
Abstract
Association mapping based on linkage disequilibrium provides a promising tool for dissecting the genetic basis underlying complex traits. To reveal the genetic variations of yield and yield components traits in upland cotton, 403 upland cotton accessions were collected and analyzed by 560 genome-wide simple sequence repeats (SSRs). A diverse panel consisting of 403 upland cotton accessions was grown in six different environments, and the yield and yield component traits were measured, and 560 SSR markers covering the whole genome were mapped. Association studies were performed to uncover the genotypic and phenotypic variations using a mixed linear model. Favorable alleles and typical accessions for yield traits were identified. A total of 201 markers were polymorphic, revealing 394 alleles. The average gene diversity and polymorphism information content were 0.556 and 0.483, respectively. Based on a population structure analysis, 403 accessions were divided into two subgroups. A mixed linear model analysis of the association mapping detected 43 marker loci according to the best linear unbiased prediction and in at least three of the six environments(− lgP > 1.30, P < 0.05). Among the 43 associated markers, five were associated with more than two traits simultaneously and nine were coincident with those identified previously. Based on phenotypic effects, favorable alleles and typical accessions that contained the elite allele loci related to yield traits were identified and are widely used in practical breeding. This study detected favorable quantitative trait loci’s alleles and typical accessions for yield traits, these are excellent genetic resources for future high-yield breeding by marker-assisted selection in upland cotton in China.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cotton (mainly upland cotton) is the world’s most important natural textile fiber and a significant oilseed crop (Zhang et al. 2012). China is the world’s largest country of cotton production, consumption and importation, and it is also the largest producer of textiles and garments. The major cotton growing regions in China are the Yangtze River Region (YtRR), Yellow River Region (YRR), Northwestern Inland Region (NIR) and Northern Specific Early Maturation Region (NSEMR). In recent years, with climate change and adjustments to the industrial development strategy in China, the cotton production and growing areas were gradually reduced in the YtRR, YRR and NSEMR, but the NIR, represented by the Xinjiang cotton growing region, has rapidly expanded, becoming the largest cotton planting area worldwide. In 2016, the total cotton growing area was 1904.3 kha in Xinjiang, accounting for 50.1% of the total growing area in China, and its total yield was 3.503 million tons, contributing 62.5% of the total yield in China. The cotton yield per unit area was 1839.6 kg/ha, which is 364.3 kg/ha higher than the national average (National Bureau of Statistics of the People’s Republic of China, http://www.stats.gov.cn, 2015). Therefore, Xinjiang has become the cotton growing area with greatest yield worldwide.
The major yield indicators of cotton include seed cotton yield per plant (SY), lint percentage (LP), boll weight (BW), bolls per plant (BN), lint index (LI) and seed index (SI), which are controlled by complicated quantitative trait loci (QTLs). In cotton breeding, the mapping of QTLs conditioning the above-mentioned traits and the identification of stable QTLs and favorable alleles are of great theoretical and practical value for the improvement of cotton yield. The classical genetic method of studying quantitative traits is based on a linkage analysis of both parents, and it enabled the mapping of multiple QTLs responsible for cotton yield and fiber quality using segregating populations from different parents (Guo et al. 2013; Hu et al. 2008; Liu et al. 2014; Ma et al. 2008; Ning et al. 2013; Qin et al. 2008, 2009; Shao et al. 2014; Shen et al. 2005; Ulloa and Meredith 2000; Yin et al. 2002; Yu et al. 2013), providing the basis for the study of the genetic architecture of quantitative traits in cotton. However, because of the limited number of markers in upland cotton populations, the accuracy of QTLs is relatively low, which impedes further studies using map-based cloning and marker-assisted selection. An association analysis is an effective method for studying complex quantitative traits, and it facilitates the fine mapping of target genes, providing information on certain candidate genes and validating their functions. Compared with a conventional linkage analysis, an association analysis exhibits three significant characteristics: (1) the development of a mapping population is not required, and all of the natural population and germplasm resources can serve as mapping materials. In addition, knowing the pedigrees of these materials is not mandatory, as long as genetic variation is present; (2) the diverse genetic materials assist the identification of greater numbers of gene alleles, which improves the resolution of the map; and (3) it can evaluate most of the QTL-linked loci and their allelic variations in multiple traits simultaneously, resulting in a greater mapping efficiency. This method has already been applied to crops, such as wheat (Maccaferri et al. 2006), rice (Iwata et al. 2010), maize (Yang et al. 2010) and soybean (Hou et al. 2011). In cotton populations, association mapping has been used to identify QTLs or determine genomic regions associated with plant type (Li et al. 2016), yield and its components (Mei et al. 2013), fiber quality traits (Nie et al. 2016; Zeng et al. 2009), seed oil and protein contents (Liu et al. 2015), and resistance traits (Mei et al. 2014; Zhao et al. 2014). These studies laid a sound foundation for the application of marker-assisted selection (MAS) and molecular design in cotton breeding.
