
Abstract
Quinolinic acid (QUIN) is an important agonist of NMDA receptors that are found at high levels in cases of brain injury and neuroinflammation. Therefore, it is necessary to investigate neuroprotection strategies capable of neutralizing the effects of the QUIN on the brain. Coenzyme Q10 (CoQ10) is a provitamin that has an important antioxidant and anti-inflammatory action. This work aims to evaluate the possible neuroprotective effect of CoQ10 against the toxicity caused by QUIN. Striatal slices from 30-day-old Wistar rats were preincubated with CoQ10 25–100 μM for 15 min; then, QUIN 100 μM was added to the incubation medium for 30 min. A dose–response curve was used to select the CoQ10 concentration to be used in the study. Results showed that QUIN caused changes in the production of ROS, nitrite levels, activities of antioxidant enzymes, glutathione content, and damage to proteins and lipids. CoQ10 was able to prevent the effects caused by QUIN, totally or partially, except for damage to proteins. QUIN also altered the activities of electron transport chain complexes and ATP levels, and CoQ10 prevented totally and partially these effects, respectively. CoQ10 prevented the increase in acetylcholinesterase activity, but not the decrease in the activity of Na+,K+-ATPase caused by QUIN. We also observed that QUIN caused changes in the total ERK and phospho-Akt content, and these effects were partially prevented by CoQ10. These findings suggest that CoQ10 may be a promising therapeutic alternative for neuroprotection against QUIN neurotoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Quinolinic acid (QUIN) is an important selective N-methyl-d-aspartate (NMDA) receptor agonist produced in the kynurenine pathway by microglial cells and activated macrophages. Physiologically, this compound is present in low concentrations in the central nervous system (CNS). However, in pathological situations, there is a considerable increase in its concentration in the CNS, with consequent cell damage (Stone 1993; Schwarcz and Stone 2017). In vivo studies have shown that the increase in QUIN concentrations is related to several diseases, such as Alzheimer’s disease, amyotrophic lateral sclerosis, and the adult and juvenile forms of Huntington’s disease (Lugo-Huitrón et al. 2013; Bakels et al. 2021).
Studies show that QUIN causes many neurotoxic effects in the CNS, being the production of reactive species, the alteration of the mitochondrial function, and the inflammatory process considered important mechanisms of action of this metabolite (Lugo-Huitrón et al. 2013). Activation of NMDA receptors by QUIN causes different effects on the brain, such as excitotoxicity and increased Ca2+ influx, leading to an increase in the production of reactive oxygen/nitrogen species (ROS/RNS, respectively), mitochondrial dysfunction, decreased levels of adenosine triphosphate (ATP), and activation of cell death pathways (La Cruz et al. 2013; Jeon and Kim 2017). The production of ATP in the inner mitochondrial membrane depends on the electron transport chain (ETC). Changes in the normal functioning of the ETC, impairment of function, and loss of mitochondrial integrity are related to neurodegenerative diseases, cancer, diabetes, obesity, and aging (Sun et al. 2016; Wilkins and Morris 2017). Studies have shown that QUIN causes a decrease in the activity of the ETC and ATP levels (Sas et al. 2007; Luis-García et al. 2017). The alterations caused by QUIN can also modulate the activity of enzymes and signaling proteins important for the maintenance of cellular functions, such as alterations in the transport of molecules and neurotransmitters and maintenance of the membrane potential performed by Na+,K+-ATPase and markers of proliferation, plasticity, and cell survival as kinases (Suhail 2010; Constantino et al. 2018; Santana-Martínez et al. 2018).
Based on these findings, it is necessary to search for strategies to protect against the damage caused by QUIN in the CNS. Coenzyme Q10 (2,3-dimethoxy-5-methyl-6-decaprenyl-1,4-benzoquinone-CoQ10) is a fat-soluble provitamin that can be synthesized endogenously or obtained through the diet. It is present in all cells of the human body, with the highest concentrations found in the brain, heart, liver, and skeletal muscle (Spindler et al. 2009). It is mostly located in the inner mitochondrial membrane, where it participates in the transfer of electrons from complexes I and II and acts as a coenzyme of complex III in the respiratory chain (Bhardwaj and Kumar 2016).
CoQ10 can be present in tissues in two forms, the oxidized (ubiquinone) and the reduced (ubiquinol) (Spindler et al. 2009). In its reduced form, it has an important antioxidant action, with the ability to prevent damage to lipids and proteins in the mitochondrial membrane and protect the DNA from oxidative damage (Spindler et al. 2009; Cornelius et al. 2017). Recently, in vivo and in vitro studies show the neuroprotective action of the CoQ10 in models of cerebral ischemia and Parkinson’s and Alzheimer’s diseases (Spindler et al. 2009; Dumont et al. 2011; El-Aal et al. 2017). Furthermore, its supplementation has been used in patients as a strategy for neurodegenerative and cardiovascular diseases (Sharifi et al. 2015; Zhu et al. 2016).
In the present study, we evaluate the neuroprotective capacity of the CoQ10 against the toxic effects of the QUIN on redox homeostasis, energy metabolism, and cell signaling in young Wistar rat striatum slices. We hypothesize that CoQ10, due to its neuroprotective characteristics, protects QUIN damage in the striatum, a structure directly impaired by QUIN damage in neurodegenerative diseases.
Materials and Methods
Animals and Reagents
Fifty male Wistar rats (30-day-old) were obtained from Central Animal House of Biochemistry Department, Institute of Basic Health Sciences at the Universidade Federal do Rio Grande do Sul, in Porto Alegre, Brazil. The animals were kept under controlled light (12/12-h light/dark cycle) and temperature conditions (22 °C ± 1), with free access to water and 20% (w/w) commercial protein chow. Animal care followed the “Principles of Laboratory Animal Care” (NIH publication 85–23, revised 1996), and the experimental protocol was approved by the University’s Ethics Committee (CEUA) under project #35442.
