
Abstract
Explicit evaluation of the accuracy and power of maximum likelihood and Bayesian methods for detecting site-specific positive Darwinian selection presents a challenge because selective consequences of single amino acid changes are generally unknown. We exploited extensive molecular and functional characterization of amino acid substitutions in the plant gene eIF4E to evaluate the performance of these methods in detecting site-specific positive selection. We documented for the first time a molecular signature of positive selection within a recessive resistance gene in plants. We then used two statistical platforms, Phylogenetic Analysis Using Maximum Likelihood and Hypothesis Testing Using Phylogenies (HyPhy), to look for site-specific positive selection. Their relative power and accuracy are assessed by comparing the sites they identify as being positively selected with those of resistance-determining amino acids. Our results indicate that although both methods are surprisingly accurate in their identification of resistance sites, HyPhy appears to more accurately identify biologically significant amino acids using our data set.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ability to detect positive selection at the molecular level has expanded rapidly as both the number of complete genome sequences and the availability of sophisticated statistical methods have increased. Maximum likelihood (ML) and Bayesian estimators have been a critical development for inferring positive Darwinian selection at the molecular level (Ford 2002; Nielsen and Yang 1998). Two commonly used statistical program packages for determining specific sites under positive selection include Phylogenetic Analysis Using Maximum Likelihood (PAML) (Yang 1997; Yang et al. 2000, 2005) and Hypothesis Testing Using Phylogenies (HyPhy) (Pond et al. 2005). These methods are attractive because they enable identification of individual codon sites under positive selection and eliminate the assumptions about population demography associated with other statistical tests of selection (McDonald and Kreitman 1991; Tajima 1989; Yang 2002). The more recently developed fixed effects likelihood (FEL) approach, applied through HyPhy, is similar but makes no previous assumptions concerning the distribution of rates across the gene. Rates are estimated independently at each site using the modified Suzuki and Gojobori method (Kosakovsky Pond and Frost 2005; Suzuki 2004; Suzuki and Gojobori 1999). There seems to be no clear consensus regarding which of these methods is more appropriate for a given data set, but it is thought generally that PAML methods are more accurate for large data sets and prone to false-positive results with small data sets (Kosakovsky Pond and Frost 2005).
A body of literature exists that examines the power and accuracy of statistical methods for detecting positive selection (Yang 2002; Yang and Bielawski 2000). Approaches to assessing these methods generally fall into two categories: those that rely on computer simulation (Anisimova et al. 2001; Anisimova et al. 2002) and those that rely on empirical testing of genes suspected of being positively selected (Sorhannus 2003; Suzuki and Nei 2004). Both approaches are problematic. The former produces modeled results that are unconvincing without accompanying empirical data, and the latter identifies positively selected amino acids but offers no experimental evidence that significant fitness consequences exist as a result of modifications in those amino acids. Although they are often able to show which methods are more or less powerful, they provide no information regarding accuracy because they have no way of distinguishing between true- and false-positive results (Suzuki and Nei 2004).
ML and Bayesian methods are frequently used to localize the action of positive selection to specific codons and to show tight association between these sites and functionally important regions of the protein they encode (Hughes and Nei 1988; Meyers et al. 1998; Savage and Miller 2006). However, positive selection acting directly on functionally significant amino acids is rarely demonstrated because the fitness-related phenotypic consequences of individual amino acids are usually unknown (Bishop 2005). Consequently, the accuracy of site-specific tests of selection remains fundamentally in question. These criticisms compelled us to seek a system in which the fitness consequences of particular amino acids are well documented. If any sites are positively selected along such a gene, it is likely that the sites involved increase fitness.
