time, anti-cardiolipin IgG and IgM antibodies, and β2-glycoprotein I IgG and IgM antibodies, noting that the presence of IgG is more indicative of antiphospholipid syndrome.
A comprehensive skin examination performed during flares as well as at regular intervals is key to the assessment of disease progression and damage. Although several disease scoring methods such as the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) and British Isles Lupus Assessment Group (BILAG) index have been used to evaluate disease in SLE patients, they lack the sensitivity necessary for the skin evaluation of disease activity within the subtypes of CLE. Accordingly, various tools have been proposed to aid clinicians and researchers in specifically assessing cutaneous disease. Developed in 2005, the Cutaneous Lupus Area and Severity Index™ (CLASI™) is a tool used to specifically measure disease activity and damage [19]. This tool takes into account anatomical location and lesional morphology and has been validated for use by both dermatologists and rheumatologists, providing an efficient means of disease measurement for use in a busy clinical setting [20]. Further studies on the CLASI™ have shown it to be a valuable means to characterize disease severity and identify clinical improvement and responsive to biomarkers of disease pathogenesis [21–23]. Importantly, this index is useful as a disease-related, quality-of-life indicator and is also consistent with patient-reported assessments of body image and health status [24, 25]. The CLASI™ remains an effective instrument across different ethnic populations, including in the assessment of cutaneous disease-related activity and damage within Indian patients as well as in quality-of-life measures in Japanese patients [26, 27]. The CLASI™ is also a reliable indicator of treatment responsiveness, with a 4-point or 20 % decrease in CLASI™ activity score being a specific criterion for classifying patients as responders or nonresponders [21]. The CLASI™ is currently used in almost all international studies that want to evaluate cutaneous lupus. Other tools have not been fully validated [28, 29], present problems with difficult parameters to ascertain by dermatologists, let alone rheumatologists, such as differentiating infiltration versus hypertrophy, subcutaneous nodules, and surface area [28], and present limitations with the scope of disease measured [28, 29].
5 Pathogenesis
CLE pathogenesis is a multifactorial process that includes contributions from genetics, environmental triggers, and innate and adaptive immune response. The mechanism is not fully elucidated, but increasing evidence points towards the involvement of ultraviolet (UV) irradiation, alteration of cells and their contents, T cell dysregulation, B cell defects and autoantibody generation, dendritic cell (DC) activation, and chemokine and cytokine imbalances [30].
5.1 Genetics
Earlier genetic studies have established human leukocyte antigen (HLA) associations with CLE subtypes; specifically, the HLA B8, DR3, DQA1, and DRB1. HLA DR3 and DR2 haplotypes are associated with positive anti-Ro/SSA autoantibodies and SCLE, while certain alleles of HLA DQA1 and DRB1 appear to be associated with DLE [31, 32]. More recent research has focused on genes outside the major histocompatibility (MHC) region. Using single nucleotide polymorphisms (SNPs), a panel of genes with strong susceptibility to SLE were applied to plasma samples from DLE and SCLE patients [33]. Tyrosine kinase 2 (TYK2), interferon regulatory factor 5 (IRF5), and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) are associated with CLE. Specifically, TYK2, a Janus kinase that binds to the interferon (IFN)-α receptor 1 and is involved in cytokine signaling, is associated with DLE. However, clinical characteristics were not significantly associated with specific genotypes of TYK2 or IRF5.
Familial CHLE is a genodermatosis with an autosomal dominant transmission and has been mapped to chromosome 3p [34]. Underlying the disease is a missense mutation in the TREX1 gene, which encodes an exonuclease that breaks down single-stranded or mispaired dsDNA. TREX1 is important in the caspase-independent apoptotic pathway, specifically, granzyme A-mediated cell death [35]. Regulated cell death is important for self-tolerance and may lead to autoreactive immune cells.
ITGAM encodes for the cell surface receptor for multiple ligands that are expressed on key immune cells such as neutrophils, macrophages, and DCs. Polymorphisms at this locus present a greater risk for DLE compared with SLE; the risk of DLE is independent of systemic involvement [36]. Defects in the ITGAM pathway may predispose patients to DLE through impaired phagocytosis, leukocyte trafficking, or immune suppression in UV-exposed skin.
5.2 The Role of UV Light
UV exposure is a common trigger for CLE, and photosensitivity is an established ACR criterion to diagnose SLE. UV light induces cytokine release and apoptosis in the skin.
5.2.1 Apoptosis
Apoptosis is a normal part of the cell cycle; however, UV light has been shown to cause keratinocyte apoptosis via pathways such as the generation of reactive oxygen species, DNA damage, and activation of Fas and FasL [37]. One study found elevated levels of apoptotic keratinocytes in UV-exposed skin within the stratum granulosum in CLE patients, as well as increased apoptotic cells in non-lesional skin. They noted that apoptotic cells continued to accumulate up to 70 h after UV exposure. This may be due to accumulation secondary to abnormal clearance of apoptotic cells, leading to necrosis, inflammation, and an autoimmune response [38]. Another study using UVB in SLE patients did not find a significant difference in apoptosis induction, clearance rate of apoptotic cells, or secondary necrosis in tested skin compared with healthy controls; however, there was a noted influx of macrophages and inflammatory lesions in the vicinity of apoptotic cells. These discordant results may be due to differences in technique. The authors believe this may represent an inflammatory clearance of apoptotic cells [39].
5.2.2 Cytokines and Immune Cells
Skin infiltration by leukocytes and other immune cells in response to UV light is key to the development of CLE lesions. UV radiation facilitates the production and release of cytokines and chemokines that stimulate inflammation and recruitment of immune cells. UVB radiation causes tumor necrosis factor (TNF)-α release from keratinocytes and dermal fibroblasts. There is a synergistic role between interleukin (IL)-1 and UVB radiation, leading to increased TNF-α in keratinocytes. This leads to the cytokine-induced inflammatory cascade that eventually contributes to CLE lesions [40].
