
Abstract
The germplasm with exotic genomic components especially from Sea Island cotton (Gossypium barbadense L. Gb) is the dominant genetic resources to enhance fiber quality of upland cotton (G. hirsutum L., Gh). Due to low efficiency of phenotypic evaluation and selection on fiber quality, genetic dissection of favorable alleles using molecular markers is essential. Genetic dissection on putative Gb introgressions related to fiber traits were conducted by SSR markers with mapping populations derived from a cross between Luyuan343 (LY343), a superior fiber quality introgression line (IL) with genomic components from Gb, and an elite Upland cotton cv. Lumianyan#22 (LMY22). Among 82 polymorphic loci screened out from 4050 SSRs, 42 were identified as putative introgression alleles. A total of 29 fiber-related QTLs (23 for fiber quality and six for lint percentage) were detected and most of which clustered on the putative Gb introgression chromosomal segments of Chr.2, Chr.16, Chr.23 and Chr.25. As expected, a majority of favorable alleles of fiber quality QTLs (12/17, not considering the QTLs for fiber fineness) came from the IL parent and most of which (11/12) were conferred by the introgression genomic components while three of the six (3/6) favorable alleles for lint percentage came from the Gh parent. Validation of these QTLs using an F8 breeding population from the same cross made previously indicated that 13 out of 29 QTLs showed considerable stability. The results suggest that fiber quality improvement using the introgression components could be facilitated by marker-assisted selection in cotton breeding program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton is the leading natural fiber crop in the world. Breeding for high cotton lint yield is still the primary goal of any breeding program, but improving fiber quality has become increasingly important with technological development in the textile industry (Meredith 1992). Upland cotton, Gossypium hirsutum L. (Gh), which dominates the world’s cotton fiber production, has high lint yield but undesirable fiber quality. However, Sea Island cotton (G. barbadense L., Gb), another cultivated allotetraploid cotton species, has superior fiber quality but lower lint yield. Therefore, combining the desirable lint yield of Gh with the superior fiber quality of Gb by interspecific hybridization has been an elusive goal of cotton breeders all over the world. Extensive efforts have been put into doing successful wide-cross between Gh and Gb in order to enhance the fiber quality of upland cotton. Unfortunately, improvement directly through interspecific hybridization has proven to be very difficult due to the excessive segregation of interspecific progenies (Percy et al. 2006). However, a series of germplasms with improved fiber quality containing chromosomal introgressions from Gb have been created. Compared with Gb, the superior introgression line (IL) has fiber quality more similar to that of Sea Island cotton and is usually more convenient to use in cotton improvement, as it has significantly less hybrid incompatibility and segregation distortion than interspecific cotton hybridization populations. However, most ILs with superior fiber quality also have some unfavorable alleles affecting lint yield introgressed into the genome. This negative association between lint yield and fiber quality is known as linkage drag (Hospital 2001; Meredith and Bridge 1971; Meredith 1992) and has proven difficult to break by traditional breeding methods. This is one of the major impediments in the synchronous improvement of fiber quality and lint yield in upland cotton breeding.
Molecular markers provide a useful approach for fine dissection and selection of economically important traits in cotton. Identification of exogenous useful genomic components from plant relatives and marker-guided introgression into cultivated crop varieties for potential application in marker-assisted selection (MAS) have been reported in tomato (Barone et al. 2009; Fernie et al. 2006; Zamir 2008), rice (Steele et al. 2006), wheat (Pestsova et al. 2006), maize (Zheng 2008), and oat (Yu and Herrmann 2006).
Several studies about QTL mapping for cotton fiber quality and lint yield have been reported in recent years. However, the majority of mapping population used in QTL detection were derived from direct interspecific (Gh × Gb) hybridization (Lacape et al. 2005; He et al. 2007; Paterson et al. 2003; Chee et al. 2005a; Mei et al. 2004; Draye et al. 2005). QTL mapping studies for fiber related traits in intraspecific population of Gh have also been reported (Ulloa et al. 2002; Ulloa and Meredith 2000; Zhang et al. 2003; Shen et al. 2005a; Zhang et al. 2009; Wu et al. 2009). However, no specific relationship between the fiber quality and introgression alleles and/or chromosomal segments were found, and most fiber related QTL mapping using intraspecific populations reported previously (Ulloa et al. 2002; Ulloa and Meredith 2000; Zhang et al. 2003; Zhang et al. 2009) were conducted using either genetic materials or outdated cultivars. In addition, some studies were carried out by non-PCR based molecular markers such as RFLP (Ulla et al. 2005; Ulloa et al. 2002) which is inconvenient for MAS due to the low throughput of marker analysis.
