
Abstract
Sugarcane mosaic virus (SCMV) one of the causative viruses of mosaic disease in sugarcane occurs in sugarcane growing countries worldwide. India is the second largest sugarcane producing country and genome of SCMV from India has not been characterized so far. Hence detailed studies were carried out to characterize the virus isolates based on its complete genome. Comparative genome analyses of five new isolates were performed with previously reported SCMV full genome sequences of isolates infecting sugarcane, maize, sorghum and Canna. Sequence identity matrix and phylogenetic analyses clearly represented that Indian isolates are closely related to sugarcane infecting isolates reported from Australia, Argentina, China and Iran and they diverged as a separate subgroup from other reported maize infecting isolates from Mexico, China, Ohio, Spain, Germany, Iran, Ethiopia, Kenya and Eucador. Selection pressure analysis clearly depicted the predomination of strong purifying selection throughout the viral genome, and strongest in CI and HC-Pro gene. Evidence for few positively selected sites was identified in all the cistrons except in 6 K1 and Nib rep. Among the genomic region, CI gene has exhibited comparatively more recombination hotspots followed by HC-Pro unlike other reported isolates. As the cultivation of sugarcane first originated in India, our results from the recombination events strongly suggest that Indian SCMV populations contribute for the emergence of upcoming new recombinant SCMV isolates not only within the sugarcane isolates but also with maize infecting isolates of SCMV in other countries irrespective of geographic origin and host type.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sugarcane (Saccharum spp. hybrid) is considered to be one of the major food and energy crops grown in many tropical and subtropical countries. Mosaic disease occurs throughout the sugarcane sugarcane growing across the globe and causes considerable yield losses in different countries (Koike and Gillespie 1989). Mosaic disease of sugarcane is associated by three viruses viz., Sugarcane mosaic virus (SCMV), Sugarcane streak mosaic virus (SCSMV) and Sorghum mosaic virus (SrMV) (Grisham 2000; Viswanathan et al. 2007). SCMV and SrMV belong to Potyvirus genus, which is the largest member of plant viruses. As sugarcane is vegetatively propagated for commercial cultivation, transporting virus infected setts favours the spread of mosaic disease in the field. Moreover aphids play a predominant role in transmission of SCMV from infected to healthy plants. SCMV is known to be most prevalent in infecting sugarcane, sorghum and maize. In India, the presence of this disease in sugarcane was first reported nearly a century ago (Barber 1921), since then it widely prolonged its adaptation in all the sugarcane growing zones of India. Despite, mosaic is less concerned in some countries, its severe epidemic outbreaks gained the attention of sugarcane researchers to focus on its genome biology owing to its substantial yield loss. The only effective way to control sugarcane mosaic or any virus diseases has been the use of resistant cultivars (Xia et al. 1999). Previously there is a general opinion among sugarcane workers that mosaic does not cause any extensive damage to the crop (Chona and Rafay 1950). However, the economic losses depend on varietal susceptibility, virus strain, its interaction with other diseases, vector population, and environmental conditions (Goodman 1999). In the mid-1920s, the disease caused frequent epiphytotics in Argentina, Cuba, USA and Brazil and threatened the sugar industry (Koike and Gillespie 1989), and 40–50% yield losses in South America were reported (Costa and Muller 1982).
Whole genome characterization of viruses help in understanding the viral evolution, diversity and the host pathogen interaction. Recent studies on plant viruses emphasize not only on determining primary sequences but also in depth study is carried out to gain insight into the selection pressure analysis and detection of recombination events. Numerous studies of viral evolution have addressed the role of positive Darwinian selection, particularly selection exerted by the host immune system favouring the evasion of immune recognition (Hughes and Hughes 2007). On the other hand, there is also abundant evidence that purifying selection which acts to eliminate deleterious mutations and that play an important role in the evolution of viruses (Hughes and Hughes 2005). Recombination refers to the formation of chimeric molecules from parental genomes of mixed origin and which can be important for evolution, commonly occurs with RNA viruses (Simon and Holmes 2011). Identifying recombination events in rapidly mutating viruses are considered to be pivotal which have a profound influence on evolutionary rates. Phylogenetic tree drawn from multiple sequence alignment of full length sequences is a powerful mirror for studying the evolution of viruses. Single phylogenetic tree cannot robustly reveal the recombination events that affect the evolution of homologous sequences. Earlier characterization based on partial coat protein (CP) sequences was done as the first attempt to identify the SCMV population representing both tropical and subtropical India by molecular approaches and observed high level of genetic variability among Indian isolates (Viswanathan et al. 2008, 2009). However, there was a need to critically analyse genetic structure and evolution of SCMV populations prevailing in India to define full length genomic sequences. Hence we have taken up detailed studies to determine full length sequences of five different SCMV isolates from India and addressed the information on evolutionary lineage and recombination hotspots in Indian population in comparison with the already available sequences in the GenBank. The outcome of the results clearly depict that Indian isolates actively participate in contributing for the emergence of new recombinant isolates not only within India but also across the countries.
