
Abstract
Sugarcane streak mosaic virus (SCSMV), an economically important causal agent of mosaic disease of sugarcane, is a member of the newly created genus Poacevirus in the family Potyviridae. In this study, we report the molecular characterization of three new SCSMV isolates from China (YN-YZ211 and HN-YZ49) and Myanmar (MYA-Formosa) and their genetic variation and phylogenetic relationship to SCSMV isolates from Asia and the type members of the family Potyviridae. The complete genome of each of the three isolates was determined to be 9781 nucleotides (nt) in size, excluding the 3′ poly(A) tail. Phylogenetic analysis of the complete polyprotein amino acid (aa) sequences (3130 aa) revealed that all SCSMV isolates clustered into a phylogroup specific to the genus Poacevirus and formed two distinct clades designated as group I and group II. Isolates YN-YZ211, HN-YZ49 and MYA-Formosa clustered into group I, sharing 96.8–99.5 % and 98.9–99.6 % nt (at the complete genomic level) and aa (at the polyprotein level) identity, respectively, among themselves and 81.2–98.8 % and 94.0–99.6 % nt (at the complete genomic level) and aa (at the polyprotein level) identity, respectively, with the corresponding sequences of seven Asian SCSMV isolates. Population genetic analysis revealed greater between-group (0.190 ± 0.004) than within-group (group I = 0.025 ± 0.001 and group II = 0.071 ± 0.003) evolutionary divergence values, further supporting the results of the phylogenetic analysis. Further analysis indicated that natural selection might have contributed to the evolution of isolates belonging to the two identified SCSMV clades, with infrequent genetic exchanges occurring between them over time. These findings provide a comprehensive analysis of the population genetic structure and driving forces for the evolution of SCSMV with implications for global exchange of sugarcane germplasm.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sugarcane streak mosaic virus (SCSMV) is an economically important pathogen of sugarcane (Saccharum spp.), causing the common mosaic disease of sugarcane [22]. The virus was first isolated from quarantined sugarcane germplasm imported from Pakistan into the USA [11, 12] and was recently assigned to a new genus, Poacevirus (family Potyviridae) with Triticum mosaic virus (TriMV) as its type species [2]. SCSMV induces pinwheel and laminated aggregate types of inclusions that are characteristic of members of the family Potyviridae [16, 33]. However, it is distinct from potyviruses in its host range, tryptic digestion profile, and lack of reactivity with a monoclonal antibody against potyviruses [46] or polyclonal antisera against potyviruses and rymoviruses [19, 20]. In addition to sugarcane, other members of the plant family Poaceae have been documented to be natural hosts of SCSMV, including sorghum (Sorghum bicolor), maize (Zea mays), millet (Pennisetum glaucum), sudan grass (Sorghum bicolor drummondii), johnsongrass (Sorghum halepense) and crabgrass (Digitaria sp.) [16, 20, 36, 37, 46]. SCSMV is transmitted naturally or experimentally by mechanical sap inoculation [15, 17], and no insect vector have been identified yet for the virus [17], although it is known that TriMV is vectored by the wheat curl mite Aceria tosichella [35].
SCSMV has flexuous filamentous particles (890 × 15 nm) with a positive-sense single-stranded RNA (ssRNA) of approximately 10 kb in size, excluding a poly-A tail at the 3′ terminus [15, 46]. It contains a large open reading frame (ORF) encoding a polyprotein of approximately 3130 amino acid (aa) residues and two (5′ and 3′) untranslated regions (UTRs), each ≈200 nt in length. The large polyprotein of SCSMV is self-cleaved into ten mature proteins, P1, HC-Pro, P3, 6K1, CI, 6K2, VPg, NIa-Pro, NIb and CP [23, 46]. Similar to other members of the family Potyviridae, a small ORF (Pretty Interesting Potyviridae ORF [PIPO]) [8] is embedded within the N-terminal part of P3 and translated in the +1 reading frame [42].
To date, several isolates of SCSMV have been identified and characterized from different sugarcane-producing Asian countries, including Pakistan, Bangladesh, Sri Lanka, Thailand, India, Vietnam, China, Japan and Indonesia [7, 9, 12, 15, 23], and shown to display a high genetic variation based on analysis of complete genome or gene-specific sequences [13, 14, 23, 43]. In China, SCSMV is widespread in Yunnan province, and at least three SCSMV genotypes are prevalent [10, 13, 14], with isolates JP1, JP2 and ID having been introduced into the Chinese sugarcane germplasm from Japan and Indonesia [23]. Recently, the genetic diversity and population structure of SCSMV isolates from China were determined and compared to those of virus isolates from India [13, 14]. These studies were based on analysis of partial virus genome sequences, and hence, very little information is available on the complete genome characteristics of SCSMV isolates from China, and neither study included virus isolates from Myanmar, a country where sugarcane is the second most important commodity after rice (FAO’s FAOSTAT database, 2014) and that is located in close proximity to major sugarcane-producing regions of China. In this study, we report the complete genome sequence of two new SCSMV isolates from China and one from Myanmar naturally infecting sugarcane. We describe the genetic variation and phylogenetic relationship of the three new isolates relative to seven Asian SCSMV isolates and 25 isolates of representative members of the family Potyviridae.