In the present study, two experimental sites were employed, Korla in southern Xinjiang and Shihezi in northern Xinjiang. Using 403 upland cotton germplasms from different cotton growing regions (199 from NIR, 49.3% of the total number) and SSR markers evenly distributed on the cotton genome, we performed a whole genome scan and an association analysis to investigate the favorable genes, allelic mutations and germplasms related to cotton yield and yield-related traits in the high-yield region of Xinjiang. This work will provide a theoretical basis for MAS in upland cotton breeding.
Materials and methods
Selection of upland cotton accessions
A total of 403 upland cotton accessions from China (378) and other countries (25) were selected from cotton germplasm collections in our laboratory for association mapping purposes (Table S1). These accessions were divided into the following five groups based on ecological areas, with 87 from the YRR in China, 63 from the YtRR in China, 199 from the NIR in China, 29 from the NSEMR in China and 25 from foreign countries, with 18 from the United States, five from the Soviet Union, one from Israel and one from Angola.
Field experiments and trait phenotyping
All of the 403 upland cotton accessions were planted at Shihezi, North Xinjiang (E85.94°, N44.27°) in 2013, 2014 and 2015 (designated SHZ13, SHZ14 and SHZ15, respectively) and at Korla, South Xinjiang (E86.06°, N41.68°, in the NIR) in 2013, 2014 and 2015 (designated KRL13, KRL14 and KRL15, respectively). Each accession was grown in a plot having 40–45 plants in two rows, with 0.10 m between the plants in each row and 0.45 m between the rows. The field planting followed a randomized complete block design with three replications in each environment. Field management followed conventional standard field practices. The 10 plants in the middle of each row were tagged for scoring and harvesting seed cotton. The yield traits evaluated included SY (g/plant), LP (%), BW (g), BN, LI (g) and SI (g).
SSR marker genotyping
Total genomic DNA of 403 accessions was extracted from the leaves as described by Paterson et al. (1993). SSR markers were selected at an average distance of 10 cM on each of the 26 chromosomes from the tetraploid cotton genetic map (Zhao et al. 2012). Additionally, previously reported markers linked to QTLs for breeding target traits of cotton (Guo et al. 2013; Ning et al. 2013; Qin et al. 2008; Shen et al. 2005) were also selected. In total, 560 SSR markers were used to screen for the genetic polymorphisms of the population. The SSR primer sequences were obtained from the Cotton Microsatellite Database (http://www.cottonmarker.org/). The polymerase chain reaction (PCR) amplification procedure was performed as described by Zhang et al. (2002). The polymorphisms detected by SSR markers were evaluated using FA™-96 Automated High Throughput SSR/Tilling Analysis System (Advanced Analytical Technology Inc, Ankeny, IA, USA), SSR genotyping was coded as “1” for present, “0” for absent and “?” for missing data.
Phenotypic data analysis
The descriptive statistics, association analyses and the frequency distributions of the phenotypic traits were carried out using SPSS 19.0 software (Li and Chen 2010). The best linear unbiased predictions (BLUP) for yield and yield component traits across the six environments were obtained using the “R” program. The ANOVA for each trait under multiple environments was performed in SAS 8.1. The variance was divided into components of germplasm, environment, and the interaction of germplasm and environment to determine the variance of each trait, and the broad sense heritability \({{h}}_{{{B}}}^{{\text{2}}}\) was calculated based on the variance.