Materials and methods
Sample collection and RNA extraction
Varying phenotypic symptom expressions of mosaic disease on different varieties, parental clones and germplasm maintained at hybridization blocks by Sugarcane Breeding Institute (SBI) at Coimbatore (NE) and Agali (Kerala). Following varieties (CoC 671, CP 5268, CoN 05072, CoSi 776) and clone (UB 1), exhibiting distinct mosaic symptoms were selected for complete genome determination. The collected leaf samples were stored in −80 °C until processing. Total RNA was extracted from the lamina and midrib using TRI reagent (Sigma, USA) following the manufacturer’s protocol. The extracted RNA was dissolved in a final volume of 40 μl sterile milliQ water and stored at −20 °C. The quality of RNA was checked in a 1.5% agarose gel.
RT-PCR assay
The RNA was reverse transcribed using RevertAid H minus First Strand cDNA Synthesis Kit (MBI Fermentas, USA). PCR was carried out using diagnostic SCMV CP primers SCMV-380F and SCMV-380R (Viswanathan et al. 2013) to confirm the presence of SCMV in the synthesized cDNA following the manufacturer’s protocol in a thermocycler (Mastercycler gradient, Eppendorf, Germany). The primers were designed by aligning the available SCMV complete genome sequences in GenBank at the time of study. Only eight full genome sequences were available during the study period. The isolates were SCMV Brisbane, SCMV Mexico, SCMV Spain, SCMV SX, SCMV Henan, SCMV SD, SCMV China1 and SCMV Beijing. Consensus sequence was generated from the eight available sequences using the software BioEdit and eight overlapping primers were designed (Supplementary Table 1). PCR reaction was performed with 2μl cDNA, 2.5 μl of 10X PCR buffer containing 15 mM MgCl2, 0.5 μl of 10mM dNTP mix, 10 pmol each of forward and reverse primers,1.25 units of Taq DNA polymerase (Merck, Mumbai, India), and sterile milliQ water to the final volume of 25 μl. The PCR reaction was performed with initial denaturation at 94°C for 3 min, 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min and a final extension of 72°C for 10 min in the same thermocycler. Amplicons were visualized by electrophoresis on 1.6% agarose gels stained with Ethidium bromide.
Cloning and sequencing of RT-PCR amplicons
RT-PCR products of each fragment were eluted from low melting agarose gel (1.6%) using Gen- Elute Gel Extraction Kit (Sigma, USA). The eluted PCR products were ligated into pTZ57R/T vector (MBI Fermentas, USA) and then used to transform in E. coli DH5α. More than twenty white colonies were obtained during cloning along with blue colonies. Ten white colonies were randomly picked and were subjected to PCR for confirmation of positive recombinant clones. Out of them five were taken for plasmid extraction using GenElute™ Plasmid Miniprep Kit (Sigma, USA). Restriction digestion was carried out for all the five samples to confirm the release of target fragment from the isolated plasmid. Positively confirmed clones of each virus isolate were sequenced by bidirectional sequencing to eliminate potential sequence heterogeneity introduced by Taq DNA polymerase (Bioserve Biotechnologies, Hyderabad, India).
Sequence alignment and phylogenetic analysis
The full-length genomic sequences of the 5 isolates were assembled from eight overlapping fragments using the software BioEdit version 7.0.4.1 (Hall 1999). Full length sequences generated in this study, were compared with 87 SCMV sequences from the database and the sequence identity matrices were generated. Multiple alignment was done using Mega X software (Kumar et al. 2018) for the construction of phylogenetic tree with the help of Neighbor-Joining method. Bootstrap analysis with 1000 replicates was performed to evaluate the significance of the interior branches.
Recombination and selection pressure analysis
Potential recombination events were identified using two algorithms: genetic algorithm for recombination detection (GARD) that yields the best Akaike Information Criteria (AICc); a measure of the goodness of fit and the single breakpoint recombination (SBP) in Datamonkey server (Pond and Frost 2005). Recombination events, likely parental isolates of recombinants, and recombination break points were analyzed using the RDP, GENECONV, Chimaera, MaxChi, BOOTSCAN, SISCAN and 3Seq methods implemented in the RDP4 program with default settings (Martin et al. 2015). The reliability of the results were cross checked with Recombination Analysis Tool (RAT) (Etherington et al. 2005). Purifying and diversifying selection affecting the sites from multiple sequence alignments were statistically tested by the fixed-effects likelihood methods such as single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), random effects likelihood (REL) and internal fixed effects likelihood (IFEL) were available from Datamonkey server (Delport et al. 2010). These methods would allow identification of codons undergoing positive selection and removes the assumptions about the demographics associated with other statistical selection tests (Cavatorta et al. 2008). Evidence of episodic diversifying selection were identified using mixed effects model of episodic selection (MEME) an approach to test the hypothesis whether the individual sites have been subject to positive or diversifying selection implemented in the HyPhy program (hypothesis testing using phylogenies) in the Datamonkey server.
Results
RT-PCR assays
RT-PCR assays of RNA isolated from five SCMV infected varieties/clones CoC 671, CP 5268, UB 1, CoN 05072 and CoSi 776 with diagnostic primers amplified the target CP region of 380bp and that confirmed the presence of SCMV in all the isolates. Eight amplicons of expected sizes of SCMV genome fragments were amplified using the overlapping primers designed for the study.
