
Abstract
We recently described a novel autosomal-dominant genodermatosis, termed familial chilblain lupus, and mapped its genetic locus to chromosome 3p21. Familial chilblain lupus manifests in early childhood with ulcerating acral skin lesions and is associated with arthralgias and circulating antinuclear antibodies. In this study, we report the identification of a heterozygous missense mutation (D18N) in TREX1 encoding the 3′-5′repair exonuclease 1 in affected individuals of the family with chilblain lupus. The homodimeric TREX1 is the most abundant intracellular DNase in mammalian cells. We have recently shown that TREX1 plays a role in apoptotic single-stranded DNA damage induced by the killer lymphocyte protease granzyme A. D18N affects a highly conserved amino acid residue critical for catalytic activity. Recombinant mutant TREX1 homodimers are enzymatically inactive, while wild type/mutant heterodimers show residual exonucleolytic activity, suggesting a heterozygous loss of function. Lymphoblastoid cells carrying the D18N mutation are significantly less sensitive to granzyme A-mediated cell death, suggesting a novel role for this caspase-independent form of apoptosis in the pathogenesis of familial chilblain lupus. Our findings also warrant further investigation of TREX1 in common forms of lupus erythematosus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
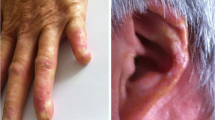
Sporadic chilblain lupus (CHBL) usually affects middle-aged females [1, 2]. We recently described the first family with autosomal-dominant CHBL and termed this novel genodermatosis familial CHBL [3]. Familial CHBL manifests in early childhood and is characterized by painful bluish-red inflammatory cutaneous lesions in acral locations such as fingers, toes, nose, cheeks, and ears, which tend to ulcerate [3]. Cutaneous lesions are precipitated by cold and wet exposure and usually improve during summer. Other cutaneous findings such as oral ulcers or photosensitivity are not seen. Histological findings of skin biopsies from affected individuals include perivascular inflammatory infiltrates and granular deposits of immunoglobulins and complement along the basement membrane [3]. In addition, some affected individuals show circulating antinuclear antibodies and immune complex formation [3]. Thus, familial CHBL represents the first monogenic form of cutaneous lupus erythematosus.
We mapped the genetic locus of CHBL in a large German family over five generations to chromosome 3p21 [3]. The CHBL critical interval overlaps with the locus for autosomal recessive Aicardi-Goutières syndrome 1 (AGS1), which is identical to pseudo-TORCH syndrome or Cree encephalitis [4, 5]. AGS is a rare infantile encephalopathy characterized by basal ganglia calcification, transient elevation of interferon alpha, and chronic lymphocytosis in cerebrospinal fluid mimicking congenital viral infection [4]. Recently, homozygous mutations of TREX1 encoding the 3′-5′repair exonuclease 1 were shown to be the cause of AGS1 [6]. AGS is genetically heterogeneous and in addition to TREX1 at the AGS1 locus, homozygous mutations in all three subunits of the RNase H2 complex (AGS2/RNASEH2B on 13q, AGS3/RNASEH2C on 11q, AGS4/RNASEH2A on 19p) were found in AGS patients [7]. It was suggested that impairment in processing of DNA and RNA structures could trigger an abnormal innate immune response.
Although CHBL is not associated with any signs of neurological disease, there is phenotypic overlap between CHBL and AGS as some children affected with AGS develop chilblain-like lesions, hypocomplementemia, or antinuclear antibodies [8–11], suggesting that AGS1 and CHBL could represent allelic phenotypes.
Genomic sequencing of TREX1 in members of the family affected with CHBL revealed a heterozygous missense mutation affecting a highly conserved amino acid residue critical for catalytic activity that segregated with the CHBL phenotype. Functional analysis of the identified mutation showed that a loss of TREX1 enzyme activity was associated with impaired granzyme A (GzmA)-mediated apoptosis, suggesting a role for this caspase-independent form of apoptosis in the pathogenesis of familial CHBL.