Quinolinic acid (purity: ≥ 98.5%), coenzyme Q10 (purity: ≥ 98%), acrylamide (purity: ≥ 99%), bisacrylamide (purity: 97%), and sodium dodecyl sulfate (SDS) (purity: ≥ 98.5%), and all other chemical analytical reagents used for analysis were obtained from Sigma-Aldrich, St. Louis, MO. USA. ATPlite luminescence assay kit was obtained from PerkinElmer and antibodies were purchased from Sigma-Aldrich, Santa Cruz Biotechnology, and Cell Signaling Technology.
Preparation and Incubation of Striatal Slices
Animals were killed by decapitation, the brain was removed, and striatum was rapidly dissected in ice-cold Krebs-HEPES buffer of the following composition: 124 mM NaCl; 4 mM KCl; 1.2 mM MgSO4; 1 mM CaCl2; 25 mM Na-HEPES; and 12 mM d-glucose, pH 7.4. Slices (0.4 mm) were prepared using a McIlwain Tissue Chopper (The Mickle Laboratory Engineering Co., Ltd., England) and separated in buffer at 4 °C. Immediately after sectioning, slices were transferred to buffer for 15 min to recover from slicing trauma, before starting the experiments. All experiments were performed at 37 °C.
Slices (a pool of 4–6 slices was used for each sample) were preincubated with CoQ10 (25–100 μM) for 15 min. Afterwards, slices were exposed for 30 min to QUIN (100 μM), co-incubated with CoQ10. Slices corresponding to the control group were incubated in a normal Krebs-HEPES buffer.
Tissue Preparation
For the determination of oxidative stress parameters and nitrite levels, the tissue was homogenized (1:10, w/v) in 20 mM sodium phosphate buffer with 140 mM KCl, pH 7.4. The sample was centrifuged at 800 × g for 10 min at 4 °C. The pellet was discarded, and the supernatant was immediately separated and used for the oxidative stress and nitrite levels analysis.
For activities of complexes I, II, and IV determination, tissue was homogenized (1:20 w/v) in SETH buffer (250 mM sucrose, 2 mM EDTA, 10 mM Trizma base, 50 UI/mL heparin), pH 7.4. The samples were centrifuged at 800 × g for 10 min, and the supernatants were used for enzyme activity determination.
Cell Viability
The cell survival was determined using mitochondrial dehydrogenase activity to reduce MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Mosmann 1983; Liu and Peterson 1997). The tetrazolium ring of MTT is cleaved by various dehydrogenase enzymes in active mitochondria and then precipitated as a blue formazan product. Striatal slices were incubated with MTT (0.5 mg/mL) in KREBS-HEPES buffer for 20 min at 37 °C. The medium was then aspirated, the precipitated formazan was solubilized with dimethyl sulfoxide, and viable cells were quantified spectrophotometrically at a wavelength of 550 nm.
2′7′-Dichlorofluorescein Fluorescence Assay
The method described by LeBel et al. (1992) was used to analyze the production of reactive species. The samples were incubated in a medium containing 100 μM of 2′7′-dichlorofluorescein diacetate (H2DCF-DA) solution, and in the 2′7′-dichlorofluorescein (H2DCF) oxidation reaction, the compound dichlorofluorescein (DCF) is produced, which is measured at λem = 488 nm and λex = 525 nm; results were represented as nanomoles of DCF per milligram of protein.
Nitrite Assay
Nitrite levels were measured using the Griess reaction: the samples were incubated in a medium containing Griess reagent (1:1 mixture of 1% sulfanilamide in 5% phosphoric acid, and 0.1% naphthylethylenediamine dihydrochloride in water) for 10 min. The absorbance was measured at a wavelength of 543 nm. Nitrite concentration was calculated using sodium nitrite standard (Green et al. 1982).
Acetylcholinesterase Activity Assay
Striatal slices were homogenized (1:10, w/v) in 0.1 mM potassium phosphate buffer (pH 7.5) and centrifuged at 1000 × g for 10 min at 4 °C. Acetylcholinesterase (AChE) activity was determined in the supernatant according to the method of Ellman et al. (1961) with some modifications (Scherer et al. 2010). Fifteen microliters of sample was added to reaction mixture (30 mM phosphate buffer; pH 7.5; and 1.0 mM 5,5′-dithiobis-2-nitrobenzoic acid (DTNB)) and incubated for 3 min for 25 °C. The hydrolysis was monitored by the formation of the thiolate di-anion of DTNB at 412 nm for 2–3 min, measured at intervals of 30 s.
Catalase Assay
The catalase (CAT) evaluation was realized according to Aebi (1984). The method is based on the disappearance of H2O2 in a reaction medium containing 20 mM H2O2, 0.1% Triton X-100, and 10 mM potassium phosphate buffer of pH 7.0 measured in a spectrophotometer at 240 nm. One CAT unit is defined as 1 mmol of H2O2 consumed per minute and the specific activity is represented as CAT units per milligram of protein.
Superoxide Dismutase Assay
The superoxide dismutase (SOD) activity assay is based on pyrogallol (1,2,3-trihydroxybenzene) autoxidation, a process highly dependent on superoxide (substrate for SOD). The inhibition of autoxidation of this compound occurs in the presence of SOD, whose activity can be then indirectly measured in a spectrophotometer at 420 nm (Marklund and Marklund 1974). A calibration curve was performed with purified SOD as standard, in order to calculate the activity of SOD present in the samples. SOD activity was expressed as the amount of enzyme necessary to inhibit 50% of pyrogallol autoxidation. The results were reported as SOD units per milligram of protein.
Glutathione Peroxidase Assay
Glutathione peroxidase (GPx) activity was measured according Wendel (1981), using tert-butyl hydroperoxide as a substrate. NADPH disappearance was monitored at 340 nm. One GPx unit is defined as 1 μmol of NADPH consumed per minute; the specific activity is represented as GPx units per milligram of protein.