5.2.3 Photoprovocation
Since the 1960s, photoprovocation has been studied to better evaluate photosensitivity in LE [41]. More recently in 2011, a multicenter European study evaluated a standardized photoprovocation protocol using UVA and UVB irradiation in 47 patients with various CLE subtypes [42]. About half of the CLE patients and none of the healthy controls responded to photoprovocation testing with inducible lesions consistent with LE type histopathology. In a larger, retrospective study using the standardized photoprovocation protocol in 431 CLE patients, more than 60 % expressed positive photoprovocation and also demonstrated that photosensitivity may vary over the course of the disease [43]. Recent work aimed at identifying biomarkers in skin and blood samples of photoprovoked CLE subjects found a specific plasma peptide signature including beta 2-microglobulin, human beta-defensin-1, CD99-derived peptides, polymeric immunoglobulin receptor, and immunoglobulin kappa light chains [44]. Furthermore, there was a marked increase of beta 2-microglobulin, which is essential for MHC class I expression and antigen presentation, in the UV-induced lesional samples. Further studies in this area may help lead to predictors of photosensitivity while providing further insight into CLE pathogenesis.
5.3 Autoantibodies
Various autoantibodies are associated with LE, as described previously, but their role in CLE pathogenesis is unclear. Recent work has focused on understanding their association with CLE subtypes and clinical manifestations. Anti-Ro/SSA antibodies were detected in 72.1 % of patients with SCLE, 47.4 % of patients with ACLE, and 22 % of patients with DLE. Anti-La/SSB antibodies were detected in 36.2 % of patients with SCLE, 27.5 % of patients with ACLE, and 7.0 % of patients with DLE [45]. Other studies have explored the role of antibodies as prognostic indicators. Cluster analysis in a mostly Chinese SLE population showed discoid rash, photosensitivity, and hematologic involvement are associated with anti-Ro/SSA and anti-La/SSB autoantibodies, while malar rash, oral ulcers, arthritis, and serositis are associated with anti-Sm, anti-ribonuclear protein (anti-RNP) and antiphospholipid (anti-PL) antibodies. Solely, anti-dsDNA is associated with renal involvement [46]. In another study performed in a mostly Caucasian population of SLE patients, anti-Sm antibodies were associated with discoid rash and photosensitivity, anti-U1RNP antibodies were associated with Raynaud’s and malar rash, and anti-Ro/SSA antibodies were associated with malar rash, oral ulcers, xerostomia, xerophthalmia, and the presence of rheumatoid factor [47]. Discrepancies between these studies could be related to differences in analysis and/or ethnic backgrounds, and further studies are needed to better elucidate the role of autoantibodies.
5.4 The Role of Signaling Molecules
5.4.1 Apoptotic Signaling
Recent work has shown that CLE lesions highly express Fas (CD95), a cell surface death receptor that mediates the extrinsic apoptotic pathway. TNF-related apoptosis-inducing ligand (TRAIL), a pro-apoptotic protein, is expressed by keratinocytes strongly expressed in the skin and the blood of patients with CLE. Similarly TRAIL-R1, the keratinocyte receptor that mediates TRAIL apoptosis, is also enriched, while TRAIL-R4, a TRAIL receptor with anti-apoptotic properties, appears to be decreased. IFN-α strongly induces TRAIL expression [48, 49].
5.4.2 IL-18
IL-18 is a proinflammatory cytokine within the IL-1 superfamily. In vitro it stimulates IFN-γ and TNF-α expression and induces the production of chemokines which promote the infiltration of type 1 T cells [50]. CLE patients express high levels of the IL-18 receptor on cell surfaces in response to stimulation by IFN-γ and TNF-α, also demonstrating an increase in TNF-α in an autocrine fashion. In addition, in the presence of IL-18, CLE keratinocytes failed to express IL-12, which has been shown to protect keratinocytes from UV-induced apoptosis [51]. These results are supported by the IL-18 promoter polymorphisms seen in some SLE patients [52].
5.4.3 Type I IFNs
Type I IFNs (such as IFN-α and IFN-β) are usually part of the viral defense in a normal immune system; however, they are made in higher levels by plasmacytoid DCs in CLE lesions. The antiviral myxovirus (MxA) protein is a specific surrogate marker for type I IFN production and has been found in CLE lesions, further supporting the role of type 1 IFN in CLE pathogenesis. Skin lesions in scarring DLE are specifically characterized by a large number of skin-homing granzyme B-positive lymphocytes closely associated with the lesional expression of the MxA protein [53]. Lesional type I IFN production is a key factor driving inflammation, promoting a T helper cell-1-biased infiltrate that may contribute to autoimmunity [54].
6 Treatment
CLE treatment remains a challenge as no medication has been approved specifically for CLE, despite several agents that have been studied and approved for SLE. Few randomized, double-blind, placebo-controlled studies are available, and systemic agents are often applied to CLE for off-label use (Table 1).
6.1 Prevention
The first aim within CLE treatment should be educating patients on their disease and avoiding potential triggers or exacerbating influences. Minimizing sun exposure and strict sunscreen adherence with chemical and/or physical blocking agents are essential [55]. The application of broad-spectrum sunscreen with a sun protection factor (SPF) of at least 50 should be applied in sufficient amounts (approximately 2 mg/cm2) about 20–30 min prior to expected exposure. This recommendation is based on the findings of a vehicle-controlled, randomized, double-blind trial of 25 photosensitive CLE patients, which provided 100 % protection from UV-induced skin lesions in patients with different subtypes of the disease [56]. Patients should also be advised to avoid activities such as tanning, sunbathing, working outdoors, and travel to areas where sun exposure is difficult to avoid or the sun is particularly strong. UV exposure via indoor lighting should also be considered. Patients may be advised to use bulbs with the lowest UV emission and effective irradiance and perhaps more realistically to cover fluorescent bulbs with an acrylic diffuser or shade and to use glass-covered halogen lamps which significantly filter out UV [57, 58]. A recent study expanded photosensitivity testing to include energy-efficient halogen (EEF) and light-emitting diode (LED) bulbs, concluding that LEDs offer a safe alternative light source for individuals with photosensitive LE without the UV risk [59].
Vitamin D deficiency is common in CLE patients, and levels may remain low throughout the year [60]. Furthermore, a cross-sectional study showed that CLE increases the odds of inadequate serum 25-hydroxyvitamin D levels in patients compared with age- and sex-matched controls, even after adjusting for sun exposure and sunscreen use. This was expanded to compare disease severity in CLE patients with baseline vitamin D insufficiency. Those treated with vitamin D3 supplementation for 1 year had a significant improvement on CLASI™ scores and a reduction in the number of days with active lesions compared with untreated CLE patients [61]. Low vitamin D levels in SLE patients have also been associated with worse disease severity, but not flare-ups [62]. Vitamin D supplementation may provide beneficial immunologic effects, as one study in SLE patients showed a decrease in memory B cells and effector T cells and an increase of regulatory T cells in patients treated with vitamin D relative to controls [63]. Although vitamin D has yet to be studied in CLE with randomized control trials, serum 25-hydroxyvitamin D levels should be monitored, and supplementation with at least 400 IU per day of vitamin D3 (cholecalciferol) is recommended [64].
