
Abstract
N-methyl D-aspartate (NMDA) preconditioning is evoked by the administration of a subtoxic dose of NMDA and is protective against neuronal excitotoxicity. This effect may involve a diversity of targets and cell signaling cascades associated to neuroprotection. Phosphatidylinositol-3 kinase/protein kinase B (PI3K/Akt) and mitogen-activated protein kinases (MAPKs) such as extracellular regulated protein kinase 1/2 (ERK1/2) and p38MAPK pathways play a major role in neuroprotective mechanisms. However, their involvement in NMDA preconditioning was not yet fully investigated. The present study aimed to evaluate the effect of NMDA preconditioning on PI3K/Akt, ERK1/2, and p38MAPK pathways in the hippocampus of mice and characterize the involvement of PI3K on NMDA preconditioning-evoked prevention of seizures and hippocampal cell damage induced by quinolinic acid (QA). Thus, mice received wortmannin (a PI3K inhibitor) and 15 min later a subconvulsant dose of NMDA (preconditioning) or saline. After 24 h of this treatment, an intracerebroventricular QA infusion was administered. Phosphorylation levels and total content of Akt, glycogen synthase protein kinase-3β (GSK-3β), ERK1/2, and p38MAPK were not altered after 24 h of NMDA preconditioning with or without wortmmanin pretreatment. Moreover, after QA administration, behavioral seizures, hippocampal neuronal degeneration, and Akt activation were evaluated. Inhibition of PI3K pathway was effective in abolishing the protective effect of NMDA preconditioning against QA-induced seizures, but did not modify neuronal protection promoted by preconditioning as evaluated by Fluoro-Jade B staining. The study confirms that PI3K participates in the mechanism of protection induced by NMDA preconditioning against QA-induced seizures. Conversely, NMDA preconditioning-evoked protection against neuronal degeneration is not altered by PI3K signaling pathway inhibition. These results point to differential mechanisms regarding protection against a behavioral and cellular manifestation of neural damage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain tolerance or resistance can be achieved by interventions before and/or after injury through potential toxic agents used in low stimulus or dose. Janoff (1964) introduced the term preconditioning to describe the tolerance response of an organism or tissue as the result of protective mechanisms towards potentially recurrent challenges. In fact, any stimulus able to generate damage to an organism or tissue can, when applied below the damage threshold, activate endogenous protective mechanisms, which may mitigate the impact of subsequent stimuli, inflicted above the damage threshold (Dirnagl et al. 2003; Constantino et al. 2015).
In neuronal cell cultures, a subtoxic concentration of N-methyl-D-aspartate (NMDA) prevents neuronal death induced by glutamate, NMDA (Chuang et al. 1992; Dickie et al. 1996; Boeck et al. 2005), or oxygen/glucose deprivation (OGD) (Pringle et al. 1999; Valentim et al. 2003). Intraperitoneal (i.p.) administration of subtoxic doses of NMDA was established as an in vivo chemical preconditioning model against further brain damage evoked by glutamatergic agonists, as the NMDA receptor agonist quinolinic acid (QA) (Ogita et al. 2003; Boeck et al. 2004; Constantino et al. 2015).
Preconditioning may induce cell signaling cascades that modulate a diversity of effectors responsible for neuroprotection. Noteworthy, this action can involve the attenuation of cell damage pathways such as excitotoxicity, ionic imbalance, oxidative stress, metabolic dysfunction, inflammation, and processes related to cell death via necrosis or apoptosis (Gidday 2006). Phosphatidylinositol-3 kinase (PI3K) and mitogen-activated protein kinases (MAPKs) signaling pathways are well known to play a significant role in neuroprotective mechanisms. However, few studies are evaluating the role of signaling pathways in NMDA preconditioning as most of the reports assess the ischemic preconditioning.
PI3K/Akt pathway mediates survival signals in various cell types, including neurons. PI3K/Akt leads to the modulation of several targets that regulate metabolism, survival, growth, and cell differentiation, as well as the traffic of intracellular vesicles (Hanada et al. 2004). The activation of PI3K/Akt occurs in response to a variety of stimuli, including growth factors, traumatic brain injury, or ischemia (Ouyang et al. 1999; Noshita et al. 2002). Miyawaki and collaborators (Miyawaki et al. 2008) showed that PI3K inhibition, which interrupts downstream events such as Akt phosphorylation, reduces the neuroprotection induced by ischemic preconditioning. Likewise, ischemic preconditioning in cortical neurons in vitro showed that ischemic tolerance could be mediated primarily by activation of PI3K/Akt pathway, without the involvement of MAPKs (Bhuiyan et al. 2011). Conversely, in vivo and in vitro studies using brain ischemia models showed that the MAPK/ERK kinase (MEK) is involved in the regulation of neuronal cell death and cell survival (Irving and Bamford 2002; Maddahi and Edvinsson 2010). Moreover, Zhan and collaborators (Zhan et al. 2013) demonstrated that MEK/ERK pathway seems to be involved in the neuroprotective mechanisms triggered by hypoxic preconditioning in a transient global cerebral ischemia model. Thus, the role of p38MAPK, via translocation of the anti-apoptotic protein Bcl-XL, has also been described in the neuroprotection induced by hypoxic preconditioning against ischemic brain damage, suggesting the anti-apoptotic mechanism as a critical event for hypoxic preconditioning (Zhao et al. 2013).