Glutathione Levels
Glutathione (GSH) levels were measured according to Browne and Armstrong (Browne and Armstrong 1998). One hundred microliters of sample was incubated with an equal volume of o-phthaldialdehyde (1 mg/mL in methanol) at room temperature for 15 min. Fluorescence was measured using excitation and emission wavelengths of 350 and 420 nm, respectively. Calibration curve was performed with standard GSH (0.001–0.1 mM), and GSH concentrations were calculated as nanomoles of GSH per milligram of protein.
Sulfhydryl Content
This assay was performed as described by Aksenov and Markesbery (2001), which is based on the reduction of DTNB by the sulfhydryl groups, generating a yellow derivative (TNB) whose absorption is measured spectrophotometrically at 412 nm. Briefly, 15 μL of homogenate was added to 275 μL of phosphate-buffered saline of pH 7.4 containing 1 mM EDTA. The reaction was started by the addition of 10 μL of 10 mM DTNB and incubated for 30 min at room temperature in a dark room. The sulfhydryl content is inversely correlated to oxidative damage to the protein. Results were reported as nanomoles of TNB per milligram of protein.
Thiobarbituric Acid Reactive Substances
Considered an index of lipid peroxidation, thiobarbituric acid reactive substances (TBARS) were measured according to Ohkawa et al. (1979). The sample was incubated in a medium containing 8.1% SDS, 20% acetic acid, and 0.8% thiobarbituric acid. The reaction was carried out in a boiling water bath for 1 h. The resulting pink-stained TBARS were determined spectrophotometrically at 535 nm. A calibration curve was generated using 1,1,3,3-tetramethoxypropane as a standard, and the results were represented as nanomoles of TBARS per milligram of protein.
Na+,K+-ATPase Activity Assay
The striatum slices were homogenized in 10 vol (1:10, w/v) of 0.32 mM sucrose solution containing 5.0 mM HEPES and 1.0 mM EDTA, pH 7.5, and were centrifuged at 1000 × g for 10 min at 4 °C. Supernatants were taken for Na+,K+-ATPase activity assay. Reaction mixture for Na+,K+-ATPase activity assay contained 5.0 mM MgCl2, 80.0 mM NaCl, 20.0 mM KCl, and 40.0 mM Tris–HCl, pH 7.4, in a final volume of 200 μL. After 10 min of preincubation at 37 °C, the reaction was started by the addition of ATP to a final concentration of 3.0 mM and was incubated for 20 min. Controls were carried out under the same conditions with the addition of 1.0 mM ouabain (enzyme inhibitor). Na+,K+-ATPase activity was calculated by the difference between the two assays (de Souza Wyse et al. 2000). Inorganic phosphate (Pi) released was measured by the method of Chan et al. (1986) and enzyme-specific activity was expressed as nanomoles of Pi per minute per milligram of protein.
Complex I Activity
Mitochondrial complex I (NADH dehydrogenase) activity was measured by the NADH-dependent ferric reduction rate at 420 nm as described by Cassina and Radi (1996). The activity was calculated in nanomoles per minute per milligram of protein.
Complex II Activity
The complex II activity was measured according to Fischer et al. (1985), by the decrease in absorbance due to the reduction of 2,6-dichloroindophenol (DCIP). Instants before this test, the samples were thawed and refrozen three times to break the mitochondrial membrane. The reaction solution was preincubated with 40–80 μg of protein homogenized at 30 °C for 20 min. Afterwards, 4 mM sodium azide and 7 μM rotenone were added. The reaction was initiated by the addition of 40 μM DCIP and was verified by 5 min. The results were expressed as nanomoles per minute per milligram of protein.
Complex IV Activity
The complex IV (cytochrome c oxidase) activity was determined according to Rustin et al. (1994). The enzymatic activity was measured by the decrease in absorbance due to the oxidation of previously reduced cytochrome c at 550 nm with 580 nm as reference wavelength (ε = 19.1 mM−1 cm−1), at 25 °C for 10 min. The results were expressed in nanomoles per minute per milligram of protein.
ATP Levels
To determine the ATP levels, striatal slices were immediately frozen in liquid nitrogen. The homogenization was performed, adding 1 mL of 0.1 M NaOH for each sample; it is not necessary to centrifuge. The samples were analyzed using ATPlite kit (PerkinElmer, Waltham, MA, USA) (Siebert et al. 2014). The measurement of chemiluminescence was performed using a PerkinElmer Microbeta Microplate Scintillation Analyzer. The ATP concentration was calculated from a standard curve expressed in micromoles per gram.
Western Blot Analysis of AChE, ERK 1/2, GSK-3β, and Phospho-Akt
Western blotting was performed as described by Biasibetti-Brendler et al. (2017). Initially, striatum slices were homogenized in 200 µL of a lysis solution (2 mM EDTA, 50 mM Tris–HCl, pH 6.8, and 4% sodium dodecyl sulfate (SDS)) for electrophoresis analysis. Then, samples were dissolved 1:1 in Laemmli buffer 2 × and boiled for 5 min. Total protein homogenate was separated by 10% SDS-PAGE gel (30 μg/lane of total protein) and transferred (Trans-Blot SD Semi-Dry Transfer Cell, Bio-Rad) to nitrocellulose membranes for 1 h at 15 V in transfer buffer. Blot was then incubated overnight at 4 °C in a blocking solution containing 5% bovine serum albumin (BSA) and the following diluted antibodies: anti-AChE (1:1000, sc-373901, Santa Cruz Biotechnology), anti-ERK 1/2 (1:1000, #4695, Cell Signaling Technology), anti-GSK-3β (1:1000, SAB5700767, Sigma-Aldrich), anti-phospho-Akt (1:1000, #4060, Cell Signaling Technology), and anti-β-actin (1:1000, #12,620, Cell Signaling Technology). Blot was washed twice for 5 min with T-TBS and twice for 5 min with TBS and incubated with peroxidase-conjugated anti-rabbit IgG (#7074, Cell Signaling Technology) diluted 1:1000. The blot was revealed using a chemiluminescence kit (Immobilon Western Chemiluminescent HRP Substrate, Millipore) and detected by ImageQuant LAS 4000 (GE Healthcare Life Sciences).