Smoking is another modifiable factor that may have an effect on disease severity and response to treatment. A study in the European Society of Cutaneous Lupus Erythematosus (EUSCLE) cohort examined the smoking behaviors of 838 CLE patients. The authors noted a high number of patients with CLE who have ever smoked (59.5 %) and who were already smoking at the date of their first diagnosis (87.2 %). Furthermore, the CLASI™ damage score was significantly higher in patients who have ever smoked, compared with nonsmokers [65]. This suggests that smoking may also induce and/or aggravate the disease and agrees with earlier studies that showed similar trends with DLE [66, 67]. In a British multicenter observational and pharmacogenetic study of 200 patients with DLE treated with hydroxychloroquine (HCQ), 39 % failed to respond or were intolerant to treatment. Further pharmacogenetic analysis of cigarette smoking and the cytochrome P450 (CYP) genotype did not have any significant influence on response to HCQ, while concomitant SLE and disseminated disease were associated with poor response to the drug. These results suggest that baseline lupus severity and SLE are predictors of response to HCQ and smoking may be more of a class effect rather than a risk factor in and of itself [68]. An American CLE cohort found that current smokers with CLE had greater disease activity and a worse quality of life than nonsmokers. Current smokers also did worse when treated with a combination of antimalarial drugs and immunomodulators, but when treated with antimalarial drugs alone, they responded better than nonsmokers. Dissimilarities in the results could be due to difference in baseline disease severity between treatment groups and the quantity of cigarettes smoked [69]. A recent meta-analysis concluded that smoking is associated with a twofold decrease in the proportion of patients with CLE achieving cutaneous improvement with antimalarials [70]. Although the true impact of tobacco on CLE response to antimalarials remains unclear without randomized control trials, patients should be assessed as to their smoking habits and offered help to quit if necessary.
6.2 Calcineurin Inhibitors
Topical calcineurin inhibitors have emerged as an alternative treatment to steroids for CLE lesions. A double-blind, randomized controlled trial in 20 CLE patients treated half the face with tacrolimus 0.1 % ointment and the other half with clobetasol propionate 0.05 % ointment. Both treatments showed similar efficacy, but areas of the face treated with clobetasol developed significantly more telangiectasias, suggesting that tacrolimus may be a better option, so as to avoid the unfavorable effects of steroids [71]. A 2011 randomized, double-blind, vehicle-controlled trial using tacrolimus 0.1 % ointment in 20 patients with various CLE subtypes showed a significant improvement after 28 and 56 days but not after 84 days. Patients with LET showed the highest degree of improvement [72]. Topical calcineurin inhibitors may enhance the therapeutic effect of antimalarials as well [73].
6.3 Antimalarials
Oral antimalarials, including HCQ, chloroquine, and quinacrine, continue to be first-line treatment for systemic CLE treatment. HCQ has a low toxicity profile that makes it a good candidate for long-term use. Early treatment with HCQ may delay the onset of systemic involvement in SLE patients [74]. A 2012 study examined HCQ levels of CLE patients on HCQ monotherapy. Those with higher median blood concentrations of the drug (910 ng/ml) were more likely to experience complete remission, versus those with partial remission (692 ng/ml) and those considered to have failed treatment (569 ng/ml). Monitoring HCQ blood concentrations might improve the management of refractory CLE and help determine if patients have an optimal level of drug or if they are non-adherent to their treatment regimen [75]. For patients who are resistant to HCQ alone, the addition of quinacrine may be beneficial, as noted by two studies that showed a significant decrease in CLASI™ scores with combination therapy [76, 77].
6.4 Immunosuppressives
In cases of recalcitrant CLE, the addition of immunosuppressives may be helpful. These include methotrexate (MTX), mycophenolate mofetil (MMF), and azathioprine. Adding an immunosuppressive to existing antimalarial therapy may be helpful.
MTX has been used in the treatment of SLE and may also be considered as a second-line treatment for CLE when antimalarials are ineffective. A retrospective analysis of 43 patients with recalcitrant CLE reported a significant improvement in cutaneous manifestations while on low-dose MTX [78]. Serious side effects such as bone marrow suppression, hepatoxicity, gastrointestinal toxicity, and pneumonitis may preclude its use, and folic acid supplementation is necessary.
An open-label retrospective review of 24 CLE patients treated with MMF showed at least some clinical improvement in all cases when patients received at least 2500 mg/day [79]. Mycophenolate sodium was also safe and effective in a study of ten patients with treatment-resistant SCLE [80]. However, MMF was not particularly effective in the treatment of severe, multi-treatment-resistant skin disease in SLE patients [81]. Randomized control trials are necessary to better understand the role of MMF.
6.5 Other Treatments
Oral retinoids are an alternative therapeutic option for refractory CLE. Acitretin was evaluated in CLE patients in a double-blind, randomized control trial, with comparable results to HCQ [82], and may be particularly useful in cases of hypertrophic forms of CLE [83]. Recently, isotretinoin has been used successfully in SCLE [84]. Alitretinoin was also used successfully in three refractory CLE patients with near to complete clearance of lesions [85]. Retinoids may cause hyperlipidemia and hepatotoxicity in addition to their teratogenicity, and extreme caution must be used in women of childbearing potential.
Dapsone (4,4′-diaminodiphenylsulfone) has shown efficacy in case reports with bullous LE, LEP, SCLE, and DLE. In a retrospective analysis of 34 patients treated with dapsone as monotherapy or combined with antimalarials, skin lesions were cured or improved in nine out of 17 patients after combined treatment and 11 out of 17 patients on dapsone monotherapy [86]. Dapsone also has the potential for significant side effects such as hemolysis, methemoglobinemia, hepatotoxicity, and agranulocytosis, and requires close monitoring. Controlled studies evaluating dapsone in CLE have not yet been performed.