Besides, protein kinase C (PKC) and MAPKs may be involved in the preconditioning mechanism since PKCε and MEK/ERK were engaged in OGD-induced neuroprotection via NMDA receptors in mice hippocampal slices (Jia et al. 2007). A previous study in the laboratory, using selective protein kinase inhibitors, has also shown that protein kinase A (PKA), PI3K, and MEK have a crucial role in the achievement of a neuroprotective state following NMDA preconditioning (de Araújo Herculano et al. 2011), suggesting the participation of these signaling pathways in the neuroprotective mechanism.
Then, it is relevant to investigate the effects of NMDA preconditioning on the modulation of PI3K/Akt and MAPKs, as well as the crosstalk between these pathways to characterize the molecular events involved in the prevention evoked by NMDA preconditioning on QA-induced seizures and cell damage.
Material and Methods
Animals
Male adult Swiss albino mice (30–40 g) were maintained on a 12-h light/12-h dark schedule (lights on at 7:00 a.m.) at 25 °C. Mice were housed in plastic cages with food and water ad libitum. All manipulations were carried on between 9:00 and 16:00 h. All experimental procedures involving animals were performed by the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) and were designed to minimize suffering and limit the number of animals used. The experiments were performed after approval of the protocol by the local Institutional Ethics Committee for Animal Research (CEUA/UFSC PP0549).
The experimental protocols conducted in the present study are summarized in Fig. 1.

Experimental protocols. a Mice were subjected to stereotaxic surgery and after 48 h were treated with PI3K pathway inhibitor wortmannin 15 min before NMDA preconditioning. After 24 h, animals received an i.c.v. infusion of QA, and they were observed during 10 min for the incidence of seizures. After 24 h, hippocampi were dissected, and samples were prepared for immunodetection of the total and phosphorylated Akt protein levels and tissue processing for Fluoro-Jade B histochemistry. b Mice were subjected to stereotaxic surgery and after 48 h were treated with PI3K pathway inhibitor wortmannin 15 min before NMDA preconditioning. After 24 h, hippocampi were dissected, and samples were prepared for immunodetection of the total and phosphorylated Akt, ERK 1/2, GSK-3β, and p38MAPK protein levels
Surgical Procedures
Animals were anesthetized with chloral hydrate 7% (700 mg/kg; 10 mL/kg; i.p.). Stereotaxic surgery and infusion techniques were carried out as previously described in (Vandresen-Filho et al. 2007). Briefly, in a stereotaxic apparatus, the skin of the skull was removed, and a 27-gauge/7-mm guide cannula was placed at 1 mm posterior to bregma, 1 mm right from midline, and 1 mm above the lateral brain ventricle. The guide cannula was implanted 1.5 mm ventral to the superior surface of the skull and fixed with jeweler acrylic cement. The tip of the 30-gauge infusion cannula protruded 1 mm beyond the guide cannula, aiming for the lateral ventricle. Mice were allowed to recover for 48 h after surgery before the beginning of treatments.
Treatment with the PI3K Pathway Inhibitor
Wortmannin (4 μL of a 0.25 mM solution, resulting in 1 nmol; i.c.v.), a selective inhibitor for intracellular signaling pathway PI3K, was infused through the guide cannula. The inhibitor was dissolved in saline solution (NaCl 0.9%) and administered 15 min before NMDA preconditioning. The dose used was based on previous studies (Stein 2001; Ozaita et al. 2007; de Araújo Herculano et al. 2011).
NMDA Preconditioning and QA Infusion
NMDA was dissolved in saline solution (NaCl 0.9%) and adjusted to pH 7.4 with NaOH. Animals were pretreated with NMDA in a low non-convulsant dose (75 mg/kg; 10 mL/kg; i.p.) or with an infusion of vehicle (saline 0.9%; 10 mL/kg; i.p.) 48 h after the cannula implantation and 24 h before QA or saline administration (Boeck et al. 2004). Animals were observed for 30 min immediately after NMDA administration. The seizures were evoked by a chemical stimulus, i.e., the intracerebroventricular QA infusion (4 μL of a 9.2 mM solution, resulting in 36.8 nmol; i.c.v.) (Schmidt et al. 2000). Mice were observed during 10 min for the occurrence of wild running, clonic, tonic, or tonic-clonic seizures lasting for more than 5 s. Mice that did not display any seizures during this 10 min were considered protected. Then, mice pre-treated with NMDA and QA that did not present behavioral seizures were deemed protected and are referred as NMDA-QA non-convulsed (NQnc) group. Animals pre-treated with NMDA and QA that showed behavioral seizures were considered as not protected and are regarded as NMDA-QA convulsed (NQc) group.
A quantitative scale to evaluate the QA-induced seizures severity was developed based on previous studies (Cruz et al. 2003; Marganella et al. 2005; Vandresen-Filho et al. 2013). Therefore, mice subjected to QA-induced seizures received the following numeric scale according to their seizure severity: 0 = no response; 1 = immobility and excessive grooming + paroxysmal scratching; 2 = circling and rearing; 3 = wild running; 4 = jumping and falling; 5 = forepaw clonus and tail hypertonus; 6 = generalized tonic-clonic convulsions; and 7 = generalized tonic convulsion followed by death. Latency and duration of generalized clonic or tonic-clonic convulsions seizures were also determined.