Recently, Zeng et al. (2009) identified associations between SSR markers and fiber traits using an exotic germplasm population derived from multiple crosses among Gossypium tetraploid species, and evaluated the germplasm for genetic variation in lint yield and fiber quality (Zeng and Meredith 2009). However, there are not any reports on direct QTL mapping and analysis on introgression genomic components.
In this study, we used a superior fiber quality IL, Luyuan343 (LY343), which was developed by wide cross between Gh and Gb (An unknown Gh cultivar cross to an Egyptian Gb cv. Ashimouni) (Su et al. 2000), to evaluate the association between the putative stably integrated introgression alleles or chromosomal segments under Gh background and the economically important fiber-related traits by PCR-based identification of SSR markers. Further validation of the effects of detected QTLs was conducted using an advanced breeding population from the same cross we previously developed. The purpose of our study was to dissect the association among the introgression alleles and fiber-related traits by using molecular markers for possible application in fiber quality improvement of upland cotton by MAS. Our findings should provide useful insights into the use of fiber quality improvement using superior fiber quality ILs in upland cotton by MAS.
Materials and methods
Population development and phenotypic evaluation
Luyuan343 (LY343) is a high fiber quality upland cotton line developed by continuous selection through 13 generations on the natural heterozygote of an Egyptian cotton cultivar (Ashimouni) with a Gh cultivar in China in the last century (Su et al. 2000). Its exact Gh parent is unknown. It had once been cultivated as a high quality variety of cotton at a small scale in China due to its earlier maturity, and higher number of boll set per plant compared with other high fiber quality lines (Wang et al. 2007). Seeds of LY343 were kindly supplied by the Institute of Application of Atomic Energy of Shandong Academy of Agricultural Sciences. Lumianyan#22 (LMY22) is a transgenic upland cotton cultivar with higher lint percentage but common fiber quality (Table 1) developed from a cross of Simian#3 × GK-12 by conventional pedigree selection. Seeds of TM-1, which is widely used as the standard genetic line of upland cotton, and Ashmouni, which is the donor Sea Island cotton cultivar of the LY343 were introduced from National Cotton Germplasm Bank in Cotton Institute of Chinese Academy of Agricultural Sciences. All seeds were propagated by self-pollination before the experiments were conducted.
The hybridization of LMY22 × LY343 was performed in 2006 and the F1 seeds were planted and self-pollinated at Hainan Island in winter of the same year. Two hundred and nine F2 individuals from a single F1 plant were grown and self-pollinated to generate F2:3 progeny families at the Experimental Station of Shandong Cotton Research Center (ESSCRC) in Linqing county, Shandong province, China. The seed cotton of each F2 individuals were harvested separately. The seeds of each F2 individual were divided into two parts and generated two similar populations of F2:3 families, which were planted at Hainan Island in the winter of 2007 and at ESSCRC in the spring of 2008, respectively.
Single fiber samples of each F2 individuals were collected directly from the gin-out of self-pollinated bolls while three fiber samples of every F2:3 line were taken by homogenizing the gin-out of bolls harvested evenly from every individual of the corresponding lines. The fiber quality parameters of F2 and F2:3 in two environments including fiber length (FL, mm), fiber strength (FS, cN Tex−1), fiber length uniformity (FU, %), fiber elongation (FE, %) and fiber fineness (FM, micronaire value) were tested by the Supervision, Inspection and Test Center of Cotton Quality, Ministry of Agriculture of China using a High-Volume Precision Instrument (HVI; Zellweger-Uster, Knoxville, Tenn). Data of other agronomic traits including lint percentage were collected and evaluated following the conventional cotton breeding methods.
Phenotypic variation was evaluated by ANOVA analysis and genetic correlations among fiber quality related traits (including lint percentage data) in both F2 and F2:3 populations planted at Hainan and Linqing locations in china were conducted using stepwise regression analysis. All statistical analyses were performed using the DPS software (Tang and Feng 2007).
Linkage map construction and identification of putative introgression genomic components
Genomic DNA of 209 F2 individuals and the mapping parents, LMY22 and LY343 together with TM-1 (genetic standard line of Gh) and Ashimouni (the donor Gb parent) was extracted as described by Paterson et al. (1993). A total of 4050 SSR primer pairs from Cotton Microsatellite Database (CMD, http://www.cottonmarker.org) (Blenda et al. 2006) were selected to screen for polymorphisms between two parents. The method for PCR reaction and PAGE/silver staining survey was followed as described by Zhang et al. (2000).
Because the exact Gh parent of LY343 was not clear (Su et al. 2000), it is impossible to identify the introgression genomic components by directly comparing the recipient and the donor parents. So, in order to identify the putative introgression alleles, all SSRs revealing polymorphism between Gh parent LMY22 and IL parent LY343 were further compared with TM-1, and Ashmouni. The marker genotypes of the LY343 which are identical to Ashmouni but different from TM-1 were considered as putative introgression alleles.