Full-length genome assembly
The Indian SCMV isolates of this study were designated as SCMV-IND (Table 1). Graphical representation of whole sequence of SCMV IND671 is given in the Fig. 1. The whole sequence lengths were 9574, 9570 and 9519 nt respectively for SCMVIND671, SCMV IND5268 and SCMV INDUB1. The isolates SCMV INDN05072 and SCMV INDSi776 had a total length of 9573 excluding polyA tail. The total amino acid length was found to be 3064 for SCMV IND671, SCMV INDN05072 and SCMV INDSi776. The respective aa length for IND5268 and INDUB1 were 3063 and 3046. Individual length of the ten cistrons and their positions were given in the Table 2. The differences in length were due to the deletions in the CP genome (Supplementary Fig. 1). The recently discovered putative protein “Pretty Interesting Potyviridae” PIPO was also found with a frame-shifting motif of G2A6 within the P3 protein gene in all the five isolates. N-terminal K54ITC57 motif is found to be conserved in all SCMV HC-Pro except in Indian isolate INDSi776, it is replaced as IIAC where lysine is substituted with isoleucine and threonine is replaced by alanine and P312TK313 motif is conserved in all the isolates (Supplementary Fig. 2).

Graphical representation of complete genome of Indian SCMV isolate IND671. The above values represent nucleotide length and the below values represent amino acid length
Phylogenetic analyses
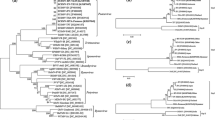
Phylogenetic analyses of full length nucleotide sequences revealed that the SCMV isolates were diverged into three distinct groups. Group I consisted of SCMV isolates infecting various crops like sugarcane, maize, sorghum and Canna. It was further divided into Group IA, Group IB and Group IC (Fig. 2). Group IA consisted of 5 Indian (SCMV IND671, SCMV INDUB1, SCMV IND5268, SCMV INDSi776 and SCMV INDN05072), 1 Australian (SCMV Brisbane), 2 Argentinean (SCMV ARG1662, SCMV ARG915, 1 Iranian (SCMV NRA) and 2 Chinese isolates (SCMV FZ-C1 and SCMV FZ-C2) infecting the host sugarcane. However, SCMV ZRA isolated from maize shared a sister taxon with SCMV NRA isolated from sugarcane formed another separate clade from the rest of the isolates within group A is the exception. Group IB consisted of all SCMV isolates infecting the host maize. Similar to the case in group A, four Chinese isolates viz., SCMV Yuhang, SCMV Xiangshan, SCMV Lingpin and SCMV China1 isolated from sugarcane were placed in group B. Group 1C consisted of two Chinese isolates viz., SCMV Canna Taian and SCMV Canna Jinan isolated from Canna. Group III and IV consisted of MDMV and SrMV isolates. The grouping of five new isolates clearly revealed that they were clustered together based on the geographic origin. Their close relationship with Australian, Argentinean, Iranian and Chinese isolates was found to be purely based on host specificity. Hence the grouping pattern of the isolates was found to be a combined effect of host specificity and geographic origin of the isolates.

Neighbor joining tree based on the complete genome sequences of five SCMV Indian and other country isolates evaluated using the interior branch test method with Mega X software. The scale bar represents a genetic distance of 200. The Indian isolates were clustered in group IA along with closely related sugarcane infecting isolates from Australia, Argentina, Iran and China. Rest of the maize infecting SCMV isolates were clustered in group IB. Two SCMV isolates infecting canna is grouped as IC. SrMV isolates and MDMV were included as an outgroup II and III respectively. The details and their accession numbers were given in Table 1
Selection pressure analyses
The rate of (dS) synonymous and (dN) non-synonymous substitutions were estimated for all the protein coding DNA sequences individually using Datamonkey server (Table 3). Codons of CP followed by P1 were under weaker purifying selection with mean dN/dS ratios 0.2912 and 0.1761, respectively when compared to the other eight proteins. Whereas, codons of CI gene undergo strong purifying selection followed by HC-Pro with mean dN/dS ratio 0.0264 and 0.0351, respectively. However, the rate of synonymous substitutions did not exceed the rate of non-synonymous substitution throughout the genomic regions indicating the presence of purifying selection pressure. The genes viz., P1, HC-Pro, P3, CI, Nia-VPg and CP were identified with the evidence of positively selected sites by four different methods SLAC, FEL, IFEL and REL. P3 gene had the highest positively selected sites numbering 20 identified by REL followed by CP with 5 positively selected sites identified by FEL and IFEL. MEME model identified 28 codon sites with evidence of diversifying selection in the CP region (Table 3). In conclusion, P3, CP and VPg had the highest positively selected sites identified by REL and MEME models. Among the 10 protein coding genes 6 K1 and Nib rep did not show any evidence of diversifying selection. Hence these genes may play a critical role in maintaining the structural and functional integrity for viral fitness in the stressful environment exerted by the host.