Western Blot Analysis
The brains were excised from the skull and hippocampi were dissected (4 °C) and placed in liquid nitrogen and then stored at − 80 °C until sample preparation day. During dissection, the hippocampi were maintained in Krebs-Ringer bicarbonate (KRB = 122 mM NaCl, 3 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 0.4 mM KH2PO4, 25 mM NaHCO3, 10 D-glucose mM) and aerated with carbogen (95% O2–5% CO2) to reach pH 7.4. Samples were prepared as previously described by Oliveira et al. (2008). Briefly, samples were mechanically homogenized in 400 μL of Tris 50 mM pH 7.0, EDTA 1 mM, NaF 100 mM, PMSF 0.1 mM, Na3VO4 2 mM, Triton X-100 1%, glycerol 10%, and Sigma Protease Inhibitor Cocktail (P2714) followed by incubation for 10 min in ice. Lysates were centrifuged (10,000×g for 10 min, at 4 °C) to eliminate cellular debris. The supernatants were diluted 1/1 (v/v) in Tris 100 mM pH 6.8, EDTA 4 mM, SDS 8% and boiled for 5 min. After that, samples were diluted (40% glicerol, 100 mM Tris, bromophenol blue, pH 6.8) in the ratio 25:100 (v/v) and β mercaptoethanol (final concentration 8%).
Protein content was estimated through the method described by Peterson (1977). The same amount of protein (70 μg per lane) for each sample was separated by SDS–PAGE in 10% minigels and transferred to nitrocellulose membranes by using a semidry blotting apparatus (1.2 mA/cm2; 1.5 h). Membranes were blocked with 5% skim milk in TBS (Tris 10 mM, NaCl 150 mM, pH 7.5). Akt, GSK-3β, ERK1/2, and p38MAPK phosphorylated and total forms were detected after overnight incubation with specific antibodies diluted in TBS-T containing BSA 2% in the dilution of 1:1000 (anti-phospho-AktSer473–Sigma® P4112, anti-total-Akt–Cell Signaling® 9272S; anti-phospho-GSK-3βSer9–Cell Signaling® 9336S; anti-total- GSK-3β–Cell Signaling® 9315S; anti-phospho-p38MAPK–Millipore® 05–1059); 1:40,000 (anti-fosfo-ERK1/2-Sigma® M8159); 1:2000 (anti-total ERK1/2–Sigma® M5670), and 1:10,000 (anti-total-p38MAPK-Sigma® M0800). Moreover, the membranes were incubated for 1 h at room temperature with horse radish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibodies (1:5000) for detection of phosphorylated sites or total form of each protein. The reactions were developed by chemiluminescence substrate (LumiGLO). All blocking and incubation steps were followed by three steps of washing of 5 min with TBS-T (Tris 10 mM, NaCl 150 mM, Tween-20 0.1%, pH 7.5). All membranes were incubated with mouse anti-β-actin (Cell Signaling; 1:1000) antibody to verify that equal amounts of proteins were loaded on the gel. The phosphorylation level of proteins was determined as a ratio of optical density (OD) of phosphorylated band/OD of the total band (Calloni et al. 2005; Posser et al. 2007). Bands were quantified by using the Scion Image® software.
Tissue Processing and Fluoro-Jade B Histochemistry
Twenty-four hours after QA-induced seizures, mice were anesthetized with chloral hydrate (700 mg/kg; i.p.) and subjected to transcardiac perfusion with 0.1 M phosphate-buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The brains were removed and post-fixed in the same fixative buffer for 4 h. Brain tissues were cryoprotected by infiltration with 30% sucrose overnight. Then, frozen tissues were serially sectioned on a cryostat (Leica, CM 1850 UV) into 40-μm coronal sections and collected into six-well plates containing PBS 1× plus sodium azide 0.1%. The Fluoro-Jade histochemistry was used as an indicative of neuronal degeneration. Histological tissue sections were immersed in 100% ethanol for 3 min, and after, into 70% ethanol for 1 min followed by distilled water for 1 min. Slices were immersed in 0.06% potassium permanganate solution for 15 min on the bed shake to suppress background and then washed with distilled water for 1 min. Slices were stained for 30 min with the addition of Fluoro-Jade B staining solution (10 mL of 0.01% Fluoro-Jade B dye aqueous solution to 90 mL of 0.1% acetic acid in distilled water). After staining, the sections were rinsed three times with distilled water. Excess water was drained off, and the slides were immersed in xylene and then coverslipped with dibutyl phthalate in xylene (D.P.X.-Aldrich Chem. Co., Milwaukee, WI) to assemble the parts. Sections were examined with an epifluorescence microscope (Olympus–Bx41 Model) by using a filter system suitable for visualizing fluorescence in mice hippocampus. Quantification was performed by using Image J software. In the whole hippocampal slices, the optical density of Fluoro-Jade B staining was measured. In the CA1 region of the hippocampus, higher magnification (40×) was used to count the number of positive cells for Fluoro-Jade B staining (Image J multi-point tool). Analyses were performed in three slices per mouse, and eight mice were analyzed by treatment.
Statistical Analysis
The incidence of seizures was analyzed by Fisher’s exact test (p < 0.05). Statistical analyses to Western blotting and neuronal degeneration were performed with the analysis of variance (ANOVA) followed by Tukey’s test when appropriate.
Results
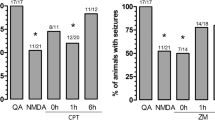
The administration of QA (36.8 nmol, i.c.v.) induced seizures in 100% of mice and NMDA preconditioning (75 mg/kg, i.p.) reduced in 47% the incidence of QA-induced seizures as it was previously reported (Boeck et al. 2004; Vandresen-Filho et al. 2007). In the first protocol used in this study (Fig. 1a), mice received the PI3K inhibitor (wortmannin, Wort, 1 nmol, i.c.v.), and after 15 min, they were preconditioned with NMDA. Twenty-four hours later, they were infused with QA. As previously published by our group, it was observed that inhibition of PI3K/Akt signaling pathway by wortmannin blocked the anticonvulsant effect triggered by NMDA preconditioning (de Araújo Herculano et al. 2011) (Fig. 2a).