Material and methods
Sample collection
Visible dewlap leaf samples with mosaic symptoms were collected from cultivated sugarcane (Saccharum spp.) hybrid varieties YZ05-211 (Kaiyuan, Yunnan province) and YZ05-49 (Linggao, Hainan province) in China, and variety Formosa in Myanmar (Mongkong). Fresh symptomatic leaf tissue samples were collected from one plant per variety, kept in cool storage in transit and stored at −80 °C until RNA isolation.
Cloning and sequencing
Total RNA for RT-PCR was isolated from SCSMV-infected sugarcane leaves, using TRIzol® Reagent (Invitrogen/Life Technologies, USA) according to the manufacturer’s instructions, and dissolved in a final volume of 50 µL of nuclease-free water. First-strand cDNA was synthesized from total RNA (1 µg) using a PrimeScript RT Reagent Kit (TaKaRa Biotechnology Co., Ltd., Dalian, China). To clone the three complete SCSMV genome sequences of YN-YZ211, HN-YZ49 and MYA-Formosa, seven overlapping fragments (designated as fragments A-G) were amplified using pairs of virus-specific primers (Table 1). PCR was performed using 1 µL of cDNA, 2.5 µL of 10× Ex Taq buffer, 2.5 µL of 2.5 mM deoxynucleoside triphosphates, 10 pmol of each upstream and downstream primer and 1.25 units of Ex Taq polymerase (TaKaRa Biotechnology Co., Ltd.) in a final volume of 25 µL. PCR conditions were as follows: an initial denaturation at 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 53 °C for 30 s and 72 °C for 2 min for fragments A, C and D; 56 °C for 30 s and 72 °C for 2 min for fragment F; 52 °C for 30 s and 72 °C for 3 min for fragments B and E; or 57 °C for 30 s and 72 °C for 1 min for fragment G; and a final extension at 72 °C for 10 min.
Amplified genomic fragments were purified using an E.Z.N.A. Gel Extraction Kit (OMEGA Bio-Tek, Inc., Norcross, GA, USA) and cloned into the pMD19-T vector (TaKaRa Biotechnology Co., Ltd.) as per manufacturer’s instructions. Three positive clones per amplified fragment were sequenced in both orientations (Sangon Biotech Co., Ltd., Shanghai, China), and sequence contigs were generated for each cloned DNA fragment. A consensus ‘unique’ sequence was obtained when sequences derived from the three clones per isolates showed ≥99 % identity to exclude in vitro PCR errors. The seven overlapping fragments were assembled using CodonCode Aligner 6.02 software (Centerville, MA, USA) to yield the complete genome sequences of the three SCSMV isolates.
Phylogenetic analysis
The complete genomic nucleotide (nt) sequence and predicted amino acid (aa) sequence of the polyprotein of three SCSMV isolates from this study and seven isolates from GenBank database were aligned using MUSCLE, implemented in MEGA6 [39], and the percent nucleotide sequence identity and corresponding values were calculated as pairwise distances among isolates. The complete polyprotein aa sequences of the ten SCSMV isolates and 25 isolates of representative members of the family Potyviridae (Table S1), SCSMV polyprotein nt sequences, and gene-specific nt sequences of SCSMV isolates were also aligned with MUSCLE, and phylogenetic trees were constructed with MEGA6 using the maximum-likelihood method. The model that best fitted each of the aa and nt sequence datasets was determined using the ‘Find the best DNA/protein models (ML)’ option of the MEGA6 software. The robustness of each node of the phylogenetic trees was assessed from 1000 bootstrap replicates, and branches not supported by ≥ 60 % of the replicates were collapsed.