The linkage map was constructed using the computer program MAPMAKER/EXP 3.0 (Lander et al. 1987). The χ2 test for goodness-of-fit was used to assess the Mendelian ratio of 1:2:1 inheritance in the F2 population. The Kosambi mapping function was used to convert recombination frequency to genetic map distance (centiMorgen, cM). A log-odds ratio score of 6.0 was set to develop the linkage map. The linkage groups were assigned to chromosomes based on the cotton genetic linkage map constructed by Guo et al. (2007).
QTL mapping
QTL analysis was carried out using the composite interval mapping (CIM) tool of the program WinQTLCart2.5 (Wang et al. 2006). QTL mapping was conducted using three phenotypic data sets: one from F2, and two from F2:3 at Linqing (LQ) location and Hainan (HN) location, respectively. A LOD score threshold of >3.0 was used to declare significant QTLs. However, since the low polymorphism between LMY22 and LY343 influenced the saturation of our linkage map, the suggestive QTLs which were detected with LOD scores between 2.0 and 3.0 were also adopted in this study.
For the polymorphic loci not assigned to any linkage groups, simple marker analysis was conducted by ANOVA. Only the QTLs detected significant in two data sets in simple marker analysis are reported in this paper.
QTL nomenclature was followed as described by Qin et al. (2008) and the effects of QTLs are considered from a product transformation perspective (Lacape et al. 2005). Accordingly, increases in fiber length (FL), fiber strength (FS), fiber length uniformity (FU), fiber elongation (FE) and lint percentage (LP) was indicative of a positive contribution conferred by either parent. We didn’t use the “positive” or “negative” nomenclature for fiber fineness because of non-linearity in the scale of micronaire values, which are considered desirable between 3.7 and 4.1 but undesirable and even unacceptable if lower than 3.7.
Validation of QTL effect by an advanced population
The F8 lines were selected from a breeding population previously developed from the same cross of LMY22 × LY343 in 2003 (Wang et al. 2007). The genetic effects of stable QTLs that linked to molecular markers should be authenticated in any breeding populations containing the specific allele. So, the F8 population that developed from an independent crossing event by the same parents of the mapping population could be used to validate the QTLs identified in the mapping population. Eighty-three lines of the F8 population were planted in trial plots at the Linqing site in 2008 and 2009. DNA was extracted from the 83 F8 lines from mixed leaf samples. And the fiber sample collection and phenotypic evaluation were conducted as that of F2:3 lines described above.
In order to validate the QTLs effect detected in F2 and F2:3 populations, associations between marker genotype and fiber-related traits in this advanced F8 population were conducted by simple marker analysis based on ANOVA.
Results
Phenotypic variations of fiber-related traits in parents and populations
The fiber quality performance and one of the yield traits, lint percentage, for parents and their F2 and F2:3 populations are presented in Table 1. Our ANOVA results showed highly significant differences between the IL parent (LY343) and the Gh parent (LMY22) for fiber length (FL) (36.31/29.34 mm), fiber strength (FS) (36.16/27.84 cN Tex−1), fiber micronaire (FM) (3.56/5.01) and lint percentage (LP) (34.58/41.26%) (see Table 1).
The values of skewness and kurtosis indicated that all variations in F2 and F2:3 populations fitted normal distributions. We did find differences in population means for fiber strength (FS) (31.42/35.41) and fiber micronaire (FM) (3.63/4.31) between Hainan and Linqing locations (Table 1). This could be a result of the shorter growth season and therefore faster fiber development at the Hainan site compared to the Linqing site.
The genetic correlation analysis among fiber-related traits
Correlation coefficients among fiber-related traits calculated in F2 and F2:3 populations are presented in Table 2. Among fiber quality traits, highly significant correlations between FL and FS (0.58 on average), FU and FS (0.54), FL and FU (0.37) were consistently observed within the three data sets and a significant negative correlation between FL and FM (0.41) was observed in two data sets from the F2:3 generation (see Table 2).
As for lint percentage and fiber quality traits, strong negative correlations between LP and FL, and LP and FS were observed in all datasets. Positive correlation between LP and FM was identified in two of three data sets. These results are consistent with previous studies (Qin et al. 2008; Ulloa et al. 2002; Ulloa and Meredith 2000). There was no significant correlation between LP and FU, LP and FE.
Construction of SSR-based linkage map and identification of putative introgression genomic components derived from G. barbadense
A total of 82 loci screened out for 4050 SSRs (2.0%) revealed polymorphism between LMY22 and LY343. The linkage map was constructed from 82 loci comprised of 17 linkage groups with 57 mapped loci and spanned 411.6 cM. Twenty-five loci failed to link to any group.