Materials and methods
Sequence analysis
The entire coding exon and part of the 3′UTR of the TREX1 gene was amplified by three overlapping polymerase chain reaction (PCR) amplicons from genomic DNA extracted from peripheral lymphocytes from members of the family affected with CHBL and from 200 unrelated blood bank donors. The following primers were used: TREX1-1s: 3′-CCTCCCCTTCGGATCTTAAC-5′ and TREX1-1as: 3′-CCCATACAGGCGAGTGTAGA-5′, TREX1-1cs: 3′-GGGCGTCAATGTTTTGATGA-5′ and TREX1-1cas: 3′-GAGGCTGTGACCCCATACAT-5′, TREX1-1ds: 3′-ACTCGCCTGTATGGGCAGTC-5′ and TREX1-1das: 3′-GGGAAAGTGAGGGACAAACA-5′. Both strands of the purified PCR amplicons were sequenced using fluorescent dye terminator chemistry on an ABI 3730 sequencer and analyzed with Sequence Navigator and Vector NTI.
Preparation of homo- and heterodimeric mutant TREX1 enzymes
For homodimers, the human TREX1 gene was expressed as a fusion with the maltose binding protein (MBP) in pLM303, a pET-27b (Novagen) derivative encoding a polyhistidine sequence on the N terminus of MBP and a rhinovirus 3C protease recognition site between the MBP and TREX1 genes. The plasmid was transformed into Escherichia coli BL21(DE3) Rosetta 2 cells (Novagen) for overexpression. The cells were grown to an O.D.600 ∼0.5 at 37°C and quickly cooled on ice to 17°C. After induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), the cells were allowed to grow for 15 h at 17°C. The MBP-TREX1 fusion protein was purified by affinity chromatography using an amylose resin (New England Biolabs). The MBP protein was removed from the fusion by treatment with PreScission Protease™ (GE Biosciences) at 4°C for 20 h. The TREX1 homodimer protein was separated from the MBP by chromatography using a phosphocellulose column.
To prepare TREX1 heterodimers, a mutant and a wild-type copy of TREX1 were cloned on separate plasmids for coexpression in E. coli. First, the pLM303 plasmid was modified to eliminate the polyhistidine sequence on the N terminus of MBP, and the TREX1 mutant genes were cloned into the resultant modified vector pLM303X retaining the rhinovirus 3C protease recognition site between the MBP and TREX1. A second plasmid containing the wt TREX1 gene was prepared as a fusion of the His-tagged NusA with TREX1 in the pCDFDuet-1 plasmid (Novagen). The pLM303X-TREX1mut and pCDFDuet-1-TREX1wt plasmids were coexpressed in E. coli BL21(DE3) Rosetta 2 cells (Novagen), resulting in the formation of (1) wt/wt homodimers, (2) wt/mut heterodimers, and (3) mut/mut homodimers. The His-tagged NusATREX1wt/His-tagged NusATREX1wt homodimers and His-tagged NusATREX1wt/MBPTREX1mut heteodimer were recovered by affinity chromatography using a nickel-NTA resin (Qiagen). The His-tagged NusATREX1wt/MBPTREX1mut heteodimer was purified by affinity chromatography using an amylose resin. The His-tagged NusA and MBP proteins were removed from the fusion by treatment with PreScission Protease™ (GE Biosciences) and purified using a phosphocellulose column.
Exonuclease activity
Enzyme activities were determined from reactions containing increasing amounts of TREX1 and 50 nM of a FAM-labeled ssDNA 30-mer oligonucleotide substrate as described [12, 13]. Products were separated on urea-polyacrylamide gels and quantified as described previously. The relative activity was calculated as relative activity=100×[(fmol of dNMP released/s/fmol of mutant enzyme)/(fmol of dNMP released/s/fmol wild type enzyme)].
GzmA-mediated cytotoxicity
Recombinant granzymes and perforin (Pfn) from the rat RNK-16 cell were purified as described previously [14, 15]. For cytotoxicity assays, mutant or control cells were radiolabeled with 51Cr for 1 h and washed before treatment with a sublytic dose of Pfn and 1 μM of GzmA or GzmB in loading buffer (HBSS with 1 mg/ml BSA, 1 mM CaCl2, and 1 mM MgCl2). After 2 h, 51Cr release in the supernatant was measured on a Packard Topcount. Specific cytotoxicity was calculated using the formula 100×[(sample release)-(spontaneous release)]/[(total release) - (spontaneous release)]. Statistical analysis was done using two-tailed Student’s t test.