Intravenous immunoglobulin (IVIG) has shown mixed results for CLE lesions, with worsening of lesions after treatment in earlier studies [87]. Subsequent case reports showed antimalarial-resistant and immunosuppressant-resistant SCLE improved in three patients with cutaneous disease after treatment with IVIG, as well as in resistant LEP [88, 89]. More recently, a case series used IVIG monotherapy in recalcitrant CLE, with some improvement in skin disease activity [90]. The widespread use of IVIG may be limited by high costs; however, controlled studies are needed to demonstrate its efficacy for resistant cases of CLE.
6.6 Thalidomide and Analogs
Thalidomide has been successfully used to treat refractory CLE; however, its use is limited by its toxicity profile. Results for a 2012 prospective study demonstrate the efficacy of low-dose thalidomide (100 mg daily then tapered to 50 mg every other day) and identified that patients with a diagnosis of SCLE were more frequently long-term responders to the drug and that relapse was more frequently associated with DLE [91]. Some patients can lower their dose of thalidomide, but this does not appear to reduce the frequency of neuropathic side effects [92].
Lenalidomide is a structural derivative of thalidomide and has a lower frequency of thalidomide’s common adverse effects, such as sedation, constipation, and neuropathy. In an early study, five patients using lenalidomide showed clinical improvement, with a significant decrease in the CLASI™ activity score. One patient developed new-onset arthralgias and proteinuria with lenalidomide [93]. Subsequently, in an open-label pilot study of 14 patients with refractory CLE, all patients experienced some clinical improvement, and complete response was achieved in 86 % of patients within 2–12 weeks after starting the drug. None of the patients showed systemic disease manifestations; however, cutaneous relapse occurred in 75 % of patients within 2–8 weeks after the tapering dosage or withdrawing medication, especially within DLE and LET subtypes [94]. Further randomized clinical trials are necessary to better grasp the efficacy and safety of this drug.
6.7 Monoclonal Antibodies
Belimumab is a human monoclonal antibody that inhibits B-lymphocyte stimulator (BLyS), an immunomodulatory cytokine that promotes B cell survival and differentiation and is approved for SLE in the USA, Canada, and Europe. In phase III clinical trials, belimumab plus standard therapy demonstrated improved overall SLE disease activity in musculoskeletal and mucocutaneous parameters, specifically rash, mucosal ulcers, and alopecia [95]. This treatment was also associated with a reduction in severe flares and corticosteroid use [96]. In long-term follow-up, belimumab plus standard therapy was generally well tolerated, and sustained disease control was maintained for up to 7 years in patients with active SLE at baseline [97]. Importantly, these studies employed SLE-specific rather than CLE-specific tools to assess activity and monitor outcomes; the use of belimumab in CLE requires further investigation.
The anti-CD20 monoclonal antibody rituximab has shown inconsistent results with mucocutaneous manifestations in SLE patients, with partial or complete response rates ranging from 33 to 71 % and significant disease relapse rates [98]. Response may vary based on CLE subtype, with one study reporting better results in patients with ACLE while noting flares in those with SCLE and CCLE after rituximab treatment [99]. Notably, these studies are limited by the lack of CLE specific assessments.
Finally anti-IFN approaches including the anti-IFN-α monoclonal antibody sifalimumab [100] and IFN receptor-blocking anifrolumab [101] are promising SLE targeted therapies that have been evaluated in phase I and II clinical trials, with an acceptable safety profile. Most recently phase II trials demonstrated reduced SLE disease activity across multiple clinical measures. Importantly, in subjects with moderate to severe baseline mucocutaneous involvement, significantly higher percentages had a ≥4-point decrease in CLASI™ on sifalimumab than placebo [102]. Also a significantly higher number had a >50 % drop in CLASI™ activity in the anifrolumab trial relative to controls [101].
7 Conclusion
Overall, CLE is a multifactorial disease that involves an interplay between genetics and environmental triggers in the setting of immune activation. CLE encompasses a range of dermatologic manifestations and is only slightly less prevalent than SLE. Despite various criteria for SLE, the systematic work to define CLE is just beginning, and diagnosis and subtype classification continue to be a challenge. Clinical tools such as the CLASI™ aid in reliable disease assessment. Although antimalarials and topical steroids remain the standard of care, there is a need for alternative systemic treatments for refractory CLE.
References
Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4(4):471–5.
Pons-Estel GJ, Alarcon GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39(4):257–68. doi:10.1016/j.semarthrit.2008.10.007.
Petri M. Epidemiology of systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2002;16(5):847–58.
Durosaro O, Davis MD, Reed KB, Rohlinger AL. Incidence of cutaneous lupus erythematosus, 1965–2005: a population-based study. Arch Dermatol. 2009;145(3):249–53. doi:10.1001/archdermatol.2009.21.
Gronhagen CM, Fored CM, Granath F, Nyberg F. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164(6):1335–41. doi:10.1111/j.1365-2133.2011.10272.x.
Jarukitsopa S, Hoganson DD, Crowson CS, Sokumbi O, Davis MD, Michet CJ Jr, et al. Epidemiology of systemic lupus erythematosus and cutaneous lupus erythematosus in a predominantly white population in the United States. Arthritis Care Res (Hoboken). 2015;67(6):817–28. doi:10.1002/acr.22502.
McCarty DJ, Manzi S, Medsger TA Jr, Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum. 1995;38(9):1260–70.
Chiu YM, Lai CH. Nationwide population-based epidemiologic study of systemic lupus erythematosus in Taiwan. Lupus. 2010;19(10):1250–5. doi:10.1177/0961203310373780.
Mok CC, To CH, Ho LY, Yu KL. Incidence and mortality of systemic lupus erythematosus in a southern Chinese population, 2000–2006. J Rheumatol. 2008;35(10):1978–82.
Albrecht J, Berlin JA, Braverman IM, Callen JP, Connolly MK, Costner MI, et al. Dermatology position paper on the revision of the 1982 ACR criteria for systemic lupus erythematosus. Lupus. 2004;13(11):839–49.
Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–86. doi:10.1002/art.34473.
Schultz HY, Dutz JP, Furukawa F, Goodfield MJ, Kuhn A, Lee LA, et al. From pathogenesis, epidemiology, and genetics to definitions, diagnosis, and treatments of cutaneous lupus erythematosus and dermatomyositis: a report from the 3rd International Conference on Cutaneous Lupus Erythematosus (ICCLE) 2013. J Investig Dermatol. 2015;135(1):7–12. doi:10.1038/jid.2014.316.