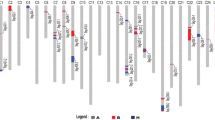
The marker genotypes revealed by PAGE/silver staining were used to determine the putative introgression alleles. The same marker genotypes of LY343 and Ashmouni (the donor Gb parent) represent the putative introgression alleles while the identical marker genotypes of TM-1 and LMY22 represent upland cotton background alleles (Fig. 1). Among 82 polymorphic loci, forty-two loci were identified as putative introgression components. Thirty-three of the 42 loci were anchored to 11 linkage groups (see Fig. 2). There were four marker-rich putative introgression chromosomal segments in the linkage map. The largest one was a segment of Chr.2 covering 57.2 cM with eight markers. Two putative introgression chromosomal segments were identified on Chr.16 covering 20.2 cM with three markers and 29.7 cM with eight markers, respectively. In addition, there was a segment on Chr.23 covering 4.7 cM with four markers. Two putative introgression segments on Chr.4 (12.7 cM) and Chr.18 (8.2 cM) with only two markers (Fig. 2) were also discovered. More markers covering these areas should be identified in order to saturate the map and to investigate the associations of the putative introgression chromosomal segment with fiber related traits.

Comparison of the amplified band patterns of some introgression SSR loci with TM-1 and the donor Sea island cotton progenitor of the introgression line. a BNL2986, b NAU751, c BNL3971, d BNL1231; 1 TM-1, the standard genetic line; 2 Lumianyan22, the higher lint percentage elite cultivar; 3 Luyuan343, the introgression line; 4 Ashimouni, the donor Sea Island cotton progenitor

The introgression genomic components and fiber related QTLs detected by CIM. Only linkage groups with introgression sites or QTL are shown in this figure. The linkage group bars filled with black color represent the introgression genomic regions and markers underlined are single introgression locus in different linkage groups. The distorted segregation loci are shown in italics. The red color bars represented the QTLs detected in two data sets of F2 and F2:3 and the pink bars represented the QTLs detected in F2 population while the green and blue bars represented the QTLs detected in F2:3 in the data sets of Hainan and Linqing in China, respectively. The data sets of which the corresponding QTLs are detected can also refer to Table 3 by the name of the QTLs. QTL bars and lines indicated 1 LOD and 2 LOD likelihood intervals. (+), (−) indicated the IL parent contributed the positive effect or negative effect to fiber quality and lint percentage. (Color figure online)
Out of the 82 mapped marker loci, fourteen loci (17.1%) exhibited skewed Mendelian segregation, ten of which (71.4%) were biased in favor of the Gh parent and nine of them located in or near the putative introgression regions, especially on the relatively large putative introgression chromosomal segments (Fig. 2).
QTL analysis for fiber related traits and the contributions of putative introgression genomic components
A total of 29 QTLs with phenotypic variance ranging from 4.24% to 19.98% were detected in all data sets (F2 and two environments of F2:3, see Tables 3, 4). Their locations and confidence intervals are shown in Fig. 2. Ten out of 29 QTLs could be detected in two data sets, six in F2 and either of the environments of F2:3, four in two environments of F2:3. Most of the QTLs (22/29) are found clustered on the putative introgression segments of Chr.2 (A2), Chr.16 (D7), Chr.23 (D9) and Chr.25 (D6). As expected, a majority (12/17, 70.6%, not considering the QTLs for fiber fineness) of fiber quality QTLs had positive effects on fiber quality traits conferred by the IL parent and almost all of which (11/12, 91.7%) mapped in or nearby the putative introgression Gb segments. Fifty percent (3/6) of favorable alleles leading to positive effects on lint percentage came from the Gh parent (Tables 3, 4).
For fiber length, three (qFL16-1, qFL25-1 and marker MUCS547) of four identified QTLs were detected in two data sets, indicating that the QTLs of fiber length were stable across different generations or environments. And, we found that all the three alleles which increased fiber length were contributed by the putative introgression components. It is worthwhile to mention that qFL-25-1, a QTL with positive effect originating from the putative introgression segment at Chr.25, was also reported by other authors (Shen et al. 2005a; Qin et al. 2008), while qFL-16-1, a QTL with positive effects originating from the Gh parent at Chr.16 (Table 3; Fig. 2), was also identified in other studies (Shen et al. 2005a; Abdurakhmonov et al. 2009).
For fiber length uniformity, five QTLs were identified. Among them, only one QTL that linked to the marker NAU3427 was detected in both F2 and F2:3 populations at Linqing location (Table 4) while others were only detected in one population. Three QTLs (qFU-15-1, qFU-16-1 and qFU-16-2) with positive effect came from the IL parent (Table 3).