Sequence analysis of SCMV isolates
The sequences determined from the five Indian isolates were deposited in the GenBank and the accession numbers were obtained (Table 2). Five Indian SCMV isolates from this study and 87 SCMV sequences reported from different countries were subjected for multiple alignments and determined sequence identity matrix (Supplementary Table 2). This study revealed that Indian isolates showed 92.7–96.2% identities at the nt level and 95.3–98% identities at the aa level. The Indian isolates revealed a very close relationship with Australian (94.2–94.9% at nt level and 96.5–98.2% at aa level), Argentinean (92.2–93.7% at nt level, and 97.1–97.9% at aa level), Chinese FZ-C1 and FZ-C2 (89.7–90.5% at nt level and 96.1–98.2% at aa level and with Iranian isolates(91.8–93.8% at nt level and 93.8–95.4% at aa level). The three Chinese isolates infecting sugarcane viz., SCMV Yuhan, SCMV Xiangshan and SCMV Linping shared very low similarity of about 78.5–79.5% at nt level and 87.6–89.5% at aa level with Indian isolates. Maize infecting Iranian isolate NRA shared 89.5–90.4% at nt level and 93.4–95.1% at aa with Indian isolates. Remaining maize infecting isolates from different countries like China of different provinces, Mexico, USA, Germany, Kenya, Ethiopia and Ecuador shared least identities with Indian isolates which ranged from 76.5–79.0% at nt level and 87.0–89.2% at aa level. Two Canna infecting SCMV isolates showed 75.0–75.2% identity at nt level and 86.0–86.4% identity at aa level. MDMV isolates shared identities of 67–67.7% at the nt and 73–74% at the aa levels with the Indian SCMV isolates. Whereas, full length SrMV isolates shared identities in the range of 67.8–69% at nt level and 73.6–75.2% at aa level with the Indian isolates.
Recombination detection and breakpoints
All the three softwares RDP4, GARD and RAT, detected recombination events in the viral genomes.
Recombination events in individual protein coding nucleotide sequences
Evidences of breakpoints were not detected in the regions of 5′ and 3’ UTRs in the RDP4 software. The results were consistent with the other two reliable softwares like RAT and GARD with no recombination breakpoints in the non-coding regions. Similarly, protein coding regions 6 K1, 6 K2, P3 and Nia-pro did not show evidence of recombination in GARD, RAT and RDP4. However GARD detected one recombination breakpoint in P3 gene but it is considered to be insignificant by the KH test report. Two breakpoints at nucleotide positions 282 and 609 in CI and 25 and 219 in CP showed evidences of recombination. Whereas other regions P1, HC-Pro, Nia VPg and Nib rep were identified with recombination breakpoints at nucleotide positions 250, 582, 219 and 585, respectively (Fig. 3).

Detection of recombination breakpoints with GARD upon complete and individual cistrons of Indian isolates. The probability of the breakpoints is evaluated by akaike information criterion (AIC) score and Kishino-Hasegawa toplogical incongruence test. No breakpoints were identified in 5’ UTR, 6 K1, 6 K2, Nia-Pro and 3’UTR when they were tested individually
Recombination analyses of full genome sequences
Recombination events were detected in Indian isolates except in INDN05072, whereas remaining four sequences generated in this study were identified with recombination events (Table 4). A recombination event was detected at the N-terminal region contributed by the major parent INDSi776 and minor parent INDUB-1 with the starting and ending zones located at 877–4014 covering HC-Pro-CI region. Recombination hotspots were detected in the CI gene of the Indian isolate IND52–68 contributed by four pairs of major and minor parental sequences. INDSi776 paired with Brisbane isolate at breakpoints 3838–4154, IND671 with unknown parent at 3785–4064, INDUB1 with INDN05072 at 4802–5340 and IND671 with INDN05072 at 4802–5340. INDUB-1 was detected with the recombination events in the HC-Pro-P3 region by the crossing over of two parents, major (unknown) and minor (IND671) at position beginning from 876 and ending at 2602. Two recombination events were detected in the CI gene of the isolate INDSi776. The parents were IND5268 (major) and unknown (minor) at position 3749–4212, and the other one with unknown (major) and SCMV Ohio (minor) at position 4815–5306. For the Argentinean isolate ARG 915, IND52–68 contributed the recombination event by acting as a minor parent at Nib rep-CP region. For the Chinese isolate SCMV FZ-C1, INDUB-1 and INDSi776 behaved as minor parents by crossing over with the major parent SCMV FZ-C2 detected the recombination events at CI, 5’UTR and P1. RAT software analysis showed Indian isolates are the major contributor for the occurrence of recombinant maize infecting isolate SCMV SX in the HC-Pro gene at position 1940 and also for SCMV ZRA in CI gene at position starting from 4365 and ending at 5335. The isolate IND671 paired with American isolate Ohio and African isolate Rwanda which resulted in contribution of two recombinant Iranian isolates SCMV NRA and SCMV ZRA, at position 1940 in the HC-Pro gene. We found that the P Value was highly significant in RDP4 and the results were consistent with the software RAT used. Similarly, Brisbane isolate also contributed to the recombinant isolate SCMV ZRA when paired with Ethiopian isolate F3-S2 at Nib-rep region and American isolate Ohio at 5’UTR-P1 region. For the recombinant Chinese isolate FZ-C1, INDSi776 and INDUB-1 acted as minor parents by crossing with FZ-C2. N-terminal region (5’UTR-CI) was identified with recombination hotspots among Indian SCMV isolates. Among the N-terminal region, CI gene was found to be prone for recombinant prone hotspots. We have further observed that when the Indian SCMV isolates contribute as major parents for other country recombinant isolates, the hotspots were highly specific in N-terminal region. The Chinese isolates Beijing, SD, SX, Henan were detected with recombination events at 5’UTR-HC-Pro, CI, 6 K1-CI and CI-3’UTR, respectively. The Mexican isolates VER1 and Jal1 showed the breakpoints at HC-Pro-P3 and HC-Pro-CI, respectively and the major and minor parents were SCMV Seehausen and SCMV Beijing. For the recombinant Ethiopian isolates, the parental isolates were Chinese isolate SCMV SD (major) and African isolate Rwanda (minor) which determined the hot spots at C-terminal region covering CI- 3’UTR. Among MDMV isolates one recombination event was detected in Golestan isolate at CI gene contributed by Bulgarian and Italian isolate. No recombination events were detected among SrMV isolates. Though recombination breakpoints were detected between MDMV and SCMV isolates, they were considered as false positives because the P Value was less than 1.0 and hence they are not significant. Hence no interspecies recombination event was detected in MDMV/SrMV or SCMV/MDMV or SrMV/SCMV.