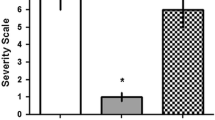
Effect of NMDA preconditioning and PI3K pathway inhibition in incidence and severity of QA-induced seizures. Mice were treated with vehicle (saline 0.9% control group) or NMDA (75 mg/kg, i.p.) 24 h before QA infusion (36.8 nM, i.c.v) and were observed for 10 min for the occurrence of seizures. Mice were treated with PI3K pathway inhibitor wortmannin (Wort) (1 nmol, 4 μL, i.c.v.) 15 min before NMDA (Wort + NMDA + QA) or 15 min before QA infusion (Wort + QA). a The percentage of animals with QA-induced behavioral seizures. The number of animals per group is defined above bars. *p < 0.05 compared with all groups according to Fisher’s exact test. b Vandresen-Filho severity scale for QA-induced seizures in NMDA preconditioned mice. Black bars represent animals with seizures and white bars represent animals without seizures. Data represent means ± SEM (n = 9) ***p < 0.001 compared with all groups (one-way ANOVA, followed by Tukey)
According to the presence or absence of tonic-clonic seizures in response to QA infusion, NMDA preconditioned mice were considered protected (NMDA + QA mice not convulsed, NMDA + QAnc) or not protected (NMDA + QA convulsed mice, NMDA + QAc). Results showed that animals treated with QA, NMDA + QA, and Wort + NMDA + QA showed behavior changes like wild running, jumping, falling and clonic, tonic, or tonic-clonic seizures, not followed by death, according to Vandresen-Filho (Vandresen-Filho et al. 2013) severity scale to QA-induced seizures. The protected animals preconditioned with NMDA (NMDA + QAnc) presented the lowest index in the severity scale analysis (1 = immobility and excessive grooming + paroxysmal scratching) when compared to QA, NMDA + QAc, and Wort + NMDA + QA groups, showing no changes in behavioral analysis, clonic or tonic-clonic seizures (Fig. 2b).
Immunodetection of phosphorylated (active) and total Akt was performed in absence or presence of the selective PI3K inhibitor wortmannin to study the involvement of cellular signaling pathways on the neuroprotective mechanism of chemical preconditioning mediated by NMDA. Therefore, 24 h after QA infusion (48 h after NMDA preconditioning), the hippocampi of mice were dissected and processed for immunodetection of proteins by Western blotting. Data showed no statistically significant changes in the level of phosphorylation (Ser473 phosphorylation) as well as in the total immunocontent of Akt between groups (Fig. 3).

Immunodetection of phosphorylated isoform and the total content of Akt protein in the hippocampus of mice preconditioned with NMDA. The animals were pretreated with wortmannin (1 nmol, 4 μL, i.c.v.) and after 15 min were preconditioned with NMDA (75 mg/kg, i.p.) or vehicle (saline 0.9%, i.p.). After 24 h, animals were infused with QA (36.8 nM, i.c.v.). Twenty four hours later, the hippocampi were dissected, and the samples were prepared for the immunodetection of the total and phosphorylated Akt protein levels. Black bars represent animals with convulsive behavior and white bars animals without seizures. NMDA + QAc indicate animals that exhibit convulsive behavior and NMDA + QAnc indicate animals that did not present seizures. The graph represents the ratio of P-Akt and T-Akt, the optical density of the bands was detected as described in experimental procedures. Data are expressed as the mean ± SEM (n = 6)
Considering that no detectable changes in Akt content or activity were evinced at the time point of 48 h after NMDA preconditioning, it was decided to analyze the phosphorylation level and content of Akt as well as its target GSK-3β along with ERK1/2 and p38MAPK in an earlier period, during the time-window of protection evoked by NMDA preconditioning, which is 24 h after inducing preconditioning (protocol depicted at Fig. 1b). Results showed that even when examined 24 h after NMDA preconditioning, Akt phosphorylation and its total content were not modified by treatments. Moreover, analysis of GSK-3β, a substrate for Akt, also showed no changes in total or phosphorylated levels 24 h after NMDA preconditioning (Fig. 4a, b).

Immunodetection of phosphorylated and total Akt, GSK3β, ERK 1/2, and p38MAPK proteins in the hippocampus of mice preconditioned with NMDA after 24 h. Animals were pretreated with wortmannin (1 nmol, 4 μL, i.c.v.). After 15 min, they were preconditioned with NMDA (75 mg/kg, i.p.) or vehicle (saline 0.9%, i.p.), and 24 h later, the hippocampi were dissected and samples were prepared for immunodetection of phosphorylated and total Akt, GSK-3β, ERK 1/2, and p38 MAPK levels. a The graph represents the ratio of P-Akt and T-Akt, the optical density of the bands was detected as described in the experimental procedures. b The figure represents the ratio of P-GSK-3β and T-GSK-3β, the optical density of the bands was detected as described in the experimental methods. c The graph represents the ratio of P-ERK1/2 and T-ERK1/2, the optical density of the bands was detected as described in the experimental procedures. d The figure represents the ratio of P-p38MAPKand T-p38MAPK, the optical density of the bands was detected as described in the experimental methods. Data are expressed as the mean ± SEM (n = 6)
It is well described that signaling through PI3K/Akt and MEK-ERK1/2 pathways may cooperate to maintain cell viability. Therefore, by using the PI3K inhibitor, levels of phosphorylated and total immunocontent of two MAPKs, ERK1/2, and p38MAPK were assessed. NMDA preconditioning and/or blockade of PI3K signaling pathway did not alter the immunocontent of total or phosphorylated forms of ERK1/2 and p38MAPK after 24 h, within the protective window of preconditioning (Fig. 4c, d).