Estimation of evolutionary divergence
The within- and between-group estimates of evolutionary divergence (d) were calculated for the complete polyprotein sequences of the ten Asian SCSMV isolates (three from this study and seven from GenBank) based on the identified SCSMV phylogroups, using MEGA6 software. The same analysis was also performed for the P1 and CP cistron datasets representing nonstructural (N-terminal) and structural (C-terminal) virus-encoded genes, respectively. Tajima’s D statistic was calculated to measure the departure from neutrality for all mutations among group-specific SCSMV polyprotein and gene-specific sequences [38], and the McDonald and Kreitman test (MKT) [27] was used to determine whether synonymous and nonsynonymous variations support the hypothesis of adaptive protein evolution between the SCSMV phylogroups and to provide evidence that divergence in SCSMV phylogroups is driven by natural selection. Both the Tajima’s D statistic and MKT were performed using DnaSP software version 5.10.01 [24].
Population genetic analysis
The degree of genetic differentiation between and within SCSMV populations was determined using three permutation-based statistical tests, Ks*, Z and Snn, based on the polyprotein, P1 and CP gene sequences. Ks*, Z and Snn represent the most powerful sequence-based statistical tests for genetic differentiation and are recommended for use in cases of high mutation rate and small sample size [18]. The degree of genetic differentiation or the level of gene flow between and within SCSMV populations was calculated from Wright’s Fst [45], for which values range from 0 to 1 for undifferentiated to fully differentiated populations, respectively. Fst values can be interpreted as the proportion of genetic diversity that is due to diversity within groups to total diversity, values of Fst > 0.05 suggest a degree of differentiation among populations, and values of Fst > 0.33 suggest an infrequent gene flow [28, 34]. These analyses were performed using DnaSP software v. 5.10.01 [24].
Recombination analysis
The aligned nt dataset of the complete genome sequences (excluding the 5′- and 3′-UTRs) of the ten SCSMV isolates (three from this study and seven from GenBank) was examined for the presence of recombination events using the suite of seven recombination detection algorithms implemented in Recombination Detection Program (RDP) version 4.16 [26]. Sequences were automatically masked to ensure optimal recombination detection before algorithm processing. A Bonferroni-corrected P-value cutoff of 0.05 and the option of “Sequences are linear” were selected. An event was considered to represent a true recombination event when detected by at least four of the seven RDP-implemented algorithms with significant (<0.05) P-values.
Results
Genomic features of YN-YZ211, HN-YZ49 and MYA-Formosa
The complete genomes of isolates YN-YZ211 (GenBank accession no. KJ187047), HN-YZ49 (KJ187048) and MYA-Formosa (KJ187049) were each determined to be 9781 nt in size, including the full-length 5′ and 3′ UTRs and excluding the 3′ poly(A) tail. The genome of each of the three isolates was found to contain a single large open reading frame (ORF) encoding a large polyprotein of 3130 aa residues. Similar to SCSMV-PAK (NC_014037), the polyproteins of isolates YN-YZ211, HN-YZ49 and MYA-Formosa were each predicted to be processed into ten mature proteins, P1 (358 aa), HC-Pro (470 aa), P3 (329 aa), 6K1 (56 aa), CI (647 aa), 6K2 (53 aa), VPg (196 aa), NIa-Pro (234 aa), NIb (492 aa) and CP (295 aa). The GGAAAAAAA (G1-2A6-7) motif at position 3085-3093 of the SCSMV genome, which is involved in expression of ORF PIPO in members of the family Potyviridae [8], was found to be embedded within the P3 gene. Further analysis revealed that similar to most SCSMV isolates, all three isolates characterized in this study lack three additional amino acids (K, G, and R) encoded in the P1 of SCSMV isolate IND671 and encode six aa residues, NLDPRN, in the C-terminus of NIa-Pro in place of four aa residues, ESGS, in isolate IND671.
Phylogenetic analysis and sequence comparisons
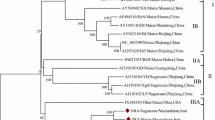
Phylogenetic analysis was performed based on the complete aa polyprotein sequences of ten SCSMV isolates (three from this study and seven from GenBank) and the corresponding sequences of 25 isolates of representative members of the family Potyviridae. The analysis revealed a clear segregation of all 35 virus isolates into genus-specific clades as shown in Fig. 1a. To further investigate phylogenetic relationships among SCSMV isolates, sequence subsets consisting of complete polyprotein sequences and P1 and CP gene sequences of the ten SCSMV isolates (three from this study and seven from GenBank) were analyzed separately. The analysis revealed the segregation of the ten SCSMV isolates into two distinct phylogroups designated as group I and group II regardless of the genomic segment under consideration (Fig. 1b–d). Eight of the ten SCSMV isolates clustered into group I and included isolates from China, Maymmar, Japan, India, Indonesia and Thailand, while group II comprised virus isolates from India and Pakistan. Based on these results, it can be concluded that SCSMV isolates from South Asia (group II) appear to be distinct from group I virus isolates from other Asian countries.