Discussion
Importance of viral diseases which impact sugarcane production and productivity has been documented in India in the recent years. Many popular varieties were infected with more than one virus and severe infection of the viruses led to varietal degeneration (Viswanathan 2016; Viswanathan and Balamuralikrishnan 2005; Viswanathan and Karuppaiah 2010; Viswanathan and Rao 2011). Viswanathan et al. (2010), confirmed the presence of three RNA viruses viz., SCMV, SCSMV and SCYLV infecting a single sugarcane host diagnosed through multiplex RT- PCR. SrMV infecting sorghum have not yet characterized at molecular level in India (Viswanathan et al. 2008). Circulation of different viral strains in a single host may increase the viral titre with increase in symptom severity. Mixed infections by different viral strains/variants is a pre-requisite for the generation of new viral strains/variants due to genetic recombination (Lai 1992). In order to combat the host defence and succeed in infection, the viruses follow a strategy of mutation and recombination, the major driving forces for evolution.
Understanding of viral evolution will help in assessing virus diversity in a country/ region in designing strategies of disease prevention and diagnosis in a better way. Earlier in India, detailed studies on SCMV diagnosis and molecular characterization based on CP genome from several infected sugarcane varieties were reported (Viswanathan et al. 2007, 2009). In continuation, full genome characterization was completed to assess genomic diversity of SCMV in the country to enrich the knowledge of selection pressure and recombination events prevailing throughout the genome of Indian SCMV population.
Phylogenetic analyses showed that Indian isolates were clustered with Australian isolate Brisbane followed by and Argentinean, Chinese and Iranian isolates. Though Iranian isolate NRA was grouped within the sugarcane specific subgroup, it diverged away from the rest of the sugarcane isolates sharing a common clade with maize infecting SCMV isolate ZRA indicating that the grouping pattern is not based on host type but by geographic origin (Moradi et al. 2016). Contrarily, the Chinese isolates both reported from sugarcane and maize did not cluster together which proved that the grouping pattern is not based on geographic origin but on host type (Achon et al. 2007). But our five new isolates showed that, their grouping pattern is a combined specificity of both geography and host when compared to the previous reports.
Our in-silico analysis clearly revealed that Indian SCMV isolates play an important role in contributing to the genetic diversity of SCMV populations in and across the geographic origin. CI and Nia pro genes showed only one site with episodic diversifying selection identified by only one method (MEME). P1, P3, Nia VPg and CP regions were identified with strong evidence of diversifying selection identified by all the five methods (SLAC, FEL, IFEL, REL and MEME) indicating that these genes are undergoing serious evolutionary constraints for adaptive evolution. Though positively selected sites were observed in all the protein coding DNA sequences, the ratio of purifying selection is highly dominating. P3 gene is identified with 20 positively selected codon sites (Table 3). Even then due to the domination of negative selection, the adaptive evolution is shaped. CI and HC-Pro were found to undergo strong purifying selection indicating that the structural and functional integrity is maintained favouring the conservation of existing genotypes.
Moury et al. (2002), reported the evidence of diversifying selection with 1 aa position in 6 k2 and 24 aa positions in CP region in Potato virus Y (PVY) of Potyvirus. Similar results were observed in our study showing the evidence of diversifying selection with 1 aa position in 6 k2 and slightly higher with 28 aa positions in CP (Table 3) which is comparatively more than PVY. Coat protein gene is found with many functional constraints by undergoing deletion mutations in the present study. Three amino acid deletions are found in the Indian isolate IND671 downstream of the position 38 and the isolate INDUB1 at position 43 showed deletion of 18aa. Long aa residue deletion in SCMV CP was observed earlier (Viswanathan et al. 2009) at 11–29 in the isolates namely CBC92061–1, CBC92061–2, CB44–101 and CB1148. Interestingly in this study, the pattern of long amino acid deletion is found at the positon 38 which is 9 bp upstream of the previous reports. This shows that the CP is actively participating in adaptive evolution without affecting the evolutionarily conserved functional domains like DAG which plays an active role in aphid transmission present in the hypervariable region.
In addition to these findings, we observed codon based model MEME determined 28 sites with evidence of positive selection. The dN/dS ratio of CP sequences analyzed in the MEGA program is 0.19 (Table 3). This value is in the range for viruses that infect plants. In our study, we strongly suggest that Indian isolates contribute to the negative selection pressure. In one hand P1 and CP gene undergo deleterious mutation, on the other hand CI and HC-pro safeguards the survival of the viral fitness. In our study, N-terminal K54ITC57 motif is found to be conserved in all SCMV HC-Pro except in Indian isolate INDSi776, it is replaced as IIAC where lysine is substituted with isoleucine and threonine is replaced by alanine and P312TK313 motif is conserved in all the isolates which are probably involved in the binding of HC-Pro to the aphid stylet (Blanc et al. 1998).