Results and discussion
We sequenced the coding region of TREX1 from genomic DNA of 26 members of the previously described large pedigree affected with autosomal dominant CHBL and identified a novel heterozygous missense change, c.52G>A, leading to the substitution of an aspartate with an asparagine at position 18 of the TREX1 peptide (D18N). This mutation completely cosegregated with all affected family members and was absent in nonaffected family members (Fig. 1). D18N was also not found in 400 control chromosomes excluding the possibility that it represents a rare polymorphism.

Identification of a TREX1 mutation in familial CHBL. a Schematic presentation of the TREX1 protein showing the exonuclease domains (Exo1–3), which coordinate the binding of 2 Mg2+-ions required for catalysis and a polyproline II motif (PII). The arrow indicates the position of the mutation identified in CHBL. b Electropherogram showing the D18N mutation with the position numbered relative to the transcriptional start site. c Multiple sequence alignment of the Exo1 domain of TREX1 and the homologous TREX2 with highly conserved residues marked in blue and the substituted amino acid shown in red above the sequence alignment. Arrows indicate Mg2+-coordinating residues; h, human; d, dog; b, bovine; m, mouse, r, rat; and dm, Drosophila melanogaster
The ubiquitously expressed TREX1 and its homolog TREX2 encode the major 3′ exonuclease activity in mammalian cells [12, 16]. Although the overall amino acid identity is only 44%, similarity is 80% within the highly conserved Exo motifs (Exo1, Exo2, and Exo3) common to proofreading exonucleases of mammalian and bacterial DNA polymerases and bacterial RNase T [12]. The Exo domains form the active site and coordinate the binding of two Mg2+ ions necessary for catalytic activity [12, 13]. TREX1 forms homodimers and displays nonprocessive 3′exonuclease activity with high specificity for single-stranded DNA [12, 13]. The finding that TREX1 can increase fidelity of an exonuclease-deficient mammalian DNA polymerase in vitro suggested a role in DNA repair [16]. However, mice lacking TREX1 do not show increased spontaneous mutations or susceptibility to cancer but develop inflammatory myocarditis consistent with an autoimmune phenotype [17].
D18N affects one of the highly conserved Mg2+-coordinating aspartate residues within the Exo1 domain required for catalysis (Fig. 1), suggesting that it could affect enzymatic function. TREX1 functions as a homodimer, and thus, dimeric TREX1 proteins in cells of heterozygous individuals could be either D18N/D18N or wild-type (wt/wt) homodimers or D18N/wt heterodimers. We prepared purified TREX1 homo- and heterodimers coexpressed in E. coli and determined 3′-5′exonuclease activity using a single-stranded 30-mer oligonucleotide (Fig. 2). D18N/D18N homodimers were enzymatically inactive, while activity of D18N/wt heterodimers was ∼40% of wt homodimers (Fig. 2), indicating independent exonucleolytic activity of the TREX1 monomers within the dimeric enzyme. These findings indicate that D18N is a loss of function allele that does not exhibit a dominant-negative effect.