Maximum-likelihood phylogenetic trees showing evolutionary relationships among members of the family Potyviridae (a) and sugarcane streak mosaic virus (SCSMV) isolates (b–d). The trees were constructed based on aligned polyprotein amino acid (aa) or nucleotide (nt) sequences (b), P1 gene nt sequences (c), and coat protein gene nt sequences of three isolates of SCSMV from this study (bold font), other SCSMV isolates (regular font) and representative isolates of selected members of the different genera in the family Potyviridae (regular font). Bootstrap values (1000 replicates) are shown at the branch internodes. The analysis involved 35 amino acid sequences (three from this study and 32 from the GenBank database) and 10 SCSMV gene-specific sequences (three from this study and seven from GenBank). All analyses were conducted in MEGA6 [39]
In pairwise comparisons, aligned complete genomic (nt) and polyprotein (aa) sequences of isolates YN-YZ211, HN-YZ49 and MYA-Formosa shared 96.8–99.5 % and 98.9–99.6 % nt and aa sequence identity, respectively, among themselves and 81.2–98.8 % and 94.0–99.6 % nt and aa sequence identity, respectively, with the corresponding sequences of seven other Asian SCSMV isolates obtained from GenBank (Table 2). Further analysis showed that the group I SCSMV isolates shared greater nt (95.2–99.5 %) genomic and aa (98.4–99.7 %) polyprotein sequence identity among themselves relative to group II isolates, which share 93.0 % and 97.5 % nt and aa sequence identity, respectively with each other (Table 2). The ranges of between group identities were 80.6–81.8 % and 94.0–95.3 %, respectively, at the complete genomic nt and polyprotein aa levels (Table 2). In contrast, lower levels of polyprotein (51.8–52.6 %) and CP (53.5–54.6 %) nt sequence identity were observed for comparisons between SCSMV isolates and TriMV, the type member of the family Poacevirus, and much lower sequence identity was found between SCSMV isolates and members of other genera in the family Potyviridae (Table S2).
Nucleotide sequence diversity and population genetic analysis
The overall mean value of nucleotide sequence diversity was 0.085 ± 0.002 for the polyprotein, 0.085 ± 0.001 for P1, and 0.072 ± 0.005 for CP (Table 3; 10 sequences: 3 from this study and 7 from GenBank). Thus, similar overall nucleotide sequence diversity values were obtained for the polyprotein and P1, while a slightly lower divergence value was obtained for the CP cistron, indicating greater sequence conservation in the capsid protein of SCSMV. Analysis of group-specific sequences revealed that members of group II are more diverged from each other relative to isolates belonging to group I, regardless of the genomic region under consideration (Table 3). The results also suggest greater sequence divergence in the P1 cistron relative to the CP and polyprotein of SCSMV (Table 3). Negative values of the Tajima’s D statistic were obtained for the polyprotein, P1 and CP gene-specific sequences, indicating an excess of low-frequency polymorphism caused by background selection, genetic hitchhiking, or population expansions [41]. However, it is also plausible that purifying selection may be acting on each of the SCSMV phylogroups, since the Tajima’s D statistic values are not statistically significant (P > 0.10) in all cases (Table 3). Notably, it was impossible to perform Tajima’s D statistic test for SCSMV group II isolates, since this analysis in the DnaSP software requires at least four sequences [24].
As expected, analysis of between-group evolutionary divergence yielded higher values for the polyprotein (0.190 ± 0.004), P1 (0.177 ± 0.010) and CP (0.152 ± 0.011) gene sequences (Table 4) than those obtained for the within-group comparisons (Table 3). These results provide additional support for the existence of the two phylogroups identified in Fig. 1b–d. Results of the McDonald and Kreitman test showed significant differences (with very good statistical support based on the P-value of the G statistic) only for the CP gene, indicating that natural selection contributed to the evolution of group-specific SCSMV isolates based on analysis of the capsid gene sequences (Table 4). In contrast, G-test statistics were not significant (P > 0.10) for the polyprotein and P1 gene sequences (Table 4), suggesting that the divergence of sequences belonging to group I from those of group II in the polyprotein and P1 cistron may be a consequence of random processes. Such contrasting gene-specific evolutionary patterns have been reported previously for other RNA viruses [4, 5].