Genotypic data analysis
The analysis of the genotypic data of the 403 germplasms was performed using PowerMarker V3.25 software (Liu and Muse 2005). The analysis of the population structure based on the genotypes was performed using Structure 2.3.1 software (Evanno et al. 2005) with the parameter settings as follows: 1–10 was selected for kinship (K) value with a replication of 5; the length of burn in period of the Markov chain monte carlo was set to 10,000 times at the beginning, followed by 100,000 times, and all other parameters were as default. The ΔK was calculated based on lnP (D), with which an appropriate K value was selected to obtain the corresponding population structure (Q) matrix. The genetic similarity coefficient (Jaccard coefficient) among germplasms was determined by NTSYS-pc V2.10 software to obtain a phylogenetic tree (Adams and Rohlf 2000). The linkage disequilibrium (LD) values among the polymorphic loci were evaluated by Tassel5.0 (Bradbury et al. 2007).
Association analysis and favorable allele exploration
Using Tassel 5.0 software (Bradbury et al. 2007), along with the K matrix produced from genotypic data, the above-mentioned marker data, the trait values under each environment, and the BLUP values and Q matrix, a mixed linear model (MLM) was employed to perform the association analysis between traits and markers, and the phenotypic variation explained (R2) by each loci at the significance of P < 0.05 (− lgP > 1.30) was calculated. After obtaining associated markers, the phenotypic effects of allelic variations at SSR loci were evaluated, and the allelic variations, phenotypic effects and typical varieties significantly associated with the traits were analyzed. The calculation method for the phenotypic effect of SSR alleles was as follows:
where a i represents the phenotypic effect of the ith allele, x ij represents the jth material’s phenotypic value of the ith allele, n i represents the number of materials having the ith allele; N k represents the nth material’s phenotypic value of all materials, and n k is the number of materials. If a i > 0, then the allele is considered to have a positive effect; if a i < 0, then the allele is considered to have a negative effect.
Results
Variations in phenotypic traits
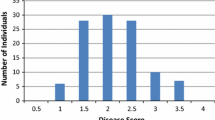
The phenotypic characteristics of yield and yield components in six environments were determined by BLUPs, the BLUPs values of each accession and phenotypic data for each environment of six yield and yield components traits were determined for association mapping. The coefficients of variance for SY, LP, BW, BN, SI and LI were 9.04, 7.01, 7.30, 8.43, 9.37 and 9.26%, respectively (Table 1), indicating that there was a broad variation under the six environmental conditions among the 403 upland cotton accessions. The \({{h}}_{{{B}}}^{{\text{2}}}\) for the six traits had a range of 47.77–91.99% among the accessions (Table 1). LP showed the greatest \({{h}}_{{{B}}}^{{\text{2}}}\) value (91.99%), indicating that LP was less impacted by environmental factors than the other five traits. An analysis of the frequency distribution based on the results of BLUP processing indicated that each trait had a normal or approximately normal distribution and was suitable for genetic analysis (Fig. 1). There were significant positive phenotypic correlations between SY and its components, and there were significant negative correlations between SI and LP, SI and BN, and BW and BN (Table 2). The correlation coefficients for SY with LP, BW, BN, SI and LI were 0.109, 0.464, 0.799, 0.234 and 0.331, respectively.

Frequency distributions of the breeding values based on the BLUPs of six yield-related agronomic traits of 403 cotton accessions in six environments. a Seed cotton yield (SY); b lint percentage (LP); c boll weight (BW); d the number of bolls per plant (BN); e seed index (SI); and f lint index (LI)
Genetic diversity of the SSR markers
From 560 SSR markers in the 403 upland cotton accessions, 201 displayed polymorphisms (Table S2), accounting for 36.1% of the total markers, with an average of 7.73 markers per chromosome. A total of 394 alleles were obtained, ranging from 1 to 4, with an average of 1.96. The number of genotypes per marker ranged from 2 to 14, with an average of 4. The gene diversity and polymorphism information content (PIC) of the 394 alleles averaged 0.556 and 0.483, respectively, with ranges of 0.142–0.669 and 0.132–0.677, respectively (Fig. 2). Thus, a large difference in the distribution frequencies existed in upland cotton accessions.