For fiber elongation, five QTLs were detected (Tables 3, 4). Among which, two QTLs (qFE-25-1 and marker MUCS547) were mapped near the putative introgression loci and their positive alleles came from the IL parent. Although qFE-2-1 was mapped in the putative introgression segment of chr.2, the positive effect was derived from the Gh parent.
For fiber strength, three QTLs (qFS-23-1, qFS-23-2 and qFS-24-1) were detected (Table 3) and among which, two QTLs (qFS-23-1 and qFS-23-2) with positive effects derived from the IL parent were mapped on the putative introgression segment of chr.23 spanning 3 cM. The qFS-23-2 was also reported as being mapped to a comparable position in the literature (Lacape et al. 2005; Shen et al. 2005a).
Lint percentage is the only lint yield trait analyzed in this work. Six QTLs of lint percentage were detected (Tables 3, 4). The positive effects of three (qLP-23-1, BNL3424 and MUSS467) out of the six QTLs were derived from Gh parent. Although IL parent has lower lint percentage than that of Gh parent, there still existed alleles (qLP-16-1, qLP-16-2 and qLP-18-1) in the IL parent, which contributes to higher lint percentage. The position and direction of qLP-16-2 and qLP-23-1 was, respectively, congruent with previous studies (Shen et al. 2005a; 2005b).
Validation of the detected QTLs in F8 breeding population
Thirteen of the 29 QTLs were detected in the F8 breeding population (Tables 3, 4). For two fiber length QTLs, qFL-16-1, which was detected in two data sets of F2 and F2:3HN, and qFL-25-1, which was detected in two data sets of F2:3HN and F2:3LQ, were also detected in the F8 breeding population with high significance (Table 3). And a lint percentage QTL, qLP-23-1 detected in both data sets of F2:3HN and F2:3LQ was validated in the F8 breeding population with moderate significance. In addition, BNL1495 associated with fiber micronaire and MUCS547 associated fiber length detected in both data sets of F2 and F2:3 LQ by single marker analysis were also validated in F8 breeding population (Table 4). We consider that these thirteen QTLs out of the total 29 QTLs have potential value for MAS in breeding, at least in this breeding population, because they can be detected in the F8 breeding population with high significance.
Among the five alleles mentioned above, particular attention should be paid to qFL-16-1, qFL-25-1 and qLP-23-1, whose similar map position and effect were previously reported (Ren et al. 2002; Shen et al. 2005a). These QTLs identified by both of the authors (Ren et al. 2002; Shen et al. 2005a) were introgression alleles as well, as the parents they used in their crosses were either substitute lines that harbored chromosome 16 and chromosome 25 from Gb (Ren et al. 2002) or germplasm derived from wide-cross (Shen et al. 2005a). These results indicate that these introgression alleles were conserved across different populations and therefore, had higher values for marker-assisted breeding program.
Discussion
Simultaneous improvement of cotton yield and fiber quality is a much-desired result in cotton breeding. However, successes of such breeding efforts are limited by the lack of favorable alleles conferring excellent fiber quality in the gene pool of cultivated upland cotton (Meredith 1992). Utilizing the superior fiber quality introgression lines is one of the key approaches to improve upland fiber quality. Since the introgression lines are more similar to the genetic background of upland cotton except for certain introgression components, nearly no problems with hybrid incompatibility and segregation are expected in breeding populations involving such ILs. It is reasonable that the superior fiber quality traits of IL are associated with the introgression genomic components or separated introgression alleles. Therefore, identifying the QTLs for fiber-related traits associated with introgression genomic components by molecular markers could provide us a better understanding of the genetic mechanism of the positive and/or negative phenotypic correlations among the fiber related traits.
In this work, we found that among the 42 putative introgression marker loci detected, twenty-three were found to be located on three chromosomal segments, Chr.2, 16, 23, respectively, while the others were dispersed throughout the genome. This indicated that several genetic recombination events had occurred in different locations of the genome in the IL, LY343 progenies. And it seemed plausible that most of the putative introgression genomic components from Gb are integrated into Gh genome through long-term selection.
A total of 29 QTLs were examined for fiber quality and lint percentage traits. As expected, for fiber quality traits except for fiber fineness, the IL parent contributed 70.6% (12/17) of the favorable alleles. And all except for one (11/12) were derived from the putative introgression genomic components from Gb. For yield related trait (lint percentage), 50% (3/6) of the favorable alleles came from the Gh parent. Although the IL parent has lower lint percentage than Gh parent, it also conferred the favorable alleles (qLP-16-1, qLP-16-2 and qLP-18-1). Likewise, the Gh parent with inferior fiber quality also had a positive influence on fiber quality traits (qFU-2-1, qFE-2-1, qFE-18-1, qFS-24-1, etc.) (Fig. 2; Tables 3, 4). These results are consistent with previously proposed ideas that there are almost always some loci for which the inferior parent contributes a superior allele, however, a great number of positive alleles are come from the superior parent (Tanksley and McCouch 1997; Chee et al. 2005b; Chee et al. 2005a).