Measurements of genetic differentiation between and within the two SCSMV phylogroups provide further support for a significant genetic differentiation between group I (East and Southeast Asia) and group II (South Asia) isolates based higher values of Wright’s Fst [45] obtained for the polyprotein, P1 or CP gene sequence comparisons (Table 5). Further support for these results could be seen in significantly high values of Snn, Ks* and Z statistics obtained for the same comparisons (Table 5), indicating that infrequent genetic exchange may have occurred between SCSMV populations from South Asia (group II) and those from East and Southeast Asia (group I). Taken together with results of the phylogenetic and nucleotide diversity analyses, it is reasonable to conclude that global SCSMV populations segregated into two groups of sequence variants that are genetically differentiated from one another and show some geographical structuring.
Recombination analysis
The RDP4-implemented recombination analysis of the polyprotein-encoding gene sequences of the ten Asian SCSMV isolates led to the identification of two significant putative recombination sites located in the regions encoding the C-terminal portions of VPg and CP in the genomes of six SCSMV isolates from China, India, Japan and Thailand (Fig. 2a). Two sites were supported by significant P-values obtained with the RDP (4.32 × 10−4), BOOTSCAN (6.31 × 10−7), MAXCHI (3.00 × 10−9), CHIMAERA (7.48 × 10−7) and SISCAN (5.45 × 10−10) programs of the RDP4.16 software (Fig. 2b). Six recombinant isolates originated from the same major parent TPT (Indian isolate) and minor parent MYA-Formosa (Myanmar isolate). Analysis of the recombination region from nt 6587 to 9391 showed that the six recombinants and two parent isolates were clustered in group I, while two non-recombinant isolates were in group II, using UPGMA, implemented in the RDP program (Fig. 2c). However, recombination signals for the Pakistani PAK and Indian IND671 isolates were not detected by the seven algorithms. Two other phylogenetic trees, constructed using non-recombinant regions or ignoring recombination, were similar to those constructed using the recombination region (data not shown).

Analysis of possible recombination in the full-length polyprotein nucleotide sequences of ten Asian sugarcane streak mosaic virus (SCSMV) isolates. Recombination events were detected simultaneously by seven methods using the RDP 4.16 program. Recombination maps, schematic illustrations of genomes and breakpoints are shown (a). The estimated nucleotide positions of recombination sites are presented relative to the genome, excluding the 5′ and 3′ untranslated regions, following the numbering of the SCSMV PAK isolate [NC_014037] sequence. Recombinant isolates, parental isolates, recombination breakpoints and statistical values were detected simultaneously by seven methods in the RDP 4.16 program (b). The phylogenetic tree of the recombination region from nucleotides 6587 to 9391 was constructed with UPGMA, implemented in the RDP program (c)
Discussion
SCSMV is a major virus associated with the common sugarcane mosaic disease, with estimated yield losses of 30–80 % [6]. Typical symptoms and host range of SCSMV are similar to those of the closely related potyviruses sugarcane mosaic virus and sorghum mosaic virus [6, 10]. To date, several SCSMV strains (genotypes) have been identified in Asian sugarcane-growing countries, including Pakistan, Bangladesh, Sri Lanka, Thailand, India, Vietnam, China, Japan and Indonesia [7, 9, 12, 15, 23], and 16 different phylogenetic groups have been proposed for the virus based on analysis of partial CP gene sequences of 60 global SCSMV isolates [43]. In the present study, the complete genome sequences of two sugarcane-derived isolates of SCSMV from China (YN-YZ211 and HN-YZ49) and one isolate from Myanmar (MYA-Formosa) were determined, and their genetic relationships were analyzed relative to genome sequences of seven Asian isolates of the virus and 25 isolates of representative members of the family Potyviridae. The levels of polyprotein (>46 %) and CP (>76–77 %) nucleotide sequence identity calculated for all 35 virus isolates analyzed in the study support the assignment of all SCSMV isolates and TriMV into the same genus, Poacevirus, and demonstrate their distinctness from members of other genera in the family Potyviridae [1–3].