Distributions of genetic diversity of 201 SSR marker loci in 403 upland cotton accessions. a Gene diversity, b polymorphism information content (PIC)
Population structure and linkage disequilibrium
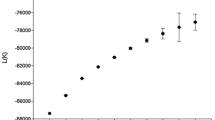
The population structure was determined using STRUCTURE software. When K = 1–10, the LnP(D) was elevated as the K value increased, and no inflection point was found in this panel (Fig. 3a). Therefore, ΔK changes were adopted to determine suitable K values. When K = 2, the ΔK reached its maximum value (Fig. 3b), suggesting that the total panel could be divided into two subpopulations (Fig. 3c), designated as Subgroups 1 and 2. Subgroup 1 contained 206 accessions, including 70 accessions (70/87, 80.5%) from the YRR, 58 accessions (58/63, 92.1%) from the YtRR, 10 accessions (10/29, 35.5%) from the NSEMR, 53 accessions (53/199, 26.6%) from the NIR and 15 accessions (15/25, 60.0%) from countries other than China. Subgroup 2 contained 197 accessions, including 17 accessions (17/87, 19.5%) from the YRR, 5 accessions (5/63, 7.9%) from the YtRR, 19 accessions (19/29, 65.5%) from the NSEMR, 146 accessions (146/199, 73.4%) from the NIR and 10 accessions (10/25, 40.0%) from countries other than China. The Neighbor-joining method was used to construct a phylogenetic tree containing the 403 upland cotton accessions based on Nei’s genetic distances calculated by PowerMarker V3.25 software, and it showed that the majorities of Subgroups 1 and 2 were clustered together in the unrooted tree (Fig. S1). Based on these results, the corresponding Q matrix at K = 2 was used for the marker-trait association mapping. The LD of this population was analyzed using 201 SSR markers. In total, 18.94% of the marker loci showed significant LD values (P < 0.05), the LD distribution was nonuniform on each chromosome, with the loci having greater LD levels being mainly concentrated on chromosomes D5, D7, D8, D11, D6 and A8 (Fig. S2).

Estimated LnP(D) and ∆K values based on the population structural analysis of 403 upland cotton accessions. a The magnitude of LnP(D) as a function of K; b magnitude of ∆K as a function of K; c population structure of 403 upland cotton accession-based SSR markers that were divided into two subpopulations: red indicates Subgroup 1 and green indicates Subgroup 2. (Color figure online)
Markers associated with yield and yield components
Based on the genotypic data, the Q, the K and the phenotypic traits data, a MLM of association mapping was performed to identify the associated SSR loci in upland cotton accessions. 43 loci were found to be related to yield and its components traits according to the BLUPs and in at least three of the six environments at the P < 0.05 (− lgP > 1.3) significance level, the range of the explained phenotypic variation observed was from 0.97 to 4.01%, with an average of 1.62% (Table 3). The numbers of marker loci associated with SY, LP, BW, BN, SI and LI were 4, 9, 6, 3, 10 and 11, respectively. Moreover, four marker loci (BNL3089a, NAU1028a, NAU3031b and NAU3881c) were simultaneously associated with two different traits, and one marker loci (NAU2984b) was simultaneously associated with three different traits.
Discovery of favorable alleles and typical accessions
The phenotypic effects of each QTL’s allele for the 21 associated marker loci were measured according to the calculation formula of the phenotypic allele effect. Phenotypic effects and typical accessions for each favorable allele are shown in Table 4. Taking LI as an example, six marker loci having positive phenotypic effects on LI were obtained, of which NAU4057a manifested the greatest positive effect on the phenotype, and increased LI by 0.14 g in the typical germplasm accessions ‘L4-13’, ‘Huiyuan14-19’ and ‘Chuang65’.
Discussion
An association analysis is an analytical method based on individual phenotypes and genotypes: thus, the accuracy of the phenotypic data has a great impact on the results of an association analysis. The BLUP method has desired characteristics, such as an unbiased estimation, minimum variance of estimation, and the ability to compensate for the bias caused by selection and elimination, and thus, it produces the best linear unbiased results when acquiring individual phenotypic data (Schenkel et al. 2002). In this study, a phenotypic trait analysis based on the BLUP results eliminated environmental effects and improved the accuracy of predicting complex quantitative traits. A genotypic analysis revealed an average genetic diversity and a polymorphism information content of 0.556 and 0.483, respectively, which were greater than those of previous studies (Nie et al. 2016; Qin et al. 2015). As a result, we explored the 403 upland cotton accessions, one of the largest yet investigated, with broad geographic and genetic diversity ranges that provided sufficient detection power for association mapping.