Protein Determination
Protein concentrations were measured by the method of Lowry et al. (1951) or Bradford (1976), using bovine serum albumin as standard.
Statistical Analysis
The data were analyzed by two-way analysis of variance (ANOVA) followed by post hoc Tukey’s test. Values of p > 0.05 were considered statistically significant. All analyzes and plots were performed using GraphPad Prism 8.0 software program in a compatible computer.
Results
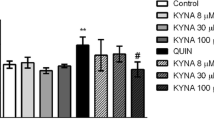
Initially, we developed a dose–response curve to verify the action of different concentrations of CoQ10, evaluating four parameters: (a) cell viability, (b) H2DCF oxidation, (c) nitrite levels, and (d) acetylcholinesterase activity, as can be seen in Fig. 1. The treatments used did not cause changes in cell viability (F(4.50) = 0.7624; p > 0.05). QUIN caused an increase in H2DCF oxidation, nitrite levels, and AChE activity (F(4.50) = 7.355, p < 0.001; F(4.70) = 5.407, p < 0.01; F(4.60) = 4.862, p < 0.001, respectively). CoQ10 100 μM prevented the effects of QUIN in the three analyses performed, being chosen to develop the subsequent experiments.

Effect of different doses of CoQ10 on QUIN-treated striatum slices on cell viability (a), H2DCF oxidation (b), nitrite levels (c), and acetylcholinesterase activity (d). Results expressed as mean ± SD of n = 6–7 per group. **p < 0.01, ***p < 0.001 compared to control group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared to QUIN group (two-way ANOVA followed by Tukey’s post hoc test)
Then, we analyzed the activity of the antioxidant enzymes SOD, CAT, GPx, and the GSH content. Figure 2 shows that QUIN treatment increased the (a) SOD/CAT ratio and decreased the (b) GPx activity and the (c) GSH levels (F(1.16) = 4.378, p < 0.05; F(1.20) = 9.589, p < 0.01; F(1.20) = 33.78, p < 0.001, respectively). CoQ10 was able to partially prevent the change in SOD/CAT ratio and GSH levels and totally prevented the decrease in GPx activity.

Effect of CoQ10 on QUIN-treated striatum slices on SOD/CAT ratio (a), GPx activity (b), and GSH content (c). Results expressed as mean ± SD of n = 6–7 per group. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control group; #p < 0.05 compared to QUIN group (two-way ANOVA followed by Tukey’s post hoc test)
To analyze possible damage to biomolecules, we evaluated the sulfhydryl content and the TBARS levels, as shown in Fig. 3. QUIN decreased the (a) sulfhydryl content and increased the (b) TBARS levels (F(1.20) = 38.50, p < 0.001; F(1.20) = 19.60, p < 0.001, respectively). CoQ10 was not able to prevent the change in sulfhydryl content, but it totally prevented the increase in TBARS levels.

Effect of CoQ10 on QUIN-treated slices on sulfhydryl content (a) and TBARS levels (b). Results expressed as mean ± SD of n = 6–7 per group. *p < 0.05, ***p < 0.001 compared to control group; ###p < 0.001 compared to QUIN group (two-way ANOVA followed by Tukey’s post hoc test)
The Na+,K+-ATPase activity was also evaluated, and as seen in Fig. 4, its activity was decreased by QUIN, and this effect did not prevent by CoQ10 (F(1.20) = 17.52, p < 0.001).

Effect of CoQ10 on QUIN-treated striatum slices on Na+,K+-ATPase activity. Results expressed as mean ± SD of n = 6–7 per group. *p < 0.05, ***p < 0.001 compared to control group (two-way ANOVA followed by Tukey’s post hoc test)
Respiratory chain enzyme activities and ATP levels were also evaluated. In Fig. 5, we observe that QUIN decreased the activity of complexes I, II, and IV (F(1.20) = 44.01, p < 0.001; F(1.20) = 15.29, p < 0.001; F(1.20) = 14.28, p < 0.01, respectively) and also the ATP levels (F(1.20) = 13.20, p < 0.01). The neuroprotective action of CoQ10 was able to completely prevent changes in enzymatic activities and partially prevented changes in ATP levels.

Effect of CoQ10 on QUIN-treated striatal slices on complex I (a), complex II (b), complex IV (c) activities, and ATP levels (d). Results expressed as mean ± SD of n = 6–7 per group. **p < 0.01, ***p < 0.001 compared to control group; #p < 0.05, ###p < 0.001 compared to the QUIN group (two-way ANOVA followed by Tukey’s post hoc test)
We also analyzed important proteins in cell signaling. Figure 6 shows that QUIN increased the ERK 1 content (Fig. 6b) (F(1.20) = 4898, p < 0.05). This effect was partially prevented by CoQ10. In addition, there is a tendency for QUIN to decrease phospho-Akt immunocontent (Fig. 6d) (p = 0.48), and when co-incubated, QUIN and CoQ10 maintain a similar profile, with a significant decrease in phospho-Akt content (F(1.20) = 13.87, p < 0.05). No changes were observed in the GSK-3β and AChE immunocontent.