A well-characterized plant virus-resistance gene encodes the eukaryotic translation initiation factor 4E (eIF4E), which functions to bind the 5′ cap of messenger RNAs and recruit them to the ribosomal complex (Gingras et al. 1999). Successful potyvirus (Potyviridae) infection of host plants requires that the virus usurp this process: viral RNA binds directly to the eIF4E plant protein, a process mediated by a viral-encoded protein (VPg) linked to the 5′ end of the viral RNA (Schaad et al. 2000; Shahabuddin et al. 1988). This binding ensures that viral RNA is recruited to the ribosomal complex and is translated. If this interaction is disrupted by natural mutations in eIF4E, the plant is resistant to infection (Leonard et al. 2000). Virus resistance has evolved independently at the eIf4E locus in several plant species, suggesting that there is a strong evolutionary pressure to resist viral pathogens. This is especially remarkable given that resistance at this locus behaves recessively, and therefore natural selection can only act to preserve beneficial mutations at this site when in the homozygous recessive state. Several of the plant species known to have evolved recessive resistance genes also contain multiple alleles, each with a unique resistance spectra, as a result of coevolution with viral pathogens (Charron et al. 2008). VPg, the gene encoding the viral protein that directly binds to eIF4E and functions as the pathogenicity determinant, was shown to be positively selected in a previous study (Moury et al. 2004). These observations suggest that the plant species investigated have experienced strong positive selection in their evolutionary history. We hypothesize that the statistical models that identify positively selected amino acids in eIF4E will also accurately predict the amino acids that are critical for the resistance phenotype. Currently, a time-consuming reverse-genetics approach is the only means by which to identify the amino acids responsible for the resistance phenotype that differ among various plant species and even among alleles within the same plant species. The identification of positively selected amino acids may be an alternative approach.
In this study, we focused on eIF4E resistance alleles in pepper (Charron et al. 2008; Kang et al. 2005a; Ruffel et al. 2002), tomato (Ruffel et al. 2005), and pea (Gao et al. 2004). The individual amino acid changes that define resistant and susceptible eIF4E alleles have been carefully detailed genetically (Kang et al. 2005a, b; Ruffel et al. 2002), biochemically (Kang et al. 2005a; Yeam et al. 2007), and by functional complementation in plants (Gao et al. 2004; Ruffel et al. 2002, 2005; Yeam et al. 2007). This precise knowledge of the effects on plant survival because of single substitutions in eIF4E provides an a priori expectation, thus allowing us to assess the accuracy of two popular methods for inferring positive selection. Both PAML Model 8 and HyPhy FEL were compared for their ability to identify positively selected amino acids that colocalize with those directly involved in providing resistance against viral pathogens. We documented for the first time that positive selection is acting on a recessive resistance gene. We found that both methods are able to detect resistance-determining sites accurately but that HyPhy FEL does so with greater precision and power.
Methods
Sequence Data and Tree Construction
The eIF4E cDNA data set used in this study was compiled from previously published sequences from pepper (Charron et al. 2008; Kang et al. 2005a; Ruffel et al. 2002), pea (Gao et al. 2004), and tomato (Ruffel et al. 2005). A total of 23 sequences were used: 11 from pepper, 8 from pea, and 4 from tomato (Fig. 1). Accession numbers are available in Supplementary Table 1. The terminal stop codon was removed, and sequences were aligned by eye using Sequencer v4.8. Three single amino acid insertions existed in Solanum relative to Capsicum and Pisum (Fig. 2). A phylogenetic tree was constructed for use in PAML and HyPhy with the program MrBayes v3.1.2 (Huelsenbeck and Ronquist 2001). MrBayes was run with 4 chains for 106 generations with a sample frequency of 100 and a 25% burn-in. The analysis was repeated 4 times to make sure that the trees generated were not clustered around local optima. A consensus of all 30,000 trees was generated using PAUP v4.0beta (Swofford 2002). The consensus tree was viewed with TreeView v1.6.6 (Page 1996). The tree was inspected before continuing with our analysis to ensure that eIF4E from the same species grouped together.