Merola JF, Nyberg F, Furukawa F, Goodfield MJ, Hasegawa M, Marinovic B, et al. Redefining cutaneous lupus erythematosus: a proposed international consensus approach and results of a preliminary questionnaire. Lupus Sci Med. 2015;2(1):e000085. doi:10.1136/lupus-2015-000085.
Walling HW, Sontheimer RD. Cutaneous lupus erythematosus: issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10(6):365–81. doi:10.2165/11310780-000000000-00000.
Tebbe B, Mansmann U, Wollina U, Auer-Grumbach P, Licht-Mbalyohere A, Arensmeier M, et al. Markers in cutaneous lupus erythematosus indicating systemic involvement. A multicenter study on 296 patients. Acta Derm Venereol. 1997;77(4):305–8.
Rothfield N, Sontheimer RD, Bernstein M. Lupus erythematosus: systemic and cutaneous manifestations. Clin Dermatol. 2006;24(5):348–62. doi:10.1016/j.clindermatol.2006.07.014.
Fonseca E, Alvarez R, Gonzalez MR, Pascual D. Prevalence of anticardiolipin antibodies in subacute cutaneous lupus erythematosus. Lupus. 1992;1(4):265–8.
Garcia-Martin P, Garcia-Garcia C, Fraga J, Garcia-Diez A. Prevalence of antiphospholipid antibodies in patients with subacute and chronic cutaneous lupus erythematosus. Actas Dermosifiliogr. 2013;104(3):232–8. doi:10.1016/j.ad.2012.10.017.
Albrecht J, Taylor L, Berlin JA, Dulay S, Ang G, Fakharzadeh S, et al. The CLASI (Cutaneous Lupus Erythematosus Disease Area and Severity Index): an outcome instrument for cutaneous lupus erythematosus. J Investig Dermatol. 2005;125(5):889–94. doi:10.1111/j.0022-202X.2005.23889.x.
Krathen MS, Dunham J, Gaines E, Junkins-Hopkins J, Kim E, Kolasinski SL, et al. The Cutaneous Lupus Erythematosus Disease Activity and Severity Index: expansion for rheumatology and dermatology. Arthritis Rheum. 2008;59(3):338–44. doi:10.1002/art.23319.
Klein R, Moghadam-Kia S, LoMonico J, Okawa J, Coley C, Taylor L, et al. Development of the CLASI as a tool to measure disease severity and responsiveness to therapy in cutaneous lupus erythematosus. Arch Dermatol. 2011;147(2):203–8. doi:10.1001/archdermatol.2010.435.
Braunstein I, Klein R, Okawa J, Werth VP. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the Cutaneous Lupus Area And Severity Index score. Br J Dermatol. 2012;166(5):971–5. doi:10.1111/j.1365-2133.2012.10825.x.
Nabatian AS, Bashir MM, Wysocka M, Sharma M, Werth VP. Tumor necrosis factor alpha release in peripheral blood mononuclear cells of cutaneous lupus and dermatomyositis patients. Arthritis Res Ther. 2012;14(1):R1. doi:10.1186/ar3549.
Vasquez R, Wang D, Tran QP, Adams-Huet B, Chren MM, Costner MI, et al. A multicentre, cross-sectional study on quality of life in patients with cutaneous lupus erythematosus. Br J Dermatol. 2013;168(1):145–53. doi:10.1111/j.1365-2133.2012.11106.x.
Jolly M, Kazmi N, Mikolaitis RA, Sequeira W, Block JA. Validation of the Cutaneous Lupus Disease Area and Severity Index (CLASI) using physician- and patient-assessed health outcome measures. J Am Acad Dermatol. 2013;68(4):618–23. doi:10.1016/j.jaad.2012.08.035.
Salphale P, Danda D, Chandrashekar L, Peter D, Jayaseeli N, George R. The study of Cutaneous Lupus Erythematosus Disease Area and Severity Index in Indian patients with systemic lupus erythematosus. Lupus. 2011;20(14):1510–7. doi:10.1177/0961203311418789.
Ishiguro M, Hashizume H, Ikeda T, Yamamoto Y, Furukawa F. Evaluation of the quality of life of lupus erythematosus patients with cutaneous lesions in Japan. Lupus. 2014;23(1):93–101. doi:10.1177/0961203313509293.
Kuhn A, Meuth AM, Bein D, Amler S, Beissert S, Bohm M, et al. Revised Cutaneous Lupus Erythematosus Disease Area and Severity Index (RCLASI): a modified outcome instrument for cutaneous lupus erythematosus. Br J Dermatol. 2010;163(1):83–92. doi:10.1111/j.1365-2133.2010.09799.x.
Wahie S, McColl E, Reynolds NJ, Meggitt SJ. Measuring disease activity and damage in cutaneous lupus erythematosus. Br J Dermatol. 2011;164(1):221–2. doi:10.1111/j.1365-2133.2010.10072.x (author reply 2–4).
Yu C, Chang C, Zhang J. Immunologic and genetic considerations of cutaneous lupus erythematosus: a comprehensive review. J Autoimmun. 2013;41:34–45. doi:10.1016/j.jaut.2013.01.007.
Osmola A, Namysl J, Jagodzinski PP, Prokop J. Genetic background of cutaneous forms of lupus erythematosus: update on current evidence. J Appl Genet. 2004;45(1):77–86.
Fischer GF, Pickl WF, Fae I, Anegg B, Milota S, Volc-Platzer B. Association between chronic cutaneous lupus erythematosus and HLA class II alleles. Hum Immunol. 1994;41(4):280–4.
Jarvinen TM, Hellquist A, Koskenmies S, Einarsdottir E, Koskinen LL, Jeskanen L, et al. Tyrosine kinase 2 and interferon regulatory factor 5 polymorphisms are associated with discoid and subacute cutaneous lupus erythematosus. Exp Dermatol. 2010;19(2):123–31. doi:10.1111/j.1600-0625.2009.00982.x.
Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, et al. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet. 2006;79(4):731–7. doi:10.1086/507848.
Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007;85(5):531–7. doi:10.1007/s00109-007-0199-9.
Jarvinen TM, Hellquist A, Koskenmies S, Einarsdottir E, Panelius J, Hasan T et al. Polymorphisms of the ITGAM gene confer higher risk of discoid cutaneous than of systemic lupus erythematosus. PLoS One. 2010;5(12):e14212. doi:10.1371/journal.pone.0014212.