Effect of CoQ10 on QUIN-treated striatal slices on AChE (a), total ERK1/2 (b), GSK-3β (c), and phospho-Akt (d) immunocontent. Results expressed as mean ± SD of n = 6–7 per group. *p < 0.05 compared to control group; #p < 0.05 compared to the QUIN group (two-way ANOVA followed by Tukey’s post hoc test)
Discussion
Quinolinic acid is a metabolite resulting from the conversion of tryptophan by the kynurenine pathway and which, under physiological conditions of the cell, participates in the formation of NAD+ (Jeon and Kim 2017); under pathological conditions, there is an increase in its concentration in the CNS, triggering deleterious effects. Studies show this substance can be related to the physiopathology of neurodegenerative diseases, causing toxic effects to cells by several mechanisms, such as excitotoxicity, increased production of free radicals, and pro-inflammatory cytokines (Guillemin 2012). Due to this, it is important to investigate neuroprotection strategies capable of neutralizing the toxic effects of this metabolite in the brain. In this context, coenzyme Q10 is a substance present in mitochondria responsible for exerting beneficial effects on cell metabolism. It has an important function as an electron acceptor in the respiratory chain and, when reduced, acts as an important antioxidant, preventing the action of lipid peroxidation in the mitochondrial membrane. Furthermore, it is responsible for the activation of mitochondrial uncoupling proteins (UCPs) (Spindler et al. 2009), triggering an anti-apoptotic effect in the cell. In addition to protecting the mitochondria, it acts as a protector against lipid peroxidation of the cellular membrane, inhibiting the action of free radicals due to its activity per se and regenerating other important cellular antioxidants, such as α-tocopherol and ascorbic acid (El-Aal et al. 2017). Its anti-inflammatory effects are also described in the literature (Sharma et al. 2006), as well as its role in modulating gene transcription and preventing the activation of apoptosis signaling cascades (Komaki et al. 2019). Therefore, the aim of this work is to evaluate the possible neuroprotective role of CoQ10 against the damage caused by QUIN on oxidative, energetic, and enzymatic parameters.
Initially, we performed a dose–response curve analyzing four parameters: cell viability, H2DCF oxidation, nitrite levels, and AChE enzyme activity. The treatments performed did not affect cell viability (as measured by the MTT reduction), and we observed an increase in H2DCF oxidation, nitrite levels, and AChE activity caused by QUIN. The results found corroborate previous studies that demonstrate the actions of QUIN in maintaining an oxidative status, with an increase in the production of ROS/RNS. Furthermore, the increase in AChE activity caused by QUIN may be related to neuroinflammation and cytotoxicity (Guillemin 2012; La Cruz et al. 2013; Mishra et al. 2014). We observed a dose-dependent effect of CoQ10 when we evaluated the DCF levels and AChE activity, which is possibly due to the already known protective effects of this compound against reactive oxygen species (El-Aal et al. 2017). In addition to acting directly on ROS, CoQ10 acts by preventing damage to signaling cascades related to AChE activity (Leclerc et al. 1997; Aitken et al. 1998). Regarding nitrite levels, we did not observe a similar effect, but the concentration of CoQ10 capable of preventing changes in the three analyses performed was 100 μM, which was then the choice for the subsequent analysis.
Analyzing the effect of QUIN on enzymatic antioxidant defenses in the striatum, we observed an increase in the SOD/CAT ratio and a decrease in GPx activity. The SOD/CAT ratio increase suggests an increase in H2O2 production, for SOD action, and this compound is not being neutralized by CAT, which has its activity reduced. In addition, the other evaluated peroxidase (GPx) activity also has decreased (Halliwell 2006). These results, together with the increase in the oxidation of H2DCF, demonstrate that QUIN leads to the maintenance of the cells’ pro-oxidant status, by increasing the production of ROS and decreasing the cellular antioxidant potential. We also observed that QUIN acts on non-cellular antioxidant defenses, decreasing the cellular GSH content. In addition, this compound causes a decrease in the sulfhydryl content, which may indicate protein damage and an increase in TBARS levels, causing lipid peroxidation. These results corroborate previous studies that support the toxic action of QUIN (Santamaría et al. 2001; Pérez-De La Cruz et al. 2005; Ferreira et al. 2018). CoQ10 was able to prevent part of these effects possibly due to its antioxidant role, partially preventing the change found in the SOD/CAT ratio, and the GSH content, exerting total prevention on the decrease in GPx activity and reinforcing its actions on membrane lipids, was also able to prevent the changes in TBARS. There was no prevention in decreasing the sulfhydryl content.
Treatment with CoQ10 was able to partially prevent changes in GSH content, possibly by preventing the change in GPx enzyme activity, because, with its regulated redox activity, this enzyme consumes the hydrogen peroxide produced in the cell and uses it for oxidation, from GSH to GSSG + H20 (Halliwell 2006). Although GSH is an important mechanism for maintaining the redox balance of proteins that contain the sulfhydryl groups, we observed that its partial prevention was not able to prevent damage to proteins that have amino acids with the SH groups (Aksenov and Markesbery 2001).
Also, in this context, we observed a decrease in the activity of the enzyme Na+,K+-ATPase caused by QUIN that was not prevented by CoQ10. This membrane enzyme is responsible for neuronal excitability, maintaining the cellular electrochemical gradient, and transporting signaling molecules and neurotransmitters. It is highly susceptible to increased free radicals, lipid peroxidation, and oxidation of the sulfhydryl groups present in its structure (Lees 1991; Wyse et al. 2002). The inhibition of its activity is related to several CNS pathologies, such as neurodegenerative diseases, ischemia, and inborn errors of metabolism (Wyse et al. 1999; Schweinberger et al. 2014; Schmitz et al. 2016; Dergousova et al. 2017). Since Na+,K+-ATPase has the cysteine groups in its active site and that the consequent irreversible oxidation of residues of cysteine may cause enzymatic damage (Wyse et al. 1999), we suggest that a decrease in Na+,K+-ATPase may be correlated to a decrease in the sulfhydryl content observed in this study, which was not prevented by CoQ10.
The action of QUIN on respiratory chain enzymes and ATP levels was also analyzed, demonstrating that the activity of complexes I, II, and IV is reduced, as well as ATP levels. Our result corroborates previous studies that demonstrate that QUIN causes toxic effects on brain energy metabolism, increasing free radical formation and leading to ATP depletion (La Cruz et al. 2013; Luis-García et al. 2017). CoQ10 was able to totally prevent the actions of QUIN on the respiratory chain complexes and partially prevented the decrease in ATP levels, reinforcing its protective effect on the mitochondria.