MrBayes tree of eIF4E. All credibility values >70 are shown. The analysis was run with 4 chains and a burn-in of 25%. The naming system of each allele consists of a two-letter abbreviation of the species, with the genotype indicated in quotes and the allele indicated in italics. Those starting with a capital letter and ending with a ‘+’ are the susceptible wild-type alleles. An asterisk denotes alleles with known resistance against at least one virus strain. Ca = Capsicum annuum; Cc = C. chinense; Ps = Pisum sativum; Sh = Solanum habrochaites; Sl = S. lycopsersicum. The scale bar is in nucleotide substitutions per site (693 nucleotides/sequence)

Alignment of pepper, pea, and tomato. P = sites detected by PAML Model 8 BEB with dN/dS > 1. H = sites detected as being positively selected using HyPhy’s FEL method. A solid circle shows which amino acids are polymorphic between the 3 species. Darkened squares show sites that are polymorphic between virus susceptible and resistant alleles within the associated genera. * Posterior p > 0.9. ** Posterior p > 0.95
Statistical Analysis Implementation
Average ω was measured in DnaSP v4.50 (Rozas and Rozas 1995) using the modified Nei-Gojobori model with Jukes-Cantor correction. Positively (dN/dS > 1) and negatively (dN/dS < 1) selected sites were detected with the program package PAML version 3.13d (Yang 1997; Yang et al. 2000), using both the M2 and M8 models, and the program package HyPhy (Pond et al. 2005), using the FEL model. PAML compares the maximum likelihood estimators of dN and dS across an alignment to a predefined distribution and uses empirical Bayes methods to identify individual positively selected sites (Nielsen and Yang 1998; Yang et al. 2000). Empirical Bayes may be performed using either the Naïve Empirical Bayes (NEB) approach or the Bayes Empirical Bayes (BEB) approach, which accounts for sampling errors in the estimates of model parameters (Yang et al. 2005). Log-likelihood scores were generated in PAML for models of neutral (M1 and M7) and positive (M2 and M8) selection and compared using likelihood ratio tests (LRTs). NEB and BEB posterior probabilities were calculated for sites with ω > 1 under M2 or M8. HyPhy FEL was run on a computer cluster through the Web-based interface, which is available at http://www.datamonkey.org (Pond and Frost 2005).
Results
The phylogenetic tree generated with MrBayes has high support at major branches and separates eIF4E perfectly by species (Fig. 1). Short branch lengths demonstrate a low degree of polymorphism within species. This is interesting given the dramatic differences in phenotypic effects of these alleles, i.e., virus resistance or susceptibility. The total tree length (the sum of all branch lengths) is 0.604. This Bayesian phylogeny was used in the PAML and HyPhy programs to investigate dN/dS ratios that would identify a molecular signature of positive selection.
Strong Evidence of Positive Selection on the Recessive Resistance Gene eIF4E
Positive selection is detected statistically as a nonsynonymous-to-synonymous nucleotide-substitution rate ratio (ω) significantly greater than 1. Across our 231 amino acid alignment of 23 complete pepper, tomato, and pea eIF4E coding sequences, ω is 0.236. This low value is consistent with previous analyses of eIF4E across eukaryotes (Athey-Pollard et al. 2002; Gao et al. 2004; Marcotrigiano et al. 1997). An excess of synonymous substitutions is not surprising because many coding sites experience purifying selection to maintain eIF4E’s pleiotropic function of translation initiation. Thus, log-LRTs were used to search for a molecular signature of positive selection acting on sites within the eIF4E coding region. Because no a priori expectation exists for the distribution of ω values for any given alignment, we compared likelihood values for two pairs of models with different assumed ω distributions: (1) M1 (a model of neutral evolution where all sites are characterized by having ω < 1 or ω = 1) versus M2 (a model of positive selection allowing sites to have ω > 1), which assumes that ω values are drawn from a normal distribution and (2) M7 versus M8, models that mirror the evolutionary constraints of M1 and M2 but assume that ω values are drawn from a beta distribution (Nielsen and Yang 1998). For each LRT, the model allowing sites to be under positive selection (M2 or M8) fit the eIF4E data from potato, pepper, and pea significantly better than the neutral model (M1 or M7) (p < 0.0001; Table 1). Under M2, most sites within the eIF4E coding sequence experience purifying selection (ω < 72.1%) or neutral evolution (ω = 1; 24.6%), whereas few codons have a signature of positive selection (ω > 1; 3.3%). However, the mean ω value for sites under positive selection is 6.85, nearly a 7-fold excess of nonsynonymous-to-synonymous substitutions, indicating that eIF4E has a molecular signature of strong positive selection targeted to a small subset of sites. The proportion of sites falling into each ω class was similar under M8 (Table 1).
Positively Selected Sites Colocalize With Resistance-Determining Amino Acids
Sixteen amino acid sites across the eIF4E coding sequence are known to be associated with virus resistance in pepper, tomato, or pea (Fig. 2). The M8 model using both NEB and BEB assigned 10 amino acid sites to the positive selection class, 7 of which were associated with resistance alleles (Fig. 2; Table 2 [heading “A”]). Sites placed into the positively selected class are nonrandomly distributed with respect to those involved in virus resistance (Table 2 [heading “B”]; Fisher’s exact test p < 0.001).
ML models including positive selection (M2 and M8) fit the data significantly better than their corresponding neutral models (M1 and M7) (Table 1). Therefore, site-specific tests for positive selection using M8 NEB and BEB with PAML and FEL with HyPhy were used to calculate posterior probabilities for each positively selected codon.