Analysis of the genetic diversity of SCSMV populations from different geographical regions can provide relevant information for understanding its emergence, epidemiology, short- and long-term movement, and gene flow, which can be important for implementing proper control strategies to limit virus dispersal. In the present study, genetic differentiation and phylogenetic analysis of ten Asian SCSMV isolates, including the three isolates from this study, revealed that the population structure of the two SCSMV phylogroups, group I and group II, correlated with their geographical origin. With the exception of isolate, TPT from India, group I consisted of members that originated from East Asia (YN-YZ211, HN-YZ49, JP1 and JP2) and Southeast Asia (MAY-Formosa, ID and THA), while group II isolates (IND671 and PAK) were exclusively from South Asia. Such geographical structuring of virus isolates may suggest bottlenecks imposed by the host plant or potential arthropod vectors, as demonstrated for other viruses [21]. SCSMV showed a spatial distribution pattern with a greater genetic deviation in the South Asian (India and Pakistan) lineage than in the subpopulation composed of virus isolates from East (China and Japan) or Southeast (Thailand, Indonesia and Myanmar) Asia. These results are in agreement with those of a recent study showing that SCSMV populations from India were more diverse than virus populations from China [14]. Additionally, the higher genetic diversity observed in this study between groups than within groups of SCSMV populations suggests that there is more frequent gene flow within groups than between groups. The SCSMV population structure might be related to virus transmission characteristics and the presence of physical and quarantine barriers between countries. However, the close genetic affinities observed between some geographically distant SCSMV isolates suggest long-distance migration, probably due to international traffic of propagative sugarcane materials. Historical accounts support the hypothesis that the SCSMV isolates were most likely introduced into China via importation of sugarcane germplasm materials [14]. For instance, the JP1, JP2 and IND SCSMV isolates were introduced into China from Japan and Indonesia by propagating germplasm material [23]. Subsequently, further spread of SCSMV within China could have occurred via movement of vegetative cuttings across provinces. In the same manner, isolate MYA-Formosa might have been inadvertently introduced into Myanmar from other sugarcane-producing Asian region (Taiwan, P. R. China) via contaminated vegetative cuttings. These results underscore the importance of rigorous inspection and implementation of quarantine measures to limit virus dispersal between different regions in the same country and across national boundaries.
Recombination, an important evolutionary force for plant viruses, can promote genome diversity and adaptability or offset fitness decrease by accumulation of deleterious mutations in bottleneck events [29]. In this study, recombination analysis led to the detection of six putative recombinants, all of which belong to group I. Previously, nine recombination events were reported among 12 Indian SCSMV isolates based on analysis of the HC-Pro gene sequences [6]. In addition, SCSMV-PAK was proposed to be a recombinant with isolates SCSMV-ID and SCSMV-IND as its parental sequences and the recombination events occurring at the 5′ UTR and P1 region [31]. Furthermore, three isolates from India, CB740, CB9217-1 and S-8, were identified as recombinants in the CP coding region [13], while other recombination events have been reported based on analysis of P1 and P3 gene sequences of several Indian SCSMV isolates [30, 42] and the P1, HC-Pro, CP and 3′ UTR of several virus isolates from China [14]. These results point to the importance of genetic recombination in the evolution of global SCSMV populations. However, no signature of recombination was detected for group II isolates identified in this study, suggesting that this potent evolutionary force has not shaped the emergence of SCSMV group II variants. It is also noteworthy that no recombination events were found between SCSMV group I and group II subpopulations, indicating significant genetic differentiation and limited gene flow between virus isolates belonging to these phylogroups, possibly due to the presence of physical and quarantine barriers between them.
Natural selection is an important evolutionary mechanism limiting variation of virus populations [32]. In the present study, adaptive molecular evolution in the polyprotein, P1, and CP protein-coding DNA sequences was detected by the G statistic of MKT between two distinct variant groups. Similarly, a strong purifying selection was detected in other genes across the SCMV genome, enhancing the speed of elimination of deleterious mutations in genes and shaping the stable genetic structure of the population [14, 42]. However, a few codons of P1, an RNA silencing suppressor protein known to play a role in enhancing the pathogenicity of a heterologous virus [40], were reported to be under positive selection [30]. Our finding also revealed one positive selection site located in a codon of P1 (data not shown). Positive selection on a few codons in the P1 protein indicated an ongoing molecular adaptation with the potential to fix selectively advantaged genotypes and enhance population diversification. The P1 gene also showed the most genetic divergence compared to the CP gene, especially in the group II subpopulation, for the PAK and IND671 isolates. Molecular adaptation due to positive selection has been described in some isolates of tomato spotted wilt virus, where an aa substitution of C to Y at position 118 in the CP protein correlated with the isolate’s ability to break resistance conferred by the Sw-5 gene in tomato [25].
The present study provides a comprehensive analysis of the population genetic structure of SCSMV and the driving forces involved in its evolution. In particular, it demonstrates that frequent gene flow, homologous recombination and strong purifying (negative) selection are driving forces acting synergistically to limit genetic variation within individual geographical subpopulations of SCSMV. Founder effects have also been suggested to play a role in shaping the genetic structure of SCSMV. Natural selection and genetic drift are two main evolutionary mechanisms limiting genetic variation of the SCSMV population. Positive selection at a few codons in the P1 protein, an RNA silencing suppressor, reflects molecular adaptation. The SCSMV population would be expanded by fixing advantaged mutations, essentially by enhancing the pathogenicity of the virus via a novel mechanism to suppress the RNA-silencing-based antiviral defense. The P1 site under selection, as identified in this study, can be used in functional genomic studies based on directed mutagenesis and reverse genetics [44]. Additional genomic sequences of SCSMV isolates from other countries are needed to fully assess the genetic variability and phylogenetic history of SCSMV.