The main mechanism of action of CoQ10 is related to participation in the electron transport chain in the mitochondrial membrane. Due to its low permeability in membranes, CoQ10 supplementation possibly does not directly reach mitochondria, but may exert important cellular effects that directly influence the functioning of ETC and mitochondria in general. Also, this molecule plays an important protective role outside the mitochondria. In non-mitochondrial membranes, CoQ10 switches between reduced and oxidized forms through the activity of CoQ reductase enzymes, and this complex is part of the plasma membrane redox system. In this context, CoQ10 is responsible for preventing lipid peroxidation of biological membranes, preventing the formation of highly reactive free radicals, and is responsible for the regeneration of α-tocopherol. It also performs intracellular signaling that influences the release of enzymatic antioxidant defenses and inflammatory markers. Furthermore, there is an important relationship between the concentration of mitochondrial CoQ10 and the plasma membrane redox system, as mitochondrial CoQ10 depletion can influence the efficiency of the external redox system (Aaseth et al. 2021; Gutierrez-Mariscal et al. 2021; Pallotti et al. 2022).
Considering that alterations in oxidative and energy parameters lead to alterations in cell signaling, we evaluated the immunocontent of proteins directly linked to the regulation of cell metabolism, as ERK1/2, GSK-3β, and phosphor-Akt. These protein kinases play a fundamental role in the maintenance of several cellular activities, such as proliferation, activation of signaling cascades, and transcription factors, in addition to being involved in cell survival/apoptosis mechanisms (Mullonkal and Toledo-Pereyra 2007; Pierozan et al. 2014, 2016; Liu et al. 2019). Previous studies demonstrate that QUIN can exert an effect on several intracellular proteins, changing cell dynamics (Constantino et al. 2018; Santana-Martínez et al. 2018). In our study, it was possible to observe the action of QUIN on the content of total ERK-1 and phospho-Akt. These proteins are closely linked to the modulation of growth, plasticity and cell survival, memory, and learning processes (Mullonkal and Toledo-Pereyra 2007; Vandresen-Filho et al. 2016). Previous studies demonstrate that the QUIN actions on these proteins lead to oxidative, cell cytoskeleton, and glutamate uptake alterations, among others (Pierozan et al. 2016; Vandresen-Filho et al. 2016; Liu et al. 2019; Zhao et al. 2019). CoQ10 was able to partially prevent the alteration of ERK-1 content, possibly due to its antioxidant actions, demonstrating the importance of this component in cell metabolism.
In conclusion, CoQ10 showed a potential protective effect against the effect of QUIN on oxidative parameters, with decreased ROS production and maintenance of antioxidant capacity, resulting in the protection to biomolecules. Furthermore, CoQ10 was able to prevent the changes caused by QUIN in AChE activity and the energy parameters evaluated. These results suggest that CoQ10 may be a promising therapeutic alternative for neuroprotection against QUIN neurotoxicity, but further studies are needed to elucidate its effects and mechanisms of action.
A summary of the results of this work can be seen in Fig. 7.

Schematic figure of the finding results: CoQ10 neuroprotective effects on redox homeostasis, energy metabolism, enzyme activities, and cell signaling on QUIN-treated striatum slices (ROS/RNS, reactive oxygen species/reactive nitrogen species; SOD/CAT, superoxide dismutase/catalase ratio; GSH, glutathione levels; GPx, glutathione peroxidase; SH, sulfhydryl content; AChE, acetylcholinesterase; TBARS, thiobarbituric acid reactive substances; CoQ, coenzyme Q10; NADH, nicotinamide adenine dinucleotide reduced; NAD + , nicotinamide adenine dinucleotide oxidized; Cyt c, cytochrome c)
References
Aaseth J, Alexander J, Alehagen U (2021) Coenzyme Q10 supplementation – in ageing and disease. Mech Ageing Dev 197:111521. https://doi.org/10.1016/j.mad.2021.111521
Aebi H (1984) [13] Catalase in vitro. Methods Enzymol 105:121–126. https://doi.org/10.1016/S0076-6879(84)05016-3
Aitken RJ, Harkiss D, Knox W et al (1998) A novel signal transduction cascade in capacitating human spermatozoa characterised by a redox-regulated, cAMP-mediated induction of tyrosine phosphorylation. J Cell Sci. https://doi.org/10.1242/jcs.111.5.645
Aksenov MY, Markesbery WR (2001) Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 302:141–145. https://doi.org/10.1016/S0304-3940(01)01636-6
Bakels HS, Roos RAC, van Roon-Mom WMC, de Bot ST (2021) Juvenile-onset Huntington disease pathophysiology and neurodevelopment: a review. Mov Disord
Bhardwaj M, Kumar A (2016) Neuroprotective mechanism of coenzyme Q10 (CoQ10) against PTZ induced kindling and associated cognitive dysfunction: possible role of microglia inhibition. Pharmacol Rep 68:1301–1311. https://doi.org/10.1016/j.pharep.2016.07.005
Biasibetti-Brendler H, Schmitz F, Pierozan P et al (2017) Hypoxanthine induces neuroenergetic impairment and cell death in striatum of young adult Wistar rats. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0634-z
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Browne RW, Armstrong D (1998) Reduced glutathione and glutathione disulfide. Methods Mol Biol 108:347–352. https://doi.org/10.1385/0-89603-472-0:347
Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328:309–316. https://doi.org/10.1006/abbi.1996.0178
Chan KM, Delfert D, Junger KD (1986) A direct colorimetric assay for Ca2+ -stimulated ATPase activity. Anal Biochem 157:375–380. https://doi.org/10.1016/0003-2697(86)90640-8