Lin JH, Dutz JP, Sontheimer RD, Werth VP. Pathophysiology of cutaneous lupus erythematosus. Clin Rev Allergy Immunol. 2007;33(1–2):85–106. doi:10.1007/s12016-007-0031-x.
Kuhn A, Herrmann M, Kleber S, Beckmann-Welle M, Fehsel K, Martin-Villalba A, et al. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006;54(3):939–50. doi:10.1002/art.21658.
Reefman E, de Jong MC, Kuiper H, Jonkman MF, Limburg PC, Kallenberg CG, et al. Is disturbed clearance of apoptotic keratinocytes responsible for UVB-induced inflammatory skin lesions in systemic lupus erythematosus? Arthritis Res Ther. 2006;8(6):R156. doi:10.1186/ar2051.
Bashir MM, Sharma MR, Werth VP. UVB and proinflammatory cytokines synergistically activate TNF-alpha production in keratinocytes through enhanced gene transcription. J Invest Dermatol. 2009;129(4):994–1001. doi:10.1038/jid.2008.332.
Kuhn A, Wenzel J, Weyd H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol. 2014;47(2):148–62. doi:10.1007/s12016-013-8403-x.
Kuhn A, Wozniacka A, Szepietowski JC, Glaser R, Lehmann P, Haust M, et al. Photoprovocation in cutaneous lupus erythematosus: a multicenter study evaluating a standardized protocol. J Investig Dermatol. 2011;131(8):1622–30. doi:10.1038/jid.2011.101.
Ruland V, Haust M, Stilling RM, Metze D, Amler S, Ruzicka T, et al. Updated analysis of standardized photoprovocation in patients with cutaneous lupus erythematosus. Arthritis Care Res (Hoboken). 2013;65(5):767–76. doi:10.1002/acr.21867.
Calderon C, Zucht HD, Kuhn A, Wozniacka A, Szepietowski JC, Nyberg F, et al. A multicenter photoprovocation study to identify potential biomarkers by global peptide profiling in cutaneous lupus erythematosus. Lupus. 2015;. doi:10.1177/0961203315596077.
Biazar C, Sigges J, Patsinakidis N, Ruland V, Amler S, Bonsmann G, et al. Cutaneous lupus erythematosus: first multicenter database analysis of 1002 patients from the European Society of Cutaneous Lupus Erythematosus (EUSCLE). Autoimmun Rev. 2013;12(3):444–54. doi:10.1016/j.autrev.2012.08.019.
Li PH, Wong WH, Lee TL, Lau CS, Chan TM, Leung AM, et al. Relationship between autoantibody clustering and clinical subsets in SLE: cluster and association analyses in Hong Kong Chinese. Rheumatology (Oxford). 2013;52(2):337–45. doi:10.1093/rheumatology/kes261.
Fredi M, Cavazzana I, Quinzanini M, Taraborelli M, Cartella S, Tincani A, et al. Rare autoantibodies to cellular antigens in systemic lupus erythematosus. Lupus. 2014;23(7):672–7. doi:10.1177/0961203314524850.
Zahn S, Rehkamper C, Ferring-Schmitt S, Bieber T, Tuting T, Wenzel J. Interferon-alpha stimulates TRAIL expression in human keratinocytes and peripheral blood mononuclear cells: implications for the pathogenesis of cutaneous lupus erythematosus. Br J Dermatol. 2011;165(5):1118–23. doi:10.1111/j.1365-2133.2011.10479.x.
Toberer F, Sykora J, Gottel D, Hartschuh W, Werchau S, Enk A, et al. Apoptotic signal molecules in skin biopsies of cutaneous lupus erythematosus: analysis using tissue microarray. Exp Dermatol. 2013;22(10):656–9. doi:10.1111/exd.12216.
Kanda N, Shimizu T, Tada Y, Watanabe S. IL-18 enhances IFN-gamma-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol. 2007;37(2):338–50. doi:10.1002/eji.200636420.
Wang D, Drenker M, Eiz-Vesper B, Werfel T, Wittmann M. Evidence for a pathogenetic role of interleukin-18 in cutaneous lupus erythematosus. Arthritis Rheum. 2008;58(10):3205–15. doi:10.1002/art.23868.
Lin YJ, Wan L, Lee CC, Huang CM, Tsai Y, Tsai CH, et al. Disease association of the interleukin-18 promoter polymorphisms in Taiwan Chinese systemic lupus erythematosus patients. Genes Immun. 2007;8(4):302–7. doi:10.1038/sj.gene.6364387.
Wenzel J, Uerlich M, Worrenkamper E, Freutel S, Bieber T, Tuting T. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br J Dermatol. 2005;153(5):1011–5. doi:10.1111/j.1365-2133.2005.06784.x.
Wenzel J, Worenkamper E, Freutel S, Henze S, Haller O, Bieber T, et al. Enhanced type I interferon signalling promotes Th1-biased inflammation in cutaneous lupus erythematosus. J Pathol. 2005;205(4):435–42. doi:10.1002/path.1721.
Fett N, Werth VP. Systemic lupus erythematosus treatment: a guide to photoprotection. CML Rheumatol. 2010;29(2):33–41.
Kuhn A, Gensch K, Haust M, Meuth AM, Boyer F, Dupuy P, et al. Photoprotective effects of a broad-spectrum sunscreen in ultraviolet-induced cutaneous lupus erythematosus: a randomized, vehicle-controlled, double-blind study. J Am Acad Dermatol. 2011;64(1):37–48. doi:10.1016/j.jaad.2009.12.053.
Klein RS, Werth VP, Dowdy JC, Sayre RM. Analysis of compact fluorescent lights for use by patients with photosensitive conditions. Photochem Photobiol. 2009;85(4):1004–10. doi:10.1111/j.1751-1097.2009.00540.x.
Klein RS, Sayre RM, Dowdy JC, Werth VP. The risk of ultraviolet radiation exposure from indoor lamps in lupus erythematosus. Autoimmun Rev. 2009;8(4):320–4. doi:10.1016/j.autrev.2008.10.003.
Fenton L, Dawe R, Ibbotson S, Ferguson J, Silburn S, Moseley H. Impact assessment of energy-efficient lighting in patients with lupus erythematosus: a pilot study. Br J Dermatol. 2014;170(3):694–8. doi:10.1111/bjd.12719.