CI gene has gained more attention among the virologists as they have multifunctional role like HC-Pro and CP gene. Indian isolates showed more recombination hotspots in CI region in both cases when subjected to full genome analysis and individual analysis of 10 separate protein coding genomic regions accompanied by strong negative selection. Statistically significant clusters of recombination sites were found in the P1 gene and in the CI/6 K2/VPg gene regions for Turnip mosaic virus (Ohshima et al. 2007). The recombination hotspots are different for different viruses though they belong to same family and genus. Our study unveiled that CI followed by HC-Pro gene has the most recombination hotspots in India for individually aligned protein coding DNA sequences and also in full genome sequences. Achon et al. (2007), identified two significant recombination signals in the NIa and NIb regions of the SCMV-Sp genome. Whereas a high level of identity was observed between the isolates from distant geographic areas (SCMV isolates from Spain and China) and could be explained by movement of maize germplasm since SCMV can be seed transmitted (Li et al. 2007; Oertel et al. 1999). Recombination events were absent in 6 K1 gene of SCMV IND isolates as reported in Papaya ring spot virus (PRSV) (Mangrauthia et al. 2008).
New Guinea is the centre of origin for S. officinarum (Daniels and Roach 1987). It was just grown as a garden crop since 8000 B.C (Fauconnier 1993). From there the cultivation spread along the human colonization routes to Southeast Asia, India and the Pacific, hybridizing with wild canes and from there it was extended to the Mediterranean around 500 B.C. Hence the historical evidence strongly suggests that from India the cane cultivation started flourishing to other parts of the world. Our recombination analysis depicts a relationship between the virus evolution and the movement of sugarcane for commercial cultivation based on two important aspects. First, the parental sequences for Indian recombinant SCMV isolates are identified to be from India only. Secondly, Indian sequences have contributed to the occurrence of many recombination isolates by participating either as major or minor parents for all the sugarcane infecting isolates and maize infecting isolates like SCMV SX and SCMV ZRA (Fig. 4a–c). Hence, with the combination of host specificity, geographic origin and recombination configuration together drive the shape of Indian SCMV evolution.



a Detection of recombination event obtained by comparing Indian and other country SCMV isolates using Auto search option in RAT software. The output for sequence maize infecting SCMV SX is shown, along with its contributing sequences of sugarcane infecting SCMV Indian isolates and maize infecting SCMV Beijing. The lines on the graph represent the genetic distance (y-axis) of each sequence in the sequence list pane (left). The x-axis represents the breakpoint location on the sequence. b Detection of recombination event obtained by comparing Indian and other country SCMV isolates using Auto search option in RAT software. The output for sequence maize infecting SCMV NRA is shown, along with its contributing sequences of sugarcane infecting SCMV Indian isolates and maize infecting SCMV Ohio. The lines on the graph represent the genetic distance (y-axis) of each sequence in the sequence list pane (left). The x-axis represents the breakpoint location on the sequence. c Detection of recombination event obtained by comparing Indian and other country SCMV isolates using Auto search option in RAT software. The output for sequence maize infecting SCMV ZRA is shown, along with its contributing sequences of sugarcane infecting SCMV Indian isolates and maize infecting SCMV Ohio. The lines on the graph represent the genetic distance (y-axis) of each sequence in the sequence list pane (left). The x-axis represents the breakpoint location on the sequence
In conclusion, our study with generation of five SCMV full genome sequences provided an insight on the knowledge of genetic diversity, selection pressure and recombination hotspots prevailing in India for the first time. With the determined SCMV primary sequences, the type of selection pressure acting throughout the whole genome helped in understanding the major part of molecular evolution and particularly concluded that recombination event is a major governing feature of SCMV evolution. Further, this full genome deposition with a good piece of evolutionary information will be helpful for investigating the future epidemics and outbreaks of virus population which will bring an effective management strategy in India probably through RNAi approach. Evidence of interspecies recombination may be detected based on the host and geographical origin. Moreover, no report on complete genome sequence of MDMV and SrMV is available in India, which may further bring new insights on the evolutionary studies and their relationships across the species as a part of future perspective.
References
Achon, M. A., Serrano, L., Alonso-Duenas, N., & Porta, C. (2007). Complete genome sequences of Maize dwarf mosaic and Sugarcane mosaic virus isolates coinfecting maize in Spain. Archives of Virology, 152, 2073–2078.
Adams, I. P., Harju, V. A., Hodges, T., Hany, U., Skelton, A., Rai, S., Deka, M. K., Smith, J., Fox, A., Uzayisenga, B., Ngaboyisonga, C., Uwumukiza, B., Rutikanga, A., Rutherford, M., Ricthis, B., Phiri, N., & Boonham, N. (2014). First report of maize lethal necrosis disease in Rwanda. New Disease Reports, 29, 22.
Barber, C. A. (1921). The mosaic mottling disease of the sugarcane. International Sugar Journal, 23, 12–19.