Constantino LC, Binder LB, Vandresen-Filho S et al (2018) Role of phosphatidylinositol-3 kinase pathway in NMDA preconditioning: different mechanisms for seizures and hippocampal neuronal degeneration induced by quinolinic acid. Neurotox Res 34:452–462. https://doi.org/10.1007/s12640-018-9903-5
Cornelius N, Wardman JH, Hargreaves IP et al (2017) Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: effect of coenzyme Q10 supplementation on these parameters. Mitochondrion 34:103–114. https://doi.org/10.1016/j.mito.2017.03.001
de Souza Wyse AT, Streck EL, Worm P et al (2000) Preconditioning prevents the inhibition of NA+, K+-ATPase activity after brain ischemia. Neurochem Res. https://doi.org/10.1023/A:1007504525301
Dergousova EA, Petrushanko IY, Klimanova EA et al (2017) Effect of reduction of redox modifications of cys-residues in the Na, K-ATPase α1-subunit on its activity. Biomolecules. https://doi.org/10.3390/biom7010018
Dumont M, Kipiani K, Yu F et al (2011) Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 27:211–223. https://doi.org/10.3233/JAD-2011-110209
El-Aal SAA, El-Fattah MAA, El-Abhar HS (2017) CoQ10 augments rosuvastatin neuroprotective effect in a model of global ischemia via inhibition of NF-κB/JNK3/Bax and activation of Akt/FOXO3A/Bim cues. Front Pharmacol. https://doi.org/10.3389/fphar.2017.00735
Ellman GL, Courtney KD, Andres V, Featherstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95. https://doi.org/10.1016/0006-2952(61)90145-9
Ferreira FS, Biasibetti-Brendler H, Pierozan P et al (2018) Kynurenic acid restores Nrf2 levels and prevents quinolinic acid-induced toxicity in rat striatal slices. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1003-2
Fischer JC, Ruitenbeek W, Berden JA et al (1985) Differential investigation of the capacity of succinate oxidation in human skeletal muscle. Clin Chim Acta 153:23–36. https://doi.org/10.1016/0009-8981(85)90135-4
Green LC, Wagner DA, Glogowski J et al (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–138. https://doi.org/10.1016/0003-2697(82)90118-X
Guillemin GJ (2012) Quinolinic acid, the inescapable neurotoxin. FEBS J 279:1356–1365
Gutierrez-Mariscal FM, de la Cruz-Ares S, Torres-Peña JD et al (2021) Coenzyme q10 and cardiovascular diseases. Antioxidants
Halliwell B (2006) Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol
Jeon SW, Kim Y-K (2017) Inflammation-induced depression: its pathophysiology and therapeutic implications. J Neuroimmunol 313:92–98. https://doi.org/10.1016/j.jneuroim.2017.10.016
Komaki H, Faraji N, Komaki A et al (2019) Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res Bull 147:14–21. https://doi.org/10.1016/j.brainresbull.2019.01.025
La Cruz VPD, Carrillo-Mora P, Santamaría A (2013) Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int J Tryptophan Res 5:1–8
LeBel CP, Ischiropoulos H, Bondy SC (1992) Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5:227–231. https://doi.org/10.1021/tx00026a012
Leclerc P, De Lamirande E, Gagnon C (1997) Regulation of protein-tyrosine phosphorylation and human sperm capacitation by reactive oxygen derivatives. Free Radic Biol Med. https://doi.org/10.1016/S0891-5849(96)00379-6
Lees GJ (1991) Inhibition of sodium-potassium-ATPase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res Rev 16:283–300
Liu P, Li Y, Qi X et al (2019) Protein kinase C is involved in the neuroprotective effect of berberine against intrastriatal injection of quinolinic acid-induced biochemical alteration in mice. J Cell Mol Med. https://doi.org/10.1111/jcmm.14522
Liu Y, Peterson D, Kimura H, Schubert D (1997) Mechanism of cellular 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) reduction. J Neurochem 69(2):581–593
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. https://doi.org/10.1016/0304-3894(92)87011-4
Lugo-Huitrón R, Ugalde Muñiz P, Pineda B et al (2013) Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longev. https://doi.org/10.1155/2013/104024
Luis-García ER, Limón-Pacheco JH, Serrano-García N et al (2017) Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J Biochem Mol Toxicol. https://doi.org/10.1002/jbt.21837
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–474. https://doi.org/10.1111/j.1432-1033.1974.tb03714.x
Mishra J, Chaudhary T, Kumar A (2014) Rosiglitazone synergizes the neuroprotective effects of valproic acid against quinolinic acid-induced neurotoxicity in rats: targeting PPARγ and HDAC pathways. Neurotox Res 26:130–151. https://doi.org/10.1007/s12640-014-9458-z
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. https://doi.org/10.1016/0022-1759(83)90303-4
Mullonkal CJ, Toledo-Pereyra LH (2007) Akt in ischemia and reperfusion. J Investig Surg
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358. https://doi.org/10.1016/0003-2697(79)90738-3
Pallotti F, Bergamini C, Lamperti C, Fato R (2022) The roles of coenzyme Q in disease: direct and indirect involvement in cellular functions
Pérez-De La Cruz V, González-Cortés C, Galván-Arzate S et al (2005) Excitotoxic brain damage involves early peroxynitrite formation in a model of Huntington’s disease in rats: protective role of iron porphyrinate 5,10,15,20-tetrakis (4-sulfonatophenyl)porphyrinate iron (III). Neuroscience 135:463–474. https://doi.org/10.1016/j.neuroscience.2005.06.027
Pierozan P, Biasibetti H, Schmitz F et al (2016) Quinolinic acid neurotoxicity: differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim Biophys Acta - Mol Cell Res 1863:3001–3014. https://doi.org/10.1016/j.bbamcr.2016.09.014