References
Adams MJ, Antoniw JF, Fauquet CM (2005) Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol 150:459–479
Adams MJ, Carstens EB (2012) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of viruses. Arch Virol 157:1411–1422
Adams MJ, Zerbini FM, French R, Rabenstein F, Stenger DC, Valkonen JPT (2011) Potyviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy. Elsevier, Oxford, pp 1069–1090
Alabi OJ, Al Rwahnih M, Mekuria TA, Naidu RA (2014) Genetic diversity of Grapevine virus A in Washington and California vineyards. Phytopathology 104:548–560
Alabi OJ, Al Rwahnih M, Karthikeyan G, Poojari S, Fuchs M, Rowhani A, Naidu RA (2011) Grapevine leafroll-associated virus 1 occurs as genetically diverse populations. Phytopathology 101:1446–1456
Bagyalakshmi K, Parameswari B, Chinnaraja C, Karuppaiah R, Ganesh KV, Viswanathan R (2012) Genetic variability and potential recombination events in the HC-Pro gene of Sugarcane steak mosaic virus. Arch Virol 157:1371–1375
Chatenet M, Mazarin C, Girard JC, Fernandez E, Gargani D, Rao GP, Royer M, Lockhart B, Rott P (2005) Detection of Sugarcane streak mosaic virus in sugarcane from several Asian countries. Sugar Cane Int 23(4):12–15
Chung BYW, Miller WA, Atkins JF, Firth AE (2008) An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci USA 105:5897–5902
Damayanti TA, Putra LK (2011) First occurrence of Sugarcane streak mosaic virus infecting sugarcane in Indonesia. J Gen Plant Pathol 77:72–74
Fu WL, Sun SR, Fu HY, Chen RK, Su JW, Gao SJ (2015) A one-step real-time RT-PCR assay for the detection and quantitation of Sugarcane streak mosaic virus. Biomed Res Int 2015:569131
Gillaspie AG, Mock RG, Smith FF (1978) Identification of sugarcane mosaic virus and characterization of strains of the virus from Pakistan, Iran, and Cameroon. Proc Int Soc Sugar Cane Technol 16:347–355
Hall JS, Adams B, Parsons TJ, French R, Lane LC, Jensen SG (1998) Molecular cloning, sequencing, and phylogenetic relationships of a new potyvirus: Sugarcane streak mosaic virus, and a reevaluation of the classification of the Potyviridae. Mol Phylogen Evol 10:323–332
He Z, Li W, Yasaka R, Huang Y, Zhang Z, Ohshima K, Li S (2014) Molecular variability of Sugarcane streak mosaic virus in China based on an analysis of the P1 and CP protein coding regions. Arch Virol 159:1149–1154
He Z, Yasaka R, Li W, Li S, Ohshima K (2016) Genetic structure of populations of Sugarcane streak mosaic virus in China: Comparison with the populations in India. Virus Res 211:103–116
Hema M, Joseph J, Gopinath K, Sreenivasulu P, Savithri HS (1999) Molecular characterization and interviral relationships of a flexuous filamentous virus causing mosaic disease of sugarcane (Saccharum officinarum L.) in India. Arch Virol 144:479–490
Hema M, Savithri HS, Sreenivasulu P (2001) Sugarcane streak mosaic virus: occurrence, purification characterization and detection. In: Rao GP, Ford RE, Tosic M, Teakle DS (eds) Sugarcane pathology, vol 2., Virus and phytoplasma diseaseScience Publishers, Enfield, pp 37–70
Hema M, Sreenivasulu P, Savithri HS (2002) Taxonomic position of Sugarcane streak mosaic virus in the family Potyviridae. Arch Virol 147:1997–2007
Hudson RR (2000) A new statistic for detecting genetic differentiation. Genetics 155:2011–2014
Jensen SG, Hall JS (1993) Characterization of a krish-sorghum infecting potyvirus. Sorghum Newslett 34:17
Jensen SG, Hall JS (1993) Identification of a new sorghum infecting species of potyvirus from sugarcane. Phytopathology 83:884
Jridi C, Martin JF, Marie Jeanne V, Labonne G, Blanc S (2006) Distinct viral populations differentiate and evolve independently in a single perennial host plant. J Virol 80:2349–2357
Koike H, Gillaspie JR (1989) Mosaic. In: Ricaud C, Egan BT, Gillaspie AG (eds) Disease of sugarcane-major disease. Elsevier Science Publisher, Amesterdam, pp 301–322
Li WF, He Z, Li SF, Huang YK, Zhang ZX, Jiang D, Wang X, Luo Z (2011) Molecular characterization of a new strain of Sugarcane streak mosaic virus (SCSMV). Arch Virol 156:2101–2104
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