Bernreiter, A., Garcia Teijeiro, R., Jarrin, D., Garrido, P., & Ramos, L. (2017). First report of Maize yellow mosaic virus infecting maize in Ecuador. New Disease Reports, 36, 11.
Blanc, S., Ammar, E. D., Garcıa-Lampasona, S., Dolja, V. V., Llave, C., Baker, J., & Pirone, T. P. (1998). Mutations in the potyvirus helper component protein: Effects on interactions with virions and aphid stylets. Journal of General Virology, 79, 3119–3122.
Cavatorta, J. R., Savage, A. E., Yeam, I., Gray, S. M., & Jahn, M. M. (2008). Positive Darwinian selection at single amino acid sites conferring plant virus resistance. Journal of Molecular Evolution, 67, 551–559.
Chaves-Bedoya, G., Espejel, F., Alcala-Briseno, R. I., Hernandez-Vela, J., & Silva-Rosales, L. (2011). Short distance movement of genomic negative strands in a host and nonhost for sugarcane mosaic virus (SCMV). Virology Journal, 8, 15.
Chen, J., Chen, J., & Adams, M. (2002). Characterisation of potyviruses from sugarcane and maize in China. Archives of Virology, 147, 1237–1246.
Chona, B. L., & Rafay, S. A. (1950). Studies on the sugarcane disease in India. Indian Journal of Agricultural Sciences, 20, 39–50.
Costa, A.S., &Muller, G.W.(1982). General evaluation of the impacts of viral diseases of economic crops on the development of Latin American countries. Proceedings of the first international conference on the impact of viral diseases in developing Latin American and Carribbean countries, Rio De Janeiro.
Daniels, J., & Roach, B. T. (1987). Taxonomy and evolution. In J. Heinz (Ed.), Sugarcane improvement through breeding (Vol. 1, pp. 7–84). Amsterdam: Elsevier.
Delport, W., Poon, A., Frost, S. D., & Kosakovsky Pond, S. L. (2010). Datamonkey: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics, 26, 2455–2457.
Etherington, G. J., Dicks, J., & Roberts, I. N. (2005). Recombination analysis tool (RAT): A program for the high-throughput detection of recombination. Bioinformatics, 21, 278–281.
Fan, Z. F., Chen, H. Y., Liang, X. M., & Li, H. F. (2003). Complete sequence of the genomic RNA of the prevalent strain of a potyvirus infecting maize in China. Archives of Virology, 148, 773–782.
Fauconnier, R. (1993). Sugar cane (pp. 1–40). UK: Macmillan Press Ltd London.
Gao, B., Cui, X. W., Li, X. D., Zhang, C. Q., & Miao, H. Q. (2011). Complete genomic sequence analysis of a highly virulent isolate revealed a novel strain of sugarcane mosaic virus. Virus Genes, 43, 390–397.
Goodman, B.S. (1999). A study of South African strains of sugarcane mosaic potyvirus (SCMV) identified by sequence analysis of the 5’ region of the coat protein gene. M.Sc thesis, Department of Biotechnology, Durban University of Technology, Durban, South Africa, 136 pp.
Grisham, M.P. (2000). A guide to sugarcane diseases. CIRAD-ISSCT, CIRAD publication services, Montpellier. (pp. 249–254).
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hughes, A. L., & Hughes, M. A. (2005). Patterns of nucleotide difference in overlapping and non-overlapping reading frames of papillomavirus genomes. Virus Research, 113, 81–88.
Hughes, A. L., & Hughes, M. A. (2007). Coding sequence polymorphism in avian mitochondrial genomes reflects population histories. Molecular Ecology, 16, 1369–1376.
Koike, H., Gillespie, A. G.(1989). Mosaic. In Ricaud, BT Egan, AG Gillespie, CG Hughes (Ed.), Disease of sugarcane – major diseases (pp. 301–322).
Kong, P., & Steinbiss, H. H. (1998). Complete nucleotide sequence and analysis of the putative polyprotein of maize dwarf mosaic virus genomic RNA (Bulgarian isolate). Archives of Virology, 143, 1791–1799.
Kumar, S., Stecher, G., Li, M., Knyas, C., & Tamura, K. (2018). Mega X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1574–1549.
Lai, M. M. C. (1992). RNA recombination in animal and plant viruses. Microbiological Reviews, 56, 61–79.
Li, L., Wang, X., & Zhou, G. (2007). Analyses of maize embryo invasion by Sugarcane mosaic virus. Plant Science, 172, 131–138.
Mahuku, G., Wangai, A., Sadessa, K., Teklewold, A., Wegary, D., Ayalneh, D., Adams, I., Smith, J., Bottomley, E., Bryce, S., Braidwood, L., Feyissa, B., Regassa, B., Wanjala, B., Kimunye, J. N., Mugambi, C., Monjero, K., & Prasanna, M. (2015). First Report of maize chlorotic mottle virus and maize lethal necrosis on Maize in Ethiopia. Plant Disease, 99, 1870.
Mangrauthia, S. K., Parameswari, B., Jain, R. K., & Praveen, S. (2008). Role of genetic recombination in the molecular architecture of Papaya ringspot virus. Biochemical Genetics, 46, 835–846.
Martin, D. P., Murrell, B., Michael, G., Khoosal, A., & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1(1), vev003. https://doi.org/10.1093/ve/vev003.