Exonuclease activity of recombinant TREX1. a Enzyme activity of increasing amounts of recombinant wild type (wt) and mutant (D18N) TREX1 homo- and heterodimers. b Relative activity of D18N/D18N and D18N/wt dimers compared to wt/wt dimers. The migration position of the ssDNA 30-mer is indicated by an arrow. The relative activity was calculated as relative activity=100×[(fmol of dNMP released/s/fmol of mutant enzyme )/(fmol of dNMP released/s/fmol wt enzyme)]
We previously identified TREX1 as the GzmA-activated DNase within the SET complex, an endoplasmic reticulum-associated complex that contains the Ape1 base excision repair endonuclease and NM23-H1, another endonuclease and nucleoside diphosphate kinase [15]. The SET complex, which translocates to the nucleus in response to oxidative stress, is postulated to repair oxidative DNA damage and activate the expression of genes required for the cellular response to oxidative stress [15, 18]. The SET complex also is responsible for apoptotic caspase-independent single-stranded DNA damage induced by the killer lymphocyte protease GzmA [14, 15]. GzmA activates mitochondrial damage and superoxide generation that induces nuclear translocation of the SET complex [15]. It then cleaves the NM23-H1 inhibitor SET, freeing NM23-H1 to make a single-stranded DNA cut that is extended by TREX1 [15]. Cells with silenced TREX1 are relatively resistant to apoptotic cell death induced by GzmA but are equally sensitive to the caspase-activating GzmB [15]. Because of the role of TREX1 in GzmA-induced DNA damage and cell death, we investigated the effect of TREX1 mutations on apoptosis of patient-derived lymphoblastoid cells treated with GzmA or GzmB and Pfn, the protein that delivers these enzymes into target cells.
Mutant or control cells demonstrated no change in background cytolysis with no additional treatment or following treatment with granzymes or Pfn alone (Fig. 3). However, cells carrying the D18N mutation were substantially less sensitive to cell death measured 2 h after treatment with GzmA and Pfn than control cells (p < 0.002, Fig. 3). GzmB-mediated cytolysis was unaffected (Fig. 3), indicating that the D18N mutation that interferes with exonuclease activity specifically interferes with GzmA-mediated cell death. The finding that cells bearing the enzymatically inactive D18N mutation are relatively resistant to GzmA confirms the importance of TREX1 in GzmA-activated DNA damage.

Cellular sensitivity to granzyme A-mediated cell death. Cells with mutations in the Exo1 domain (D18N) of TREX1 are significantly (p < 0.002) more resistant than control cells to cell death assayed by 51Cr release and induced by 1 μM granzyme A (GzmA; a) but not granzyme B (GzmB; b) and a sublytic dose of perforin (Pfn). Data shown are the means±S.D. of at least three independent experiments
A mutation in TREX1 was recently reported in three siblings of Bangladeshi origin affected with CHBL [19]. Our results support the findings by Rice et al. [19] who demonstrated that lymphoblastoid cells of patients affected with CHBL, carrying a heterozygous TREX1 mutation, display a reduced exonucleolytic activity [19]. As a novel finding, we show for the first time that a heterozygous loss of TREX1 function can lead to impairment of caspase-independent apoptosis.
Apoptosis is highly relevant to the pathogenesis of autoimmune diseases such as lupus erythematosus. First, defective apoptosis may lead to impaired antigen-mediated B-cell maturation [20] and selection of autoreactive T-cells, causing loss of self-tolerance as shown by mutations in Fas, FasL, and caspase 10, which cause human autoimmune lymphoproliferative syndrome [21]. Secondly, self-antigens displayed on cell surfaces as a result of deficient disposal of extracellular nuclear waste can induce an autoimmune response. Thus, mutations in DNase I, the most abundant extracellular DNase, cause lupus erythematosus in humans and mice [22, 23], and DNase II-/- mice develop a rheumatoid arthritis-type syndrome attributed to the lack of this enzyme in macrophages that engulf apoptotic bodies [24]. Finally, uncontrolled apoptosis is an effector mechanism responsible for autoimmune destruction of healthy cells.
A mutation in the exonuclease responsible for degrading DNA during apoptosis initiated by the most abundant cytotoxic T lymphocyte and natural killer cell protease may lead to improper clearance of altered DNA and contribute to the pathogenesis of familial CHBL. The environmental trigger may well be a viral infection activating these killer lymphocytes. Many viruses have evolved mechanisms to evade caspase and GzmB-mediated apoptosis. In a patient with heterozygous mutant TREX1 leading to relative resistance to GzmA, elimination of caspase-resistant viruses might be impaired, leading to chronic inflammation, a hallmark of both Trex-/- mice and AGS patients [6, 17].