To assess whether i.c.v. QA infusion induces neuronal degeneration in the hippocampus and to evaluate the neuroprotective effect of NMDA preconditioning, and PI3K inhibitor treatment, histological analysis of Fluoro Jade-B staining was performed. Figure 5a shows an increase in Fluoro-Jade B staining as measured in the whole hippocampus of non-protected convulsed mice (QA and NMDA + QAc groups). However, NMDA preconditioned mice, protected from seizures (NMDA + QAnc group) and animals that received the PI3K inhibitor wortmannin showed a reduction in Fluoro-jade B staining. It was possible to count Fluoro-Jade B stained neurons in the CA1 region of the hippocampus to detail such observation and also observed that PI3K inhibition did not abolish the protective effect of NMDA preconditioning against neuronal degeneration (Fig. 5b, c). Therefore, PI3K pathway seems to be involved in NMDA preconditioning-evoked protection against QA-induced seizures, although it has no involvement in prevention of cellular degeneration.

Effect of NMDA preconditioning and PI3K pathway inhibition on QA-induced neuronal degeneration. Animals were treated with vehicle (0.9% saline–control group) or NMDA (75 mg/kg, i.p.) 24 h before QA infusion (36.8 nM, i.c.v) and observed for 10 min for the occurrence of seizures. The animals received wortmannin (Wort) (1 nmol, 4 μL, i.c.v.), an inhibitor of the PI3K pathway or saline (control group) 15 min before NMDA injection (Wort + NMDA + QA) or infusion of QA (Wort + QA). Animals were perfused for posterior histological preparation of tissue and detection of neuronal degeneration by Fluoro-Jade B staining 24 h after seizure induction with QA. a The figure represents the measurement of optical density of Fluoro-Jade B staining in the whole hippocampal slice. b Representative images of Fluoro-Jade B staining in the CA1 region of the hippocampus (40× magnification). c The graph represents the number of positive cells for Fluoro-Jade B staining in the CA1 region of the hippocampus. Black bars represent animals with seizures and white bars animals that did not present seizures. Values are expressed as mean ± SEM of n = 8. *p < 0.05 compared with all groups
Discussion
Preconditioning is an adaptive response capable of preparing brain tissue to protect itself against future damage (Shpargel et al. 2008). Therefore, the elucidation of pathways involved in the neuroprotective process triggered by preconditioning may clarify the endogenous mechanisms of brain protection and justify the necessity of further studies. It is known that one significant mediator involved in neuroprotection induced by NMDA receptor activation is the PI3K pathway (Soriano 2006). It was previously published that inhibition of PKA or PI3K completely blocked the protective effect mediated by NMDA preconditioning against QA-induced seizures in mice (de Araújo Herculano et al. 2011). It was also demonstrated that a selective MEK inhibitor reduced in 75% the protective effect of preconditioning. For this reason, the involvement of PI3K pathway was evaluated by analyzing neuronal survival and phosphorylation of target proteins downstream PI3K.
This study also confirmed that inhibition of PI3K by wortmannin blocked the protective effect of NMDA preconditioning against QA-induced behavioral seizures. Moreover, a recent study showed that ischemic preconditioning was also abolished by wortmannin (Vélez et al. 2016). Besides, the use of morphine preconditioning in an ischemic model also demonstrated that wortmannin pretreatment abolished preconditioning protective effects (He et al. 2015).
Surprisingly, results demonstrated that inhibition of PI3K was ineffective in blocking NMDA preconditioning-induced prevention of hippocampal neuronal degeneration promoted by QA. Similarly, a previous study showed that blockade of adenosine A1 receptor by its antagonist CPT (8-cyclopentyltheophylline) did not block NMDA preconditioning protection regarding cell damage (cellular membrane permeabilization); however, it prevented the protection promoted by NMDA preconditioning against QA-induced seizures (Boeck et al. 2004). Our group also showed that NMDA preconditioning promotes an increased neuronal excitability as measured by EEG recording, in animals that do not manifest convulsive behavior. These results suggest that increased electrical activity decreases the probability of animals exhibiting convulsive behavior after infusion of QA (Vandresen-Filho et al. 2013). Altogether, these data suggest that PI3K pathway might be related to the initial stages of achievement of NMDA preconditioning when an increased neuronal excitation is observed (Vandresen-Filho et al. 2013), although a direct correlation between electrical activity and PI3K pathway modulation was not yet evaluated. The novelty of this study is showing that mechanism of protection evoked by NMDA preconditioning against neuronal degeneration involves different signaling from protection against QA-induced seizures. Then, a behavioral effect (seizures) and a molecular effect (neuronal degeneration) may not have the same intracellular signaling mechanism. One might speculate that the effect of NMDA preconditioning on neuronal degeneration involves additional cellular modifications (as restoring ionic unbalance, mitochondrial dysfunction, oxidative stress, and inflammation) that are independent of PI3K inhibition only.