In the association mapping, the population structure always affects the LDs of loci, which further influences the accuracy of the association analysis, resulting in false positives (Pritchard et al. 2000). The MLM (Q + K), which employs both population structure (Q value) and the genetic relationship among varieties (K value), is better than GLM (Q) and MLM (K) that are solely based on Q and K values, respectively (Zhao et al. 2007). In this study, we evaluated the genotypic data of polymorphic loci using STRUCTURE software to analyze and calibrate the population structure, to calculate the probability of classifying materials into subgroups, and to perform the MLM association analysis with it as a covariate. This effectively corrected the false association caused by the existence of subgroups. In addition, based on the subgrouping from STRUCTURE, the clustering analysis was carried out using Nei’s genetic distance calculation method, and results of both methods showed good consistency. Based on the pedigree, we demonstrated that the cotton from different growing regions in China originated from different sources. The genetic components of the cotton grown in YRR and YtRR are mainly from American cotton germplasms ‘Stoneville’, ‘Lone star’, ‘Foster’, ‘Acala’ and ‘Deltapine’, and they exhibit large BNs, high LPs, loose architecture and mid-to-late maturity. The cotton grown in NIR is mainly based on genetic components that originated from germplasms from the former Soviet Union, such as ‘108Fu’, ‘KK1543’ and ‘611Bo’, which show compact statures, short fruit branches, early maturity, and fast and centered boll openings. The cotton varieties of the NSEMR are based on the American cotton ‘King’ with the integration of genetic components of a former Soviet Union variety, and they show early maturity and disease resistance (Ai et al. 2005; Huang 2007). The tested germplasms in the present study were classified into two populations, and varieties originating from various cotton growing regions were present in each population, but the varieties in the YtRR and YRR were mainly classified into Subgroup 1, while most of the NIR and NSEMR varieties were classified into Subgroup2, which was consistent with the pedigrees of these germplasms.
The association analysis was based on the LD values among markers. Thus, understanding the genomic LDs of the targeted population can result in estimates of the density and quantity requirements of markers in the analysis. As an often cross-pollinated crop, cotton exhibits a relatively high recombination rate among genomic loci, so its LD is relatively low. The LD decay analysis showed that the average decay distances in the upland cotton population were 8–25 and 3–7 cM, with LD coefficients r2 ≥ 0.1 and r2 ≥ 0.2, respectively, and 100–400 SSR markers were used for statistical analysis in upland cotton (Cai et al. 2014; Mei et al. 2013; Nie et al. 2016; Qin et al. 2015). In the present study, among the 560 uniformly distributed SSR markers, 201 were polymorphic, and was sufficient to perform association analysis of QTLs in upland cotton. however, to perform a genome-wide association analysis, we need to increase the marker density to identify more markers linked to the targeted traits.
Identifying the favorable allelic variations and desired germplasms related to yield are important requirements for the breeding of high-yield upland cotton. In the present study, we performed phenotypic identifications of six yield-related traits in 403 upland cotton germplasms over 3 years at two locations in the high-yield environment of Xinjiang, a total of 43 marker loci were identified as associated with the six traits (− lgP > 1.30, P < 0.05). Of these, five marker loci were associated with multiple traits. For example, BNL3089a was related to SY and LI, NAU1028a was related to SY and BW, and NAU2984b was linked to BW, SI and LI, indicating that these markers can be used for the synchronous improvement of multiple traits. In addition, among the loci identified as associated with yield-related traits in the present study, nine were associated with the same traits based on the QTLs previously obtained by linkage analysis (Table 5), indicating the repeatability and stability of these markers. The remaining 34 markers may be novel loci related to yield and yield-related traits, which provide a theoretical basis for further understanding the genetic mechanisms of cotton yield. Moreover, in this study, the 21 stably detected associations provided 2, 5, 2, 2, 4 and 6 favorable alleles for SY, LP, BW, BN, SI and LI, respectively (Table 4). These favorable alleles can be used in MAS to improve the cotton output. Typical carrier accessions based on the favorable alleles for yield traits could be selected as predominant parents.