We show that a defect in a single TREX1 allele that impairs GzmA-mediated apoptosis causes familial CHBL. Patients with AGS1, who have defects in both TREX1 alleles, also sometimes develop CHBL. Thus, both of these genetic diseases establish a firm link between TREX1 and CHBL. Apart from arthralgias, none of the family members with CHBL, some of whom are in their late seventies, are known to have progressed to systemic lupus erythematosus. However, as CHBL is one component of systemic lupus erythematosus [25, 26], our findings raise the possibility that defects in cellular pathways involving TREX1 may contribute to the pathogenesis of common complex forms of systemic lupus erythematosus.
References
Hutchinson J (1888) Harveian lectures on lupus. On the various forms of lupus vulgaris and erythematosus. Br Med J 1:113–118
Franceschini F, Calzavara-Pinton P, Quinzanini M, Cavazzana I, Bettoni L, Zane C, Facchetti F, Airo P, McCauliffe DP, Cattaneo R (1999) Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus 8:215–219
Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, Ballarini A, Gahr M, Hubner N, Linne M (2006) Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet 79:731–737
Aicardi J, Goutieres F (1984) A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15:49–54
Black DN, Watters GV, Andermann E, Dumont C, Kabay ME, Kaplan P, Meagher-Villemure K, Michaud J, O’Gorman G, Reece E (1988) Encephalitis among Cree children in northern Quebec. Ann Neurol 24:483–489
Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van BH, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T (2006) Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38:917–920
Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP (2006) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38:910–916
Dale RC, Tang SP, Heckmatt JZ, Tatnall FM (2000) Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics 31:155–158
De Laet C, Goyens P, Christophe C, Ferster A, Mascart F, Dan B (2005) Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutieres syndrome. Neuropediatrics 36:399–402
Rasmussen M, Skullerud K, Bakke SJ, Lebon P, Jahnsen FL (2005) Cerebral thrombotic microangiopathy and antiphospholipid antibodies in Aicardi-Goutieres syndrome-report of two sisters. Neuropediatrics 36:40–44
Aicardi J, Goutieres F (2000) Systemic lupus erythematosus or Aicardi-Goutieres syndrome? Neuropediatrics 31:113
Mazur DJ, Perrino FW (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′->5′ exonucleases. J Biol Chem 274:19655–19660
Mazur DJ, Perrino FW (2001) Excision of 3′ termini by the Trex1 and TREX2 3′->5′ exonucleases. Characterization of the recombinant proteins. J Biol Chem 276:17022–17029
Beresford PJ, Zhang D, Oh DY, Fan Z, Greer EL, Russo ML, Jaju M, Lieberman J (2001) Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem 276:43285–43293
Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 23:133–142
Hoss M, Robins P, Naven TJ, Pappin DJ, Sgouros J, Lindahl T (1999) A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J 18:3868–3875
Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE (2004) Gene-targeted mice lacking the Trex1 (DNase III) 3′->5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24:6719–6727
Martinvalet D, Zhu P, Lieberman J (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22:355–370
Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ (2007) Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres Syndrome. Am J Hum Genet 80:811–815
Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenzweig MC (2005) Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med 201:703–711
Rieux-Laucat F (2006) Inherited and acquired death receptor defects in human Autoimmune Lymphoproliferative Syndrome. Curr Dir Autoimmun 9:18–36
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T (2000) Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 25:177–181
Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y (2001) Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 28:313–314
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443:998–1002
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271–1277
Sontheimer RD (1997) The lexicon of cutaneous lupus erythematosus—a review and personal perspective on the nomenclature and classification of the cutaneous manifestations of lupus erythematosus. Lupus 6:84–95
Acknowledgment
We thank the members of the family for their participation in this study. We thank Maja Linné for clinical assistance and Angela Rösen-Wolff and Joachim Rösler for helpful discussions. This work was supported by a Marie Curie Development Host Fellowship from the European Commission to M.L.-K., US grant GM069962 from the NIH to F.W.P., US grant AI45587 from the NIH to J.L., and a grant-in-aid from the National Genome Research Network (NGFN2) of the German Ministry of Science and Education (BMBF) to N.H.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Lee-Kirsch, M., Chowdhury, D., Harvey, S. et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med 85, 531–537 (2007). https://doi.org/10.1007/s00109-007-0199-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0199-9