QA-induced seizures are evoked by NMDA receptor activation as they are blocked by NMDA receptor antagonists (Perkins and Stone 1982). QA i.c.v. infusion is known to increase glutamate release and decrease glutamate uptake, and both effects contribute for increased extracellular levels of glutamate that further activates NMDA receptors (Piermartiri et al. 2009; Vandresen-Filho et al. 2015, 2016). Furthermore, QA increases calcium influx through modulation of L- and N-type voltage dependent-calcium channels (Vandresen-Filho et al. 2014). Interestingly, these effects of QA on glutamatergic transmission and calcium homeostasis are prevented by NMDA preconditioning only in mice that do not display seizures. It indicates that prevention of seizures by NMDA preconditioning may be related to modulation of glutamate receptors, glutamate transporters, and voltage-dependent calcium channels. We previously demonstrated a decrease in Akt phosphorylation immediately after QA infusion (Vandresen-Filho et al. 2016), which could be related to these effects on ion channels and glutamatergic homeostasis. However, there was no evaluation of the effects of NMDA preconditioning on Akt phosphorylation levels at this time point (10 min) after QA infusion because the interest was to see the impact of PI3K inhibition at the time-window of protective effect of NMDA preconditioning (from 24 h) (Boeck et al. 2004). The inhibition of PI3K pathway did not interfere with later cellular protection mechanisms, suggesting that different mechanisms might be involved in the prevention of behavioral seizures and maintenance of cellular survival.
It was previously demonstrated that transient cerebral ischemia activates both cell death and survival pathways. The activation of Akt in the first 24 h but not after 48 h of reperfusion is suggested as one of the factors responsible for the delay in neuronal death after global ischemia (Ouyang et al. 1999). These findings are in line with the observed difference in the effect of wortmannin of blocking protection mediated by NMDA preconditioning against QA-induced seizures; however, it does not affect the neuroprotective effect of NMDA preconditioning against neuronal death. Nevertheless, in our protocol, there are no changes in Akt phosphorylation 24 h after PI3K blockade and NMDA preconditioning were observed. Similarly, no alteration was observed to GSK-3β, a target of Akt, as well as ERK1/2 and p38MAPK. A recent in vivo study confirmed that neuroprotection induced by preconditioning with dexmedetomidine, in a model of cerebral ischemia, was mediated by activation of PI3K/Akt and phosphorylation of GSK-3β, as well as activation of ERK1/2 after 24 h of reperfusion injury (Zhu et al. 2013). However, this effect was not observed with NMDA preconditioning. Since the involvement of PI3K in protection against seizures induced by QA has already been observed, it is worth suggesting that NMDA preconditioning can activate this signaling pathway in an early period. Therefore, additional studies are necessary to address other signaling pathways involved in NMDA preconditioning as PKA and MAPKs pathways.
In conclusion, the study confirms that PI3K pathway participates in the mechanisms of protection induced by NMDA preconditioning against QA-induced behavioral seizures and suggests that protection against neuronal degeneration evoked by NMDA preconditioning is not dependent on PI3K signaling pathway (Fig. 6). Future studies are necessary to unravel different mechanisms and signaling pathways evoked by NMDA preconditioning that prevent behavioral alterations and neuronal degeneration.

Schematic overview of NMDA preconditioning effects. NMDA preconditioning prevents seizures and hippocampal cell degeneration induced by QA. The PI3K pathway inhibition prevents NMDA preconditioning-evoked protection of behavioral seizures, but not hippocampal neuronal degeneration induced by QA
Abbreviations
- Akt:
-
Protein kinase B
- ERK:
-
Extracellular signal–regulated kinases
- GSK-3β:
-
Glycogen synthase kinase-3 beta
- MAPK:
-
Mitogen-activated protein kinases
- NMDA:
-
N-methyl D-aspartate
- NQnc:
-
NMDA-QA non-convulsed group
- NQc:
-
NMDA-QA convulsed group
- OGD:
-
Oxygen/glucose deprivation
- PI3K:
-
Phosphatidylinositol-3 kinase
- p38MAPK :
-
P38 mitogen-activated protein kinases
- QA:
-
Quinolinic Acid
- Wort:
-
Wortmannin
References
Bhuiyan MI, Jung SY, Kim HJ, Lee YS, Jin C (2011) Major role of the PI3K/Akt pathway in ischemic tolerance induced by sublethal oxygen-glucose deprivation in cortical neurons in vitro. Arch Pharm Res 34:1023–1034. https://doi.org/10.1007/s12272-011-0620-3
Boeck CR, Ganzella M, Lottermann A, Vendite D (2004) NMDA preconditioning protects against seizures and hippocampal neurotoxicity induced by quinolinic acid in mice. Epilepsia 45:745–750. https://doi.org/10.1111/j.0013-9580.2004.65203.x
Boeck CR, Kroth EH, Bronzatto MJ, Vendite D (2005) Adenosine receptors co-operate with NMDA preconditioning to protect cerebellar granule cells against glutamate neurotoxicity. Neuropharmacology 49:17–24. https://doi.org/10.1016/j.neuropharm.2005.01.024