References
Adams DC, Rohlf FJ (2000) Ecological character displacement in Plethodon: biomechanical differences found from a geometric morphometric study. Proc Natl Acad Sci USA 97:4106–4111
Ai XT, Li XY, Qin WB, Sun GQ, Tuer XJ, Mo M (2005) The extend study on genetic composition of upland cotton breeding in Xinjiang. Mol Plant Breed 3:575–578
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cai CP, Ye WX, Zhang TZ, Guo WZ (2014) Association analysis of fiber quality traits and exploration of elite alleles in upland cotton cultivars/accessions (Gossypium hirsutum L.). J Integr Plant Biol 56:51–62
Evanno G, Regnauts S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Guo X, Guo YP, Ma J, Wang F, Sun MZ, Gui LJ, Zhou JJ, Song XL, Sun XZ, Zhang TZ (2013) Mapping heterotic loci for yield and agronomic traits using chromosome segment introgression lines in cotton. J Integr Plant Biol 55:759–774
Hou JF, Wang CL, Hong XJ, Zhao JM, Xue CC, Guo N, Gai JY, Xing H (2011) Association analysis of vegetable soybean quality traits with SSR markers. Plant Breed 130:444–449
Hu WJ, Zhang XY, Zhang TZ, Guo WZ (2008) Molecular tagging and source analysis of QTL for elite fiber quality in Upland cotton. Acta Agron Sin 34:578–586
Huang ZK (2007) The cultivars and their pedigree of cotton in China. China Agriculture Press, Beijing
Iwata H, Ebana K, Uga Y, Hayashi T, Jannink JL (2010) Genome-wide association study of grain shape variation among Oryza sativa L. germplasms based on elliptic Fourier analysis. Mol Breed 25:203–215
Lacape JM, Gawrysiak G, Cao TV, Viot C, Llewellyn D, Liu SM, Jacobs J, Becker D, Vianna Barroso PA, Assunçãog JH et al (2013) Mapping QTLs for traits related to phenology, morphology and yield components in an inter-specific Gossypium hirsutum × G. barbadense cotton RIL population. Field Crop Res 144:256–267
Li XS, Chen ZZ (2010) Correctly using SPSS software for principal components analysis. Stat Res 27:105–108
Li CQ, Ai NJ, Zhu YJ, Wang YQ, Chen XD, Li F, Hu QY, Wang QL (2016) Association mapping and favorable allele exploration for plant architecture traits in Upland cotton (Gossypium hirsutum L.) accessions. J Agr Sci Camb 154:567–583
Liu KJ, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu RZ, Ai NJ, Zhu XX, Liu FJ, Guo WZ, Zhang TZ (2014) Genetic analysis of plant height using two immortalized populations of “CRI12 × J8891” in Gossypium hirsutum L. Euphytica 196:51–61
Liu GZ, Mei HX, Wang S, Li XH, Zhu XF, Zhang TZ (2015) Association mapping of seed oil and protein contents in upland cotton. Euphytica 205:637–645
Ma XX, Ding YZ, Zhou BL, Guo WZ, Li YH, Zhang TZ (2008) QTL mapping in A-genome diploid Asiatic cotton and their congruence analysis with AD-genome tetraploid cotton in genus Gossypium. J Genet Genom 35:751–762
Maccaferri M, Sanguineti MC, Natoli V, Ortega JLA, Salem MB, Bort J, Chenenaoui C, Ambrogio DE, Moral LGD, Montis AD et al (2006) A panel of elite accessions of durum wheat (Triticum durum Desf.) suitable for association mapping studies. Plant Genet Resour 4:79–85
Mei HX, Zhu XF, Zhang TZ (2013) Favorable QTL alleles for yield and its components identified by association mapping in Chinese upland cotton cultivars. PLoS ONE 8:e82193
Mei HX, Ai NJ, Zhang X, Ning ZY, Zhang TZ (2014) QTLs conferring FOV7 resistance detected by linkage and association mapping in upland cotton. Euphytica 197:237–249
Nie XH, Huang C, You CY, Li W, Zhao WX, Shen C, Zhang BB, Wang HT, Yan ZH, Dai BS et al (2016) Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China. BMC Genom 17:352
Ning ZY, Zhao R, Chen H, Ai NJ, Zhang X, Zhao J, Mei HX, Wang P, Guo WZ, Zhang TZ (2013) Molecular tagging of a major quantitative trait locus for broad-spectrum resistance to verticillium wilt in upland cotton cultivar Prema. Crop Sci 53:2304–2312
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep 11:122–127
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Qin HD, Guo WZ, Zhang YM, Zhang TZ (2008) QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor Appl Genet 117:883–894
Qin YS, Liu RZ, Mei HX, Zhang TZ, Guo WZ (2009) QTL mapping for yield traits in upland cotton (Gossypium hirsutum L). Acta Agron Sin 35:1812–1821