López C, Aramburu J, Galipienso L, Soler S, Nuez F, Rubio L (2011) Evolutionary analysis of tomato Sw-5 resistance-breaking isolates of Tomato spotted wilt virus. J Gen Virol 92:210–215
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654
Meirmans PG (2006) Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60:2399–2402
Nagy PD (2008) Recombination in plant RNA viruses. In: Roossinck MJ (ed) Plant virus evolution. New York: Springer, Berlin, New York, pp 133–156
Parameswari B, Bagyalakshmi K, Chinnaraja C, Viswanathan R (2014) Molecular characterization of Indian Sugarcane streak mosaic virus isolates reveals recombination and negative selection in the P1 gene. Gene 552:199–203
Parameswari B, Bagyalakshmi K, Viswanathan R, Chinnaraja C (2013) Molecular characterization of Indian Sugarcane streak mosaic virus isolate. Virus Genes 46:186–189
Pérez-Losada M, Porter M, Crandall KA (2008) Methods for analyzing viral evolution. In: Roossinck MJ (ed) Plant virus evolution. Springer, Berlin, pp 165–204
Riechmann JL, Lain S, Garcia JA (1992) Highlights and prospects of potyvirus molecular biology. J Gen Virol 73:1–16
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Seifers DL, Martin TJ, Harvey TJ, Fellers JP, Michaud JP (2009) Identification of the wheat curl mite as the vector of Triticum mosaic virus. Plant Dis 93:25–29
Singh D, Rao GP (2010) Sudan grass (Sorghum sudanense Stapf): a new Sugarcane streak mosaic virus mechanical host. Guangxi Agri Sci 41:436–438
Srinivas KP, Subba RCV, Ramesh B, Lava KP, Sreenivasulu P (2010) Identification of a virus naturally infecting sorghum in India as Sugarcane streak mosaic virus. Eur J Plant Pathol 127:13–19
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tatineni S, Qu F, Li R, Morris TJ, French R (2012) Triticum mosaic poacevirus enlists P1 rather than HC-Pro to suppress RNA silencing-mediated host defense. Virology 433:104–115
Tsompana M, Abad J, Purugganan M, Moyer JW (2005) The molecular population genetics of the Tomato spotted wilt virus (TSWV) genome. Mol Ecol 14:53–66
Viswanathan C, Prabu G (2015) Molecular diversity analysis of pretty interesting Potyviridae ORF (PIPO) coding region in Indian isolates of Sugarcane streak mosaic virus. Sugar Tech. doi:10.1007/s12355-015-0376-z
Viswanathan R, Balamuralikrishnan M, Karuppaiah R (2008) Characterization and genetic diversity of Sugarcane streak mosaic virus causing mosaic in sugarcane. Virus Genes 36:553–564
Walia JJ, Willemsen A, Elci E, Caglayan K, Falk BW, Rubio L (2014) Genetic variation and possible mechanisms driving the evolution of Fig mosaic virus. Phytopathology 104:108–114
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Xu DL, Zhou GH, Xie YJ, Mock R, Li R (2010) Complete nucleotide sequence and taxonomy of Sugarcane streak mosaic virus, member of a novel genus in the family Potyviridae. Virus Genes 40:432–439
Acknowledgments
This research was funded by an Earmark Fund from the China Agriculture Research System (CARS-20-2-4) and grants from the National Natural Science Foundation of China (No. 31170345) and the Major Science and Technology Project of Fujian Province (No. 2015NZ0002-2) in China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2016_2810_MOESM1_ESM.docx
Supplementary material 1 Table S1 Sequence information for sugarcane streak mosaic virus and representative members of the family Potyviridae used in the present study (DOCX 35 kb)
705_2016_2810_MOESM2_ESM.docx
Supplementary material 2 Table S2 Nucleotide sequence identity ( %) of open reading frame (ORF) (above the diagonal line) and coat protein (CP) (below the diagonal line) sequences of isolates of typical viruses of different genera in the family Potyviridae (DOCX 22 kb)
Rights and permissions
About this article

Cite this article
Liang, SS., Alabi, O.J., Damaj, M.B. et al. Genomic variability and molecular evolution of Asian isolates of sugarcane streak mosaic virus. Arch Virol 161, 1493–1503 (2016). https://doi.org/10.1007/s00705-016-2810-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2810-2