Heine G, Lahl A, Muller C, Worm M. Vitamin D deficiency in patients with cutaneous lupus erythematosus is prevalent throughout the year. Br J Dermatol. 2010;163(4):863–5. doi:10.1111/j.1365-2133.2010.09948.x.
Cutillas-Marco E, Marquina-Vila A, Grant WB, Vilata-Corell JJ, Morales-Suarez-Varela MM. Vitamin D and cutaneous lupus erythematosus: effect of vitamin D replacement on disease severity. Lupus. 2014;23(7):615–23. doi:10.1177/0961203314522338.
Schoindre Y, Jallouli M, Tanguy ML, Ghillani P, Galicier L, Aumaitre O, et al. Lower vitamin D levels are associated with higher systemic lupus erythematosus activity, but not predictive of disease flare-up. Lupus Sci Med. 2014;1(1):e000027. doi:10.1136/lupus-2014-000027.
Terrier B, Derian N, Schoindre Y, Chaara W, Geri G, Zahr N, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther. 2012;14(5):R221. doi:10.1186/ar4060.
Kuhn A, Ruland V, Bonsmann G. Cutaneous lupus erythematosus: update of therapeutic options part I. J Am Acad Dermatol. 2011;65(6):e179–93. doi:10.1016/j.jaad.2010.06.018.
Kuhn A, Sigges J, Biazar C, Ruland V, Patsinakidis N, Landmann A, et al. Influence of smoking on disease severity and antimalarial therapy in cutaneous lupus erythematosus: analysis of 1002 patients from the EUSCLE database. Br J Dermatol. 2014;171(3):571–9. doi:10.1111/bjd.13006.
Gallego H, Crutchfield CE 3rd, Lewis EJ, Gallego HJ. Report of an association between discoid lupus erythematosus and smoking. Cutis. 1999;63(4):231–4.
Miot HA, Bartoli Miot LD, Haddad GR. Association between discoid lupus erythematosus and cigarette smoking. Dermatology. 2005;211(2):118–22. doi:10.1159/000086440.
Wahie S, Daly AK, Cordell HJ, Goodfield MJ, Jones SK, Lovell CR, et al. Clinical and pharmacogenetic influences on response to hydroxychloroquine in discoid lupus erythematosus: a retrospective cohort study. J Investig Dermatol. 2011;131(10):1981–6. doi:10.1038/jid.2011.167.
Piette EW, Foering KP, Chang AY, Okawa J, Ten Have TR, Feng R, et al. Impact of smoking in cutaneous lupus erythematosus. Arch Dermatol. 2012;148(3):317–22. doi:10.1001/archdermatol.2011.342.
Chasset F, Frances C, Barete S, Amoura Z, Arnaud L. Influence of smoking on the efficacy of antimalarials in cutaneous lupus: a meta-analysis of the literature. J Am Acad Dermatol. 2015;72(4):634–9. doi:10.1016/j.jaad.2014.12.025.
Tzung TY, Liu YS, Chang HW. Tacrolimus vs. clobetasol propionate in the treatment of facial cutaneous lupus erythematosus: a randomized, double-blind, bilateral comparison study. Br J Dermatol. 2007;156(1):191–2. doi:10.1111/j.1365-2133.2006.07595.x.
Kuhn A, Gensch K, Haust M, Schneider SW, Bonsmann G, Gaebelein-Wissing N, et al. Efficacy of tacrolimus 0.1 % ointment in cutaneous lupus erythematosus: a multicenter, randomized, double-blind, vehicle-controlled trial. J Am Acad Dermatol. 2011;65(1):54–64, e1–2. doi:10.1016/j.jaad.2010.03.037.
Avgerinou G, Papafragkaki DK, Nasiopoulou A, Arapaki A, Katsambas A, Stavropoulos PG. Effectiveness of topical calcineurin inhibitors as monotherapy or in combination with hydroxychloroquine in cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2012;26(6):762–7. doi:10.1111/j.1468-3083.2011.04161.x.
James JA, Kim-Howard XR, Bruner BF, Jonsson MK, McClain MT, Arbuckle MR, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus. 2007;16(6):401–9. doi:10.1177/0961203307078579.
Frances C, Cosnes A, Duhaut P, Zahr N, Soutou B, Ingen-Housz-Oro S, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: a French multicenter prospective study. Arch Dermatol. 2012;148(4):479–84. doi:10.1001/archdermatol.2011.2558.
Chang AY, Piette EW, Foering KP, Tenhave TR, Okawa J, Werth VP. Response to antimalarial agents in cutaneous lupus erythematosus: a prospective analysis. Arch Dermatol. 2011;147(11):1261–7. doi:10.1001/archdermatol.2011.191.
Cavazzana I, Sala R, Bazzani C, Ceribelli A, Zane C, Cattaneo R, et al. Treatment of lupus skin involvement with quinacrine and hydroxychloroquine. Lupus. 2009;18(8):735–9. doi:10.1177/0961203308101714.
Wenzel J, Brahler S, Bauer R, Bieber T, Tuting T. Efficacy and safety of methotrexate in recalcitrant cutaneous lupus erythematosus: results of a retrospective study in 43 patients. Br J Dermatol. 2005;153(1):157–62. doi:10.1111/j.1365-2133.2005.06552.x.
Gammon B, Hansen C, Costner MI. Efficacy of mycophenolate mofetil in antimalarial-resistant cutaneous lupus erythematosus. J Am Acad Dermatol. 2011;65(4):717–21. doi:10.1016/j.jaad.2010.08.011.
Kreuter A, Tomi NS, Weiner SM, Huger M, Altmeyer P, Gambichler T. Mycophenolate sodium for subacute cutaneous lupus erythematosus resistant to standard therapy. Br J Dermatol. 2007;156(6):1321–7. doi:10.1111/j.1365-2133.2007.07826.x.
Pisoni CN, Obermoser G, Cuadrado MJ, Sanchez FJ, Karim Y, Sepp NT, et al. Skin manifestations of systemic lupus erythematosus refractory to multiple treatment modalities: poor results with mycophenolate mofetil. Clin Exp Rheumatol. 2005;23(3):393–6.
Ruzicka T, Sommerburg C, Goerz G, Kind P, Mensing H. Treatment of cutaneous lupus erythematosus with acitretin and hydroxychloroquine. Br J Dermatol. 1992;127(5):513–8.
Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32(6):482–6.