Qin HD, Chen M, Yi XD, Bie S, Zhang C, Zhang YC, Lan JY, Meng YY, Yuan YL, Jiao CH (2015) Identification of associated SSR markers for yield component and fiber quality traits based on frame map and upland cotton collections. PLoS ONE 10:e0118073
Schenkel FS, Schaeffer LR, Boettcher PJ (2002) Comparison between estimation of breeding values and fixed effects using Bayesian and empirical BLUP estimation under selection on parents and missing pedigree information. Genet Sel Evol 34:41–59
Shao QS, Zhang FJ, Liu Y, Fang XM, Liu DJ, Zhang J, Teng ZH, Paterson AH, Zhang ZS (2014) Identifying QTL for fiber quality traits with three upland cotton (Gossypium hirsutum L.) populations. Euphytica 198:43–58
Shen XL, Guo WZ, Zhu XF, Yuan YL, Yu JZ, Kohel RJ, Zhang TZ (2005) Molecular mapping of QTLs for fiber qualities in three diverse lines in upland cotton using SSR markers. Mol Breed 15:169–181
Ulloa M, Meredith WR (2000) Genetic linkage map and QTL analysis of agronomic and fiber quality traits in an intraspecific population. J Cotton Sci 4:161–170
Wang BH, Guo WZ, Zhu XF, Wu YT, Huang NT, Zhang TZ (2007) QTL mapping of yield and yield components for elite hybrid derived-RILs in upland cotton. Acta Genet Sin 34:35–45
Wang FR, Gong YC, Zhang CY, Liu GD, Wang LM, Xu ZZ, Zhang J (2011) Genetic effects of introgression genomic components from Sea Island cotton (Gossypium barbadense L.) on fiber related traits in upland cotton (G. hirsutum L.). Euphytica 181:41–53
Yang XH, Yan JB, Shah T, Warburton ML, Li Q, Li L, Gao YF, Chai YC, Fu ZY, Zhou Y et al (2010) Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor Appl Genet 121:417–431
Yin JM, Wu YT, Zhang J, Zhang TZ, Guo WZ, Zhu XF (2002) Tagging and mapping of QTLs controlling lint yield and yield components in upland cotton (Gossypium hirsutum L.) using SSR and RAPD markers. Chin J Biotechnol 18:162–166
Yu JW, Yu SX, Gore M, Wu M, Zhai HH, Li XL, Fan SL, Song MZ, Zhang JF (2013) Identification of quantitative trait loci across interspecific F2, F2:3 and testcross populations for agronomic and fiber traits in tetraploid cotton. Euphytica 191:375–389
Zeng L, Meredith WR Jr, Gutierrez OA, Boykin DL (2009) Identification of associations between SSR markers and fiber traits in an exotic germplasm derived from multiple cross among Gossypium tetraploid species. Theor Appl Genet 119:93–103
Zhang J, Guo WZ, Zhang TZ (2002) Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. × Gossypium barbadense L.) with a haploid population. Theor Appl Genet 105:1166–1174
Zhang K, Zhang J, Ma J, Tang SY, Liu DJ, Teng ZH, Liu DX, Zhang ZS (2012) Genetic mapping and quantitative traitlocus analysis of fiber quality traits using a three-parent composite population in upland cotton (Gossypium hirsutum L.). Mol Breed 29:335–348
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P et al (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:e4
Zhao L, Lv YD, Cai CP, Tong XC, Chen XD, Zhang W, Du H, Guo XH, Guo WZ (2012) Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genom 13:539
Zhao YL, Wang HM, Wei CW, Li YH (2014) Genetic structure, linkage disequilibrium and association mapping of verticillium wilt resistance in elite cotton (Gossypium hirsutum L.) germplasm population. PLoS ONE 9:e86308
Acknowledgements
This program was financially supported by National Natural Science Foundation of China (31260340; 31560410) and National Key R&D Program of China (2017YFD0101600). We thank Nie Xinhui from Shihezi University for help with the analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest. Chengguang Dong declares that he does not have conflict of interest. Juan Wang declares that he does not have conflict of interest. Quanjia Chen declares that she does not have conflict of interest. Yu Yu declares that he does not have conflict of interest. Baocheng Li declares that he does not have conflict of interest.
Research involving human and animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dong, C., Wang, J., Chen, Q. et al. Detection of favorable alleles for yield and yield components by association mapping in upland cotton. Genes Genom 40, 725–734 (2018). https://doi.org/10.1007/s13258-018-0678-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-0678-0