Calloni GW, Penno CA, Cordova FM, Trentin AG, Neto VM, Leal RB (2005) Congenital hypothyroidism alters the phosphorylation of ERK1/2 and p38MAPKin the hippocampus of neonatal rats. Dev Brain Res 154:141–145. https://doi.org/10.1016/j.devbrainres.2004.10.005
Chuang DM, Gao XM, Paul SM (1992) N-methyl-D-aspartate exposure blocks glutamate toxicity in cultured cerebellar granule cells. Mol Pharmacol 42:210–216
Constantino LC, Pamplona FA, Matheus FC, Ludka FK, Gomez-Soler M, Ciruela F, Boeck CR, Prediger RD, Tasca CI (2015) Adenosine A1receptor activation modulates N-methyl-d-aspartate (NMDA) preconditioning phenotype in the brain. Behav Brain Res 282:103–110. https://doi.org/10.1016/j.bbr.2014.12.056
Cruz SL, Gauthereau MY, Camacho-Muñoz C, López-Rubalcava C, Balster RL (2003) Effects of inhaled toluene and 1,1,1-trichloroethane on seizures and death produced by N-methyl-D-aspartic acid in mice. Behav Brain Res 140:195–202. https://doi.org/10.1016/S0166-4328(02)00323-6
de Araújo Herculano B, Vandresen-Filho S, Martins WC, Boeck CR, Tasca CI (2011) NMDA preconditioning protects against quinolinic acid-induced seizures via PKA, PI3K and MAPK/ERK signaling pathways. Behav Brain Res 219:92–97. https://doi.org/10.1016/j.bbr.2010.12.025
Dickie BGM, Holmes C, Greenfield SA (1996) Neurotoxic and neurotrophic effects of chronic N-methyl-D-aspartate exposure upon mesencephalic dopaminergic neurons in organotypic culture. Neuroscience 72:731–741. https://doi.org/10.1016/0306-4522(95)00611-7
Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26:248–254
Gidday JM (2006) Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 7:437–448
Hanada M, Feng J, Hemmings BA (2004) Structure, regulation and function of PKB/AKT—a major therapeutic target. Biochim Biophys Acta Proteins Proteomics 1697:3–16. https://doi.org/10.1016/j.bbapap.2003.11.009
He SF, Jin SY, Wu H, Wang B, Wu YX, Zhang SJ, Irwin MG, Wong TM, Zhang Y (2015) Morphine preconditioning confers cardioprotection in doxorubicin-induced failing rat hearts via ERK/GSK-3β pathway independent of PI3K/Akt. Toxicol Appl Pharmacol 288:349–358. https://doi.org/10.1016/j.taap.2015.08.007
Irving EA, Bamford M (2002) Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab 22:631–647. https://doi.org/10.1097/00004647-200206000-00001
Jia J, Wang X, Li H, Han S, Zu P, Li J (2007) Activations of nPKCε and ERK1/2 were involved in oxygen-glucose deprivation-induced neuroprotection via NMDA receptors in hippocampal slices of mice. J Neurosurg Anesthesiol 19:18–24. https://doi.org/10.1097/01.ana.0000211020.88431.e2
Maddahi A, Edvinsson L (2010) Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation 7:14. https://doi.org/10.1186/1742-2094-7-14
Marganella C, Bruno V, Matrisciano F, Reale C, Nicoletti F, Melchiorri D (2005) Comparative effects of levobupivacaine and racemic bupivacaine on excitotoxic neuronal death in culture and N-methyl-D-aspartate-induced seizures in mice. Eur J Pharmacol 518:111–115. https://doi.org/10.1016/j.ejphar.2005.06.022
Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh K-M, Fujisawa S, Bonanni L, Bennett MVL, Zukin RS, Jonas EA (2008) Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci 105:4892–4897. https://doi.org/10.1073/pnas.0800628105
Noshita N, Lewén A, Sugawara T, Chan PH (2002) Akt phosphorylation and neuronal survival after traumatic brain injury in mice. Neurobiol Dis 9:294–304. https://doi.org/10.1006/nbdi.2002.0482
Ogita K, Okuda H, Yamamoto Y, Nishiyama N, Yoneda Y (2003) In vivo neuroprotective role of NMDA receptors against kainate-induced excitotoxicity in murine hippocampal pyramidal neurons. J Neurochem 85:1336–1346. https://doi.org/10.1046/j.1471-4159.2003.01778.x
Oliveira CS, Rigon AP, Leal RB, Rossi FM (2008) The activation of ERK1/2 and p38 mitogen-activated protein kinases is dynamically regulated in the developing rat visual system. Int J Dev Neurosci 26:355–362
Ouyang YB, Tan Y, Comb M, Liu CL, Martone ME, Siesjö BK, Hu BR (1999) Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome C and activation of caspase-like proteases. J Cereb Blood Flow Metab 19:1126–1135. https://doi.org/10.1097/00004647-199910000-00009
Ozaita A, Puighermanal E, Maldonado R (2007) Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem 102:1105–1114. https://doi.org/10.1111/j.1471-4159.2007.04642.x
Perkins MN, Stone TW (1982) An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res 247:184–187. https://doi.org/10.1016/0006-8993(82)91048-4
Peterson GL (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83:346–356. https://doi.org/10.1016/0003-2697(77)90043-4
Piermartiri TCB, Vandresen-Filho S, De Araújo Herculano B, Martins WC, Dal’Agnolo D, Stroeh E, Carqueja CL, Boeck CR, Tasca CI (2009) Atorvastatin prevents hippocampal cell death due to quinolinic acid-induced seizures in mice by increasing akt phosphorylation and glutamate uptake. Neurotox Res 16:106–115. https://doi.org/10.1007/s12640-009-9057-6
Posser T, De Aguiar CBNM, Garcez RC, Rossi FM, Oliveira CS, Trentin AG, Moura Neto V, Leal RB (2007) Exposure of C6 glioma cells to Pb(II) increases the phosphorylation of p38MAPK and JNK1/2 but not of ERK1/2. Arch Toxicol 81:407–414. https://doi.org/10.1007/s00204-007-0177-6