D’Erme AM, Milanesi N, Difonzo EM, Lotti T, Gola M. Treatment of refractory subacute cutaneous lupus erythematosus with oral isotretinoin: a valid therapeutic option. Dermatol Ther. 2012;25(3):281–2. doi:10.1111/j.1529-8019.2012.01461.x.
Kuhn A, Patsinakidis N, Luger T. Alitretinoin for cutaneous lupus erythematosus. J Am Acad Dermatol. 2012;67(3):e123–6. doi:10.1016/j.jaad.2011.10.030.
Klebes M, Wutte N, Aberer E. Dapsone as second-line treatment for cutaneous lupus erythematosus? A retrospective analysis of 34 patients and a review of the literature. Dermatology. 2015. doi:10.1159/000441054.
De Pita O, Bellucci AM, Ruffelli M, Girardelli CR, Puddu P. Intravenous immunoglobulin therapy is not able to efficiently control cutaneous manifestations in patients with lupus erythematosus. Lupus. 1997;6(4):415–7.
Lampropoulos CE, Hughes GR. DP DC. Intravenous immunoglobulin in the treatment of resistant subacute cutaneous lupus erythematosus: a possible alternative. Clin Rheumatol. 2007;26(6):981–3. doi:10.1007/s10067-006-0222-5.
Espirito Santo J, Gomes MF, Gomes MJ, Peixoto L, Pereira SC, Acabado A, et al. Intravenous immunoglobulin in lupus panniculitis. Clin Rev Allergy Immunol. 2010;38(2–3):307–18. doi:10.1007/s12016-009-8162-x.
Ky C, Swasdibutra B, Khademi S, Desai S, Laquer V, Grando SA. Efficacy of intravenous immunoglobulin monotherapy in patients with cutaneous lupus erythematosus: results of proof-of-concept study. Dermatol Rep. 2015;7(1):5804. doi:10.4081/dr.2015.5804.
Cortes-Hernandez J, Torres-Salido M, Castro-Marrero J, Vilardell-Tarres M, Ordi-Ros J. Thalidomide in the treatment of refractory cutaneous lupus erythematosus: prognostic factors of clinical outcome. Br J Dermatol. 2012;166(3):616–23. doi:10.1111/j.1365-2133.2011.10693.x.
Frankel HC, Sharon VR, Vleugels RA, Merola JF, Qureshi AA. Lower-dose thalidomide therapy effectively treats cutaneous lupus erythematosus but is limited by neuropathic toxicity. Int J Dermatol. 2013;52(11):1407–9. doi:10.1111/j.1365-4632.2011.05200.x.
Braunstein I, Goodman NG, Rosenbach M, Okawa J, Shah A, Krathen M, et al. Lenalidomide therapy in treatment-refractory cutaneous lupus erythematosus: histologic and circulating leukocyte profile and potential risk of a systemic lupus flare. J Am Acad Dermatol. 2012;66(4):571–82. doi:10.1016/j.jaad.2011.01.015.
Cortes-Hernandez J, Avila G, Vilardell-Tarres M, Ordi-Ros J. Efficacy and safety of lenalidomide for refractory cutaneous lupus erythematosus. Arthritis Res Ther. 2012;14(6):R265. doi:10.1186/ar4111.
Manzi S, Sanchez-Guerrero J, Merrill JT, Furie R, Gladman D, Navarra SV, et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis. 2012;71(11):1833–8. doi:10.1136/annrheumdis-2011-200831.
Strand V, Levy RA, Cervera R, Petri MA, Birch H, Freimuth WW, et al. Improvements in health-related quality of life with belimumab, a B-lymphocyte stimulator-specific inhibitor, in patients with autoantibody-positive systemic lupus erythematosus from the randomised controlled BLISS trials. Ann Rheum Dis. 2014;73(5):838–44. doi:10.1136/annrheumdis-2012-202865.
Ginzler EM, Wallace DJ, Merrill JT, Furie RA, Stohl W, Chatham WW, et al. Disease control and safety of belimumab plus standard therapy over 7 years in patients with systemic lupus erythematosus. J Rheumatol. 2014;41(2):300–9. doi:10.3899/jrheum.121368.
Cobo-Ibanez T, Loza-Santamaria E, Pego-Reigosa JM, Marques AO, Rua-Figueroa I, Fernandez-Nebro A, et al. Efficacy and safety of rituximab in the treatment of non-renal systemic lupus erythematosus: a systematic review. Semin Arthritis Rheum. 2014;44(2):175–85. doi:10.1016/j.semarthrit.2014.04.002.
Vital EM, Wittmann M, Edward S, Md Yusof MY, MacIver H, Pease CT, et al. Brief report: responses to rituximab suggest B cell-independent inflammation in cutaneous systemic lupus erythematosus. Arthritis Rheumatol. 2015;67(6):1586–91. doi:10.1002/art.39085.
Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, et al. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013;65(4):1011–21. doi:10.1002/art.37824.
Furie R MJ, Werth V, Khamashta M, Kalunian K, Brohawn P, Illei G, Drappa J, Wang L, Yoo S. Anifrolumab, an anti-interferon alpha receptor monoclonal antibody, in moderate to severe systemic lupus erythematosus (SLE) [abstract]. Arthritis Rheumatol. 2015;67:(suppl 10).
Khamashta MMJ, Werth VP, Furie R, Kalunian K, Illeis GG, Drappas J, Wang L, Greth W. Safety and efficacy of sifilmumab, an anti IFN-alpha monoclonal antibody, in a phase 2b study of moderate to severe systemic lupus erythematosus (SLE) [abstract]. Arthritis Rheum. 2014;66:3530–1 (L4).
Acknowledgments
This project is supported by the Department of Veterans Affairs Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development.
We acknowledge Majid Zeidi, MD, for providing the H & E figure.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received in the preparation of this review.
Conflict of interest
VPW has received grants from Celgene and Janssen; consulting fees and/or honoraria from Celgene, Janssen, Medimmune, Pfizer, Biogen and Sanofi-Aventis; stock in UV Therapeutics; and royalties from licensing of the CLASI™ from the University of Pennsylvania. EZH has no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Hejazi, E.Z., Werth, V.P. Cutaneous Lupus Erythematosus: An Update on Pathogenesis, Diagnosis and Treatment. Am J Clin Dermatol 17, 135–146 (2016). https://doi.org/10.1007/s40257-016-0173-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-016-0173-9