The aim of QTLs analysis by molecular markers is for MAS, so the stability and facility of QTLs were really important. In this study, an F8 breeding population from the same cross made previously was applied in validating the QTLs detected by early generations of the mapping populations. Thirteen of the 29 QTLs could be detected in F8 breeding population. Five QTLs could be detected in three of the four data sets of F2, F2:3 and F8, and showed high stability across different generations. Three (qFL-16-1, qFL-25-1 and qLP-23-1) of the five QTLs were also identified by other groups (Ren et al. 2002; Shen et al. 2005a). These QTLs could therefore be valuable for the marker-assisted breeding program. The potential value for MAS of other ten QTLs that validated in the F8 breeding population should be further validated by using different populations.
Co-localization of QTLs for different traits in cotton has been described previously (Lacape et al. 2005; Qin et al. 2008). In this study, several QTL-rich genomic regions located on Chr.2, 16, 23, and 25, together with a single marker locus, MUCS547, which controls two or more fiber related traits, were observed. Notably, two of the four QTLs for fiber length correspond with QTLs for fiber fineness and fiber elongation, respectively, and the direction of genetic effects of QTLs for the two traits was consistent, confirming the significant phenotypic correlations between fiber length and fiber fineness, and fiber length and fiber elongation (Table 2). However, the phenotypic correlations observed between fiber strength and fiber length, and fiber strength and fiber elongation (Table 2) could not be reflected at the genotypic level since we did not detect any QTLs for fiber strength, fiber length or fiber elongation that were linked or co-located in this study (Fig. 2; Table 3). The co-location of QTLs and positive phenotypic correlations among different fiber quality parameters can be very useful in the simultaneous improvement of traits that define fiber quality.
The co-location of QTLs between fiber quality and lint percentage was also observed in our study. Four of six QTLs for lint percentage correspond with more than one fiber quality trait. Three of them imparted opposite effects, except for one locus in the interval of NAU2999–NAU2432, which showed the same additive effect direction (Fig. 2; Table 3). The negative effects of the putative introgression higher fiber quality loci on lint yield components (especially the lint percentage), known as linkage drags (Meredith and Bridge 1971; Naveira and Barbadilla 1992), were the main hindrance of synchronous improvement on fiber quality and lint yield. The linked or overlapped QTLs for fiber quality and lint percentage with opposite genetic effects explained the negative correlation between these traits and why linkage drag has been so difficult to overcome in the synchronous improvement of fiber quality and lint yield in cotton breeding. We have selected some lines with synchronous improvement of fiber quality and lint yield from the validation breeding population (data not shown), which indicated that we had partly broken the linkage drag through recombination, however, the effect was limited. To further break the introgression genomic region, more crosses (including intercrosses and backcrosses) should be conducted within relatively larger segregating breeding populations and, if possible, meticulous selection guided by markers should be applied. However, in any case, the synchronous improvement of fiber quality and lint yield would be limited in a certain extent because no introgression chromosomal fragment is easily to be thoroughly broken.
Interestingly, however, no significant negative effects of fiber quality QTLs on Chr.2 (qFU-2-1, qFM-2-1 and qFE-2-1) and Chr.25 (qFL-25-1, qFM-25-1, qFE-25-1) on lint percentage were detected (Fig. 2; Table 3). In the other hand, no negative effects of the two lint percentage related markers BNL3424 and MUSS467 on fiber quality was observed. It suggests that these loci are valuable for synchronous improvement on fiber quality and lint percentage by MAS.
In conclusion, the introgression genomic components from Gb of the IL conferred the majority of favorable alleles of high fiber quality and co-localization of QTLs and positive correlations for different fiber-related traits are benefited to the simultaneous improvement of fiber quality. Some QTLs detected in all data sets and met with the reports of other groups are considerably stable indicated their potential higher valuable for MAS. In addition, our data also suggest that superior alleles can be revealed from inferior parents. Our results suggest that synchronous improvement of fiber quality and lint yield might be achieved by pyramiding the favorable alleles (including the partly broken introgression genomic components) by MAS in cotton breeding.