Moradi, Z., Mehrvar, M., Nazifi, E., & Mohammad, Z. (2016). The complete genome sequences of two naturally occurring recombinant isolates of Sugarcane mosaic virus from Iran. Virus Genes, 52, 270–280.
Moury, B., Morel, C., Johansen, E., & Jacquemond, M. (2002). Evidence for diversifying selection in potato virus Y and in the coat protein of other potyviruses. Journal of General Virology, 83, 2563–2573.
Oertel, U., Fuchs, E., & Hohmann, F. (1999). Differentiation of isolates of sugarcane mosaic potyvirus (SCMV) on the basis of molecular, serological and biological investigations. Z PflKrankh Pfl Schutz, 106, 304–313.
Ohshima, K., Tomitaka, Y., Wood Jeffery, T., Minematsu, Y., Kajiyama, H., Tomimura, K., & Gibbs Adrian, J. (2007). Patterns of recombination in turnip mosaic virus genomic sequences indicate hotspots of recombination. Journal of General Virology, 88, 298–315.
Pond, S. L. K., & Frost, S. (2005). Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics, 21, 2531–2533.
Simon-Loriere, E., & Holmes, E. C. (2011). Why do RNA viruses recombine? Natural Reviews Microbiology, 9, 617–626.
Stewart, L. R., Bouchard, R., Redinbaugh, M. G., & Meulia, T. (2012). Complete sequence and development of a full-length infectious clone of an Ohio isolate of maize dwarf mosaic virus (MDMV). Virus Research, 165, 219–224.
Tang, W., Yan, Z. Y., Zhu, T. S., Xu, X. J., Li, X. D., & Tian, Y. P. (2018). The complete genomic sequence of Sugarcane mosaic virus from Canna spp. in China. Virology Journal, 15, 147.
Viswanathan, R. (2016). Varietal degeneration in sugarcane and its management in India. Sugar Tech, 18, 1–7.
Viswanathan, R., & Balamuralikrishnan, M. (2005). Impact of mosaic infection on growth and yield of sugarcane. Sugar Tech, 7, 61–65.
Viswanathan, R., & Karuppaiah, R. (2010). Distribution pattern of RNA viruses causing mosaic symptoms and yellow leaf in Indian sugarcane varieties. Sugar Cane International, 28, 202–205.
Viswanathan, R., & Rao, G. P. (2011). Disease scenario and management of major sugarcane diseases in India. Sugar Tech, 13, 336–353.
Viswanathan, R., Balamuralikrishnan, M., & Karuppaiah, R. (2007). Sugarcane mosaic in India: A cause of combined infection of Sugarcane mosaic virus and Sugarcane streak mosaic virus. Sugar Cane International, 25, 6–14.
Viswanathan, R., Balamuralikrishnanan, M., & Karuppaiah, R. (2008). Sugarcane mosaic complex in India: Cause of different viruses/strains. Indian Journal of Virology, 19, 94–94.
Viswanathan, R., Karuppaiah, R., & Balamuralikrishnan, M. (2009). Identification of new variants of SCMV causing sugarcane mosaic in India and assessing their genetic diversity in relation to SCMV type strains. Virus Genes, 39, 375–386.
Viswanathan, R., Karuppaiah, R., & Balamuralikrishnan, M. (2010). Detection of three major RNA viruses infecting sugarcane by multiplex reverse transcription polymerase chain reaction (multiplex RT PCR). Australasian Plant Pathology, 39, 79–84.
Viswanathan, R., Ganesh Kumar, V., Karuppaiah, R., Scindiya, M., & Chinnaraja, C. (2013). Development of duplex immunocapture (duplex IC) RT-PCR for the detection of Sugarcane streak mosaic virus and Sugarcane mosaic virus in sugarcane. Sugar Tech, 15, 399–405.
Wamaitha, M. J., Nigam, D., Maina, S., Stomeo, F., Wangai, A., Njuguna, J. N., Holton, T. A., Wanjala, B. W., Wamalwa, M., Lucas, T., Djikeng, A., & Ruiz, H. G. (2018). Metagenomic analysis of viruses associated with maize lethal necrosis in Kenya. Virology Journal, 15, 90.
Xia, X. C., Melchinger, A. E., Kuntze, L., & Lubberstedt, T. (1999). Quantitative trait loci mapping of resistance to Sugarcane mosaic virus in maize. Phytopathology, 89, 660–667.
Yang, Z. N., & Mirkov, T. E. (1997). Sequence and relationships of sugarcane mosaic and sorghum mosaic virus strains and development of RT-PCR–based RFLPs for strain discrimination. Phytopathology, 87, 932–939.
Zhong, Y., Guo, A., Li, C., Zhuang, B., Lai, M., Wei, C., Luo, J., & Li, Y. (2005). Identification of a naturally occurring recombinant isolate of sugarcane mosaic virus causing maize dwarf mosaic disease. Virus Genes, 30, 75–83.
Acknowledgements
The financial support received from the Department of Biotechnology, New Delhi (BTPR4978-AGR-36712-2012) is greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bagyalakshmi, K., Parameswari, B. & Viswanathan, R. Phylogenetic analysis and signature of recombination hotspots in sugarcane mosaic virus infecting sugarcane in India. Phytoparasitica 47, 275–291 (2019). https://doi.org/10.1007/s12600-019-00726-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12600-019-00726-1