Pringle AK, Thomas SJ, Signorelli F, Iannotti F (1999) Ischaemic pre-conditioning in organotypic hippocampal slice cultures is inversely correlated to the induction of the 72 kDa heat shock protein (HSP72). Brain Res 845:152–164. https://doi.org/10.1016/S0006-8993(99)01916-2
Schmidt AP, Lara DR, De Faria Maraschin J, Da Silveira Perla A, Souza DO (2000) Guanosine and GMP prevent seizures induced by quinolinic acid in mice. Brain Res 864:40–43. https://doi.org/10.1016/S0006-8993(00)02106-5
Shpargel KB, Jalabi W, Jin Y, Dadabayev A, Penn MS, Trapp BD (2008) Preconditioning paradigms and pathways in the brain. Cleve Clin J Med 75:77–82. https://doi.org/10.3949/ccjm.75.Suppl_2.S77
Soriano FX (2006) Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci 26:4509–4518. https://doi.org/10.1523/JNEUROSCI.0455-06.2006
Stein RC (2001) Prospects for phosphoinositide 3-kinase inhibition as a cancer treatment. Endocr Relat Cancer 8:237–248
Valentim LM, Rodnight R, Geyer AB, Horn AP, Tavares A, Cimarosti H, Netto CA, Salbego CG (2003) Changes in heat shock protein 27 phosphorylation and immunocontent in response to preconditioning to oxygen and glucose deprivation in organotypic hippocampal cultures. Neuroscience 118:379–386. https://doi.org/10.1016/S0306-4522(02)00919-3
Vandresen-Filho S, de Araújo Herculano B, Franco JL, Boeck CR, Dafre AL, Tasca CI (2007) Evaluation of glutathione metabolism in NMDA preconditioning against quinolinic acid-induced seizures in mice cerebral cortex and hippocampus. Brain Res 1184:38–45. https://doi.org/10.1016/j.brainres.2007.09.091
Vandresen-Filho S, Hoeller AA, Herculano BA, Duzzioni M, Duarte FS, Piermartiri TCB, Boeck CC, De Lima TCM, Marino-Neto J, Tasca CI (2013) NMDA preconditioning attenuates cortical and hippocampal seizures induced by intracerebroventricular quinolinic acid infusion. Neurotox Res 24:55–62. https://doi.org/10.1007/s12640-012-9359-y
Vandresen-Filho S, Martins WC, Bertoldo DB, Mancini G, De Bem AF, Tasca CI (2015) Cerebral cortex, hippocampus, striatum and cerebellum show differential susceptibility to quinolinic acid-induced oxidative stress. Neurol Sci 36:1449–1456. https://doi.org/10.1007/s10072-015-2180-7
Vandresen-Filho S, Martins WC, Bertoldo DB, Rieger DK, Maestri M, Leal RB, Tasca CI (2016) Atorvastatin prevents glutamate uptake reduction induced by Quinolinic acid via MAPKs signaling. Neurochem Res 41:2017–2028. https://doi.org/10.1007/s11064-016-1913-1
Vandresen-Filho S, Severino PC, Constantino LC, Martins WC, Molz S, Dal-Cim T, Bertoldo DB, Silva FRMB, Tasca CI (2014) N-methyl-d-aspartate preconditioning prevents Quinolinic acid-induced deregulation of glutamate and calcium homeostasis in mice Hippocampus. Neurotox Res 27:118–128. https://doi.org/10.1007/s12640-014-9496-6
Vélez DE, Hermann R, Frank MB, Cordero VEM, Savino EA, Varela A, Marina Prendes MG (2016) Effects of wortmannin on cardioprotection exerted by ischemic preconditioning in rat hearts subjected to ischemia-reperfusion. J Physiol Biochem 72:83–91. https://doi.org/10.1007/s13105-015-0460-6
Zhan L, Yan H, Zhou H, Sun W, Hou Q, Xu E (2013) Hypoxic preconditioning attenuates neuronal cell death by preventing MEK/ERK signaling pathway activation after transient global cerebral ischemia in adult rats. Mol Neurobiol 48:109–119
Zhao L, Liu X, Liang J, Han S, Wang Y, Yin Y, Luo Y, Li J (2013) Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice. Brain Res 1503:78–88. https://doi.org/10.1016/j.brainres.2013.01.051
Zhu J, Rebecchi MJ, Glass PSA, Brink PR, Liu L (2013) Interactions of GSK-3β with mitochondrial permeability transition pore modulators during preconditioning: age-associated differences. J Gerontol A Biol Sci Med Sci 68:395–403. https://doi.org/10.1093/gerona/gls205
Acknowledgements
This work was supported by grants from the following Brazilian funding agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Programa de Apoio aos Núcleos de Excelência (PRONEX–NENASC Project), Fundação de Apoio à Pesquisa do Estado de Santa Catarina (FAPESC), FINEP (Financiadora de Estudos e Projetos-IBN-Net #01.06.0842-00) and INCT (Instituto Nacional de Ciência e Tecnologia) for Excitotoxicity and Neuroprotection. RBL and CIT are supported by research fellowships from CNPq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experimental procedures involving animals were performed by the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) and were designed to minimize suffering and limit the number of animals used. The experiments were performed after approval of the protocol by the local Institutional Ethics Committee for Animal Research (CEUA/UFSC PP0549).
Conflict of Interest
The authors state no conflicts of interest. All authors have materially participated in the research and/or article preparation.
Rights and permissions
About this article

Cite this article
Constantino, L.C., Binder, L.B., Vandresen-Filho, S. et al. Role of Phosphatidylinositol-3 Kinase Pathway in NMDA Preconditioning: Different Mechanisms for Seizures and Hippocampal Neuronal Degeneration Induced by Quinolinic Acid. Neurotox Res 34, 452–462 (2018). https://doi.org/10.1007/s12640-018-9903-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-018-9903-5