Abbreviations
- Gh:
-
Gossypium hirsutum
- Gb:
-
Gossypium barbadense
- IL:
-
Introgression line
- QTL:
-
Quantitative trait locus
- LMY22:
-
Lumianyan #22
- LY343:
-
Luyuan 343
- FL:
-
Fiber length
- FS:
-
Fiber strength
- FU:
-
Fiber length uniformity
- FE:
-
Fiber elongation
- FM:
-
Fiber fineness indicated by the micronaire value
- LP:
-
Lint percentage
- HVI:
-
High-volume precision instrument for testing fiber quality
- SSR:
-
Simple sequence repeat
- MAS:
-
Marker-assisted selection
References
Abdurakhmonov I, Saha S, Jenkins J, Buriev Z, Shermatov S, Scheffler B, Pepper A, Yu J, Kohel R, Abdukarimov A (2009) Linkage disequilibrium based association mapping of fiber quality traits in G. hirsutum L. variety germplasm. Genetica 136(3):401–417
Barone A, Di Matteo A, Carputo D, Frusciante L (2009) High-throughput genomics enhances tomato breeding efficiency. Curr Genomics 10(1):1–9
Blenda A, Scheffler J, Scheffler B, Palmer M, Lacape J-M, Yu J, Jesudurai C, Jung S, Muthukumar S, Yellambalase P, Ficklin S, Staton M, Eshelman R, Ulloa M, Saha S, Burr B, Liu S, Zhang T, Fang D, Pepper A, Kumpatla S, Jacobs J, Tomkins J, Cantrell R, Main D (2006) CMD: a cotton microsatellite database resource for Gossypium genomics. BMC Genomics 7(1):132
Chee P, Draye X, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005a) Molecular dissection of interspecific variation between Gossypium hirsutum and Gossypium barbadense (cotton) by a backcross-self approach: I. Fiber elongation. Theor Appl Genet 111(4):757–763
Chee PW, Draye X, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005b) Molecular dissection of phenotypic variation between Gossypium hirsutum and Gossypium barbadense (cotton) by a backcross-self approach: III. Fiber length. Theor Appl Genet 111(4):772–781
Draye X, Chee P, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum and G. barbadense (cotton) by a backcross-self approach: II. Fiber fineness. Theor Appl Genet 111(4):764–771
Fernie AR, Tadmor Y, Zamir D (2006) Natural genetic variation for improving crop quality. Curr Opin Plant Biol 9(2):196–202
Guo W, Cai C, Wang C, Han Z, Song X, Wang K, Niu X, Wang C, Lu K, Shi B, Zhang T (2007) A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium. Genetics 176(1):527–541
He DH, Lin ZX, Zhang XL, Nie YC, Guo XP, Zhang YX, Li W (2007) QTL mapping for economic traits based on a dense genetic map of cotton with PCR-based markers using the interspecific cross of Gossypium hirsutum × Gossypium barbadense. Euphytica 153(1):181–197
Hospital F (2001) Size of donor chromosome segments around introgressed loci and reduction of linkage drag in marker-assisted backcross programs. Genetics 158(3):1363–1379
Lacape J-M, Nguyen T-B, Courtois B, Belot J-L, Giband M, Gourlot J-P, Gawryziak G, Roques S, Hau B (2005) QTL analysis of cotton fiber quality using multiple × backcross generations. Crop Sci 45(1):123–140. doi:10.2135/cropsci2005.0123
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newberg LA (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1(2):174–181
Mei M, Syed NH, Gao W, Thaxton PM, Smith CW, Stelly DM, Chen ZJ (2004) Genetic mapping and QTL analysis of fiber-related traits in cotton (Gossypium). Theor Appl Genet 108(2):280–291
Meredith WR (1992) Improving fiber strength through genetics and breeding. In: Savannah, GA (ed) Proc. cotton fiber cellulose: structure, function and utilization conference. Natl. Cotton Council Am., Cotton Incorporated, USDA-ARS, p. 289–302, October 1992
Meredith WR Jr, Bridge RR (1971) Breakup of linkage blocks in cotton, Gossypium hirsutum L. Crop Sci 11(5):695–698
Naveira H, Barbadilla A (1992) The theoretical distribution of lengths of intact chromosome segments around a locus held heterozygous with backcrossing in a diploid species. Genetics 130(1):205–209
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep 11(2):122–127
Paterson AH, Saranga Y, Menz M, Jiang CX, Wright RJ (2003) QTL analysis of genotype x environment interactions affecting cotton fiber quality. Theor Appl Genet 106(3):384–396
Percy RG, Cantrell RG, Zhang J (2006) Genetic variation for agronomic and fiber properties in an introgressed recombinant inbred population of cotton. Crop Sci 46:1311–1317
Pestsova EG, Borner A, Roder MS (2006) Development and QTL assessment of Triticum aestivum-Aegilops tauschii introgression lines. Theor Appl Genet 112(4):634–647
Qin H, Guo W, Zhang YM, Zhang T (2008) QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor Appl Genet 117(6):883–894