Pierozan P, Fernandes CG, Feireira F, Pessoa-Pureur R (2014) Acute intrastriatal injection of quinolinic acid provokes long-lasting misregulation of the cytoskeleton in the striatum, cerebral cortex and hippocampus of young rats. Brain Res 1577:1–10. https://doi.org/10.1016/j.brainres.2014.06.024
Rustin P, Chretien D, Bourgeron T et al (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 228:35–51. https://doi.org/10.1016/0009-8981(94)90055-8
Santamaría A, Jiménez-Capdeville ME, Camacho A et al (2001) In vivo hydroxyl radical formation after quinolinic acid infusion into rat corpus striatum. NeuroReport 12:2693–2696. https://doi.org/10.1097/00001756-200108280-00020
Santana-Martínez RA, León-Contreras JC, Barrera-Oviedo D et al (2018) Sustained activation of JNK induced by quinolinic acid alters the BDNF/TrkB axis in the rat striatum. Neuroscience 383:22–32. https://doi.org/10.1016/j.neuroscience.2018.04.034
Sas K, Robotka H, Toldi J, Vécsei L (2007) Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci 257:221–239. https://doi.org/10.1016/j.jns.2007.01.033
Scherer EBS, da Cunha MJ, Matté C et al (2010) Methylphenidate affects memory, brain-derived neurotrophic factor immunocontent and brain acetylcholinesterase activity in the rat. Neurobiol Learn Mem 94:247–253. https://doi.org/10.1016/j.nlm.2010.06.002
Schmitz F, Pierozan P, Rodrigues AF et al (2016) Methylphenidate decreases ATP levels and impairs glutamate uptake and Na+, K+-ATPase activity in juvenile rat hippocampus. Mol Neurobiol. https://doi.org/10.1007/s12035-016-0289-1
Schwarcz R, Stone TW (2017) The kynurenine pathway and the brain: challenges, controversies and promises. Neuropharmacology 112:237–247. https://doi.org/10.1016/j.neuropharm.2016.08.003
Schweinberger BM, Schwieder L, Scherer E et al (2014) Development of an animal model for gestational hypermethioninemia in rat and its effect on brain Na+, K+-ATPase/Mg2+-ATPase activity and oxidative status of the offspring. Metab Brain Dis 29:153–160. https://doi.org/10.1007/s11011-013-9451-x
Sharifi MH, Eftekhari MH, Ostovan MA, Rezaianazadeh A (2015) Effects of a therapeutic lifestyle change diet and supplementation with Q10 plus L-carnitine on quality of life in patients with myocardial infarction: a randomized clinical trial. Tabriz Univ Med Sci 7:113–117. https://doi.org/10.15171/jcvtr.2017.03
Sharma SK, El Refaey H, Ebadi M (2006) Complex-1 activity and 18F-DOPA uptake in genetically engineered mouse model of Parkinson’s disease and the neuroprotective role of coenzyme Q10. Brain Res Bull 70:22–32. https://doi.org/10.1016/j.brainresbull.2005.11.019
Siebert C, Kolling J, Scherer EBS et al (2014) Effect of physical exercise on changes in activities of creatine kinase, cytochrome c oxidase and ATP levels caused by ovariectomy. Metab Brain Dis. https://doi.org/10.1007/s11011-014-9564-x
Spindler M, Flint Beal M, Henchcliffe C (2009) Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr Dis Treat 5:597–610
Stone TW (1993) Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev 45:309–379
Suhail M (2010) Na+, K+-ATPase: ubiquitous multifunctional transmembrane protein and its relevance to various pathophysiological conditions. J Clin Med Res. https://doi.org/10.4021/jocmr2010.02.263w
Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61:654–666
Vandresen-Filho S, Martins WC, Bertoldo DB et al (2016) Atorvastatin prevents glutamate uptake reduction induced by quinolinic acid via MAPKs signaling. Neurochem Res. https://doi.org/10.1007/s11064-016-1913-1
Wendel A (1981) Glutathione peroxidase. Methods Enzymol 77:325–333. https://doi.org/10.1016/S0076-6879(81)77046-0
Wilkins HM, Morris JK (2017) New therapeutics to modulate mitochondrial function in neurodegenerative disorders. Curr Pharm Des 23:731–752. https://doi.org/10.2174/1381612822666161230144517
Wyse ATS, Noriler ME, Borges LF et al (1999) Alanine prevents the decrease of Na+, K+-ATPase activity in experimental phenylketonuria. Metab Brain Dis 14:95–101. https://doi.org/10.1023/A:1020705713548
Wyse ATS, Zugno AI, Streck EL et al (2002) Inhibition of Na+, K+-ATpase activity in hippocampus of rats subjected to acute administration of homocysteine is prevented by vitamins E and C treatment. Neurochem Res 27:1685–1689. https://doi.org/10.1023/A:1021647329937
Zhao Q, Ma YM, Jing L et al (2019) Coenzyme Q10 protects astrocytes from ultraviolet B-induced damage through inhibition of ERK 1/2 pathway overexpression. Neurochem Res. https://doi.org/10.1007/s11064-019-02812-6
Zhu Z-G, Sun M-X, Zhang W-L et al (2016) The efficacy and safety of coenzyme Q10 in Parkinson’s disease: a meta-analysis of randomized controlled trials. Neurol Sci 215–224. https://doi.org/10.1007/s10072-016-2757-9
Funding
This study was supported by INCT (EN 465671/2014–4)/Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS)—Brazil.
Author information
Authors and Affiliations
Contributions
Fernanda Silva Ferreira: conceptualization; methodology; formal analysis; investigation; data curation; writing—original draft; writing—review and editing; Tiago Marcon dos Santos, Osmar Vieira Ramires Junior, Josiane Silva Silveira, and Felipe Schmitz: methodology; investigation; technical help; Angela T. S. Wyse: conceptualization; methodology; resources; data curation; writing—original draft; writing—review and editing; supervision; project administration; funding acquisition.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ferreira, F.S., Dos Santos, T.M., Ramires Junior, O.V. et al. Quinolinic Acid Impairs Redox Homeostasis, Bioenergetic, and Cell Signaling in Rat Striatum Slices: Prevention by Coenzyme Q10. Neurotox Res 40, 473–484 (2022). https://doi.org/10.1007/s12640-022-00484-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-022-00484-9