Ren L, Guo W, Zhang T (2002) Identification of quantitative trait loci (QTLs) affecting yield and fiber properties in chromosome 16 in cotton using substitution line. Acta Bot Sinica 44:815–820
Shen X, Guo W, Zhu X, Yuan Y, Yu JZ, Kohel RJ, Zhang T (2005a) Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol breed 15(2):169–181
Shen X, Zhang T, Guo W, Zhu X, Zhang X (2005b) Mapping fiber and yield QTLs with main, epistatic, and QTL × environment interaction effects in recombinant inbred lines of Upland cotton. Crop Sci 46(1):61–66
Steele KA, Price AH, Shashidhar HE, Witcombe JR (2006) Marker-assisted selection to introgress rice QTLs controlling root traits into an Indian upland rice variety. Theor Appl Genet 112(2):208–221
Su X, Gao G, Shi X, Zhu D (2000) Breeding of a terrestrial long-staple cotton—Luyuan 343. Acta Agric Nucleatae Sinica 14(3):180–183
Tang QY, Feng MG (2007) DPS data processing system: experimental design, statistical analysis, and data mining. Science Press, Beijing
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277(5329):1063–1066
Ulloa M, Meredith WR Jr (2000) Genetic linkage map and QTL analysis of agronomic and fiber quality traits in an intraspecific population. J Cotton Sci 4(3):161–170
Ulloa M, Meredith WR Jr, Shappley ZW, Kahler AL (2002) RFLP genetic linkage maps from four F(2.3) populations and a join map of Gossypium hirsutum L. Theor Appl Genet 104(2–3):200–208
Ulloa M, Saha S, Jenkins JN, Meredith WR Jr, McCarty JC Jr, Stelly DM (2005) Chromosomal assignment of RFLP linkage groups harboring important QTLs on an intraspecific cotton (Gossypium hirsutum L.) Joinmap. J Hered 96(2):132–144. doi:10.1093/jhered/esi020
Wang S, Basten C, Zeng Z (2006) Windows QTL Car-tographer 2.5 Department of Statistics, North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Wang FR, Zhang CY, Liu GD, Wang LM, Gao JP, Gong YC, Zhang J (2007) Identification of exotic genetic components of upland cotton (Gossypium hirsutum L.) introgression line with higher fiber qualities from Sea Island cotton (Gossypium barbadense L.). J Plant Genet Resour 8:265–270
Wu J, Gutierrez O, Jenkins J, McCarty J, Zhu J (2009) Quantitative analysis and QTL mapping for agronomic and fiber traits in an RI population of upland cotton. Euphytica 165(2):231–245. doi:10.1007/s10681-008-9748-8
Yu J, Herrmann M (2006) Inheritance and mapping of a powdery mildew resistance gene introgressed from Avena macrostachya in cultivated oat. Theor Appl Genet 113(3):429–437
Zamir D (2008) Plant breeders go back to nature. Nat Genet 40(3):269–270
Zeng L, Meredith WR Jr (2009) Associations among lint yield, yield components, and fiber properties in an introgressed population of cotton. Crop Sci 49(5):1647–1654. doi:10.2135/cropsci2008.09.0547
Zeng L, Meredith WR Jr, Gutierrez OA, Boykin DL (2009) Identification of associations between SSR markers and fiber traits in an exotic germplasm derived from multiple crosses among Gossypium tetraploid species. Theor Appl Genet 119(1):93–103
Zhang J, Wu YT, Guo WZ, Zhang TZ (2000) Fast screening of microsatellite markers in cotton with PAGE/silver staining. Acta Gossypii Sinica 12(5):267–269
Zhang T, Yuan Y, Yu J, Guo W, Kohel RJ (2003) Molecular tagging of a major QTL for fiber strength in Upland cotton and its marker-assisted selection. Theor Appl Genet 106(2):262–268
Zhang Z-S, Hu M-C, Zhang J, Liu D-J, Zheng J, Zhang K, Wang W, Wan Q (2009) Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol breed 24(1):49–61
Zheng P (2008) A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat Genet 40:367–372
Acknowledgments
This work was financially supported by the National Key Project of Scientific and Technical Supporting Programs of China during the 11th Five-year Plan (2006BAD13B04-1-07), National Major Project on Transgenic Breeding (2009ZX08005-18B and 2008ZX08005-003). We would like to thank Dr. O Lloyd May and Dr. Xinlian Shen for their suggestions to make this a better manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
F. Wang, Y. Gong contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wang, F., Gong, Y., Zhang, C. et al. Genetic effects of introgression genomic components from Sea Island cotton (Gossypium barbadense L.) on fiber related traits in upland cotton (G. hirsutum L.). Euphytica 181, 41–53 (2011). https://doi.org/10.1007/s10681-011-0378-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-011-0378-1