All three of the sites identified using FEL with posterior probabilities > 0.9 (sites 76, 77, and 110) colocalize with resistance-determining amino acids (Table 2). Site 76 was identified by NEB, BEB, and FEL and corresponds to an alanine-to-aspartic acid mutation in pepper resistance alleles pvr1 5, pvr1 6, and pvr1 9 (Charron et al. 2008), an alanine-to-aspartic acid mutation in the pea allele sbm1 1, and an alanine-to-proline mutation in the pea allele sbm1 2 relative to the wild type (Gao et al. 2004). Site 77 is an alanine-to-aspartic acid mutation in the pepper resistance allele pvr1 6 (Charron et al. 2008), an alanine-to-aspartic acid mutation in the pea resistance alleles sbm1 1 and sbm1 2 (Gao et al. 2004), and an alanine-to-aspartic acid mutation in the tomato resistance allele pot-1 (Ruffel et al. 2005). Finally, site 110 corresponds to a glycine-to-arginine mutation in pepper resistance alleles pvr1 from Capsicum chinense and pvr1 9 from Capsicum annuum (Kang et al. 2005a; Charron et al. 2008) and a glycine-to-arginine mutation in the pea resistance allele sbm1 1 (Gao et al. 2004).
One of the two codon sites with M8 NEB or BEB posterior probabilities > 0.9 (equivalent to p < 0.1; sites 76 and 209) is a resistance-determining amino acid (Table 2). Position 76, discussed previously, is present in several resistance alleles. Although site 209 is not associated with a resistance allele, site 208 directly adjacent is an aspartic acid-to-glycine mutation found in pepper resistance alleles pvr1 3, pvr1 6, pvr1 7, and pvr1 9 (Charron et al. 2008).
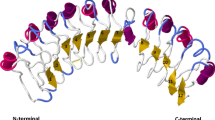
Using the estimated three-dimensional crystal structure of Capsicum eIF4E generated by Yeam et al. (2007), we were able to map positively selected sites on the protein tertiary structure. The sites detected as positively selected cluster in the region of the protein that contains resistance-determining sites and is known interact with the virus (Fig. 3). This region is also involved in binding the 5′ cap of the messenger RNA, raising interesting questions regarding maintaining eIF4E’s ability to function in host RNA translation initiation (Yeam et al. 2007).

Positively selected sites mapped onto the hypothesized three-dimensional structure reported in Yeam et al. (2007). Spacefill red = resistance-associated amino acids. Spacefill green = positively selected site identified by PAML. Spacefill purple = positively selected site identified by both HyPhy and PAML. Red stippling shows sites that are positively selected and resistance associated. The first 60 amino acids are not included because the crystal structure of that region is unresolved. Amino acid positions of positively selected sites are shown in white text.
* Posterior p (either PAML or HYPHY) > 0.9. ** Posterior p (either PAML or HYPHY) >0.95
HyPhy FEL More Accurately Detects Resistance Sites Than PAML Model 8
Both PAML Model 8 and HyPhy FEL identify positively selected amino acids known to be associated with virus resistance. PAML places 10 amino acids into the positively selected class with dN/dS >1, 7 of which are sites involved in virus resistance. Two of the remaining 3 amino acids not known to be involved in virus resistance are directly adjacent to ones that are involved (Fig. 2).
When posterior probabilities are applied using empirical Bayes, only one of the two statistically significant sites detected is a resistance-determining site. HyPhy FEL, in contrast, identifies three sites with posterior probability >0.9, all of which are resistance-determining sites (Table 2 and Fig. 2). Considering only the sites with posterior probability > 0.9, we therefore conclude that although both methods accurately identify functionally significant sites as being under positive selection, HyPhy FEL does so with higher power (three sites identified rather than two) and accuracy (three of three opposed to one of two known resistance sites) for our data set.
Discussion
Strong Evidence That the Recessive Resistance Gene eIF4E Is Under Positive Selection
In this study, we showed that the plant resistance gene eIF4E is under positive selection based on statistical analyses that consider the rate of accumulation of synonymous and nonsynonymous nucleotide substitutions across an alignment consisting of susceptible and resistant alleles from pepper, tomato, and pea. Positive selection on a recessive resistance gene has never before been documented. Previous studies examining plant defense have focused on genes involved in pathogen detection, intercellular signaling, or genes antagonistic to the pathogen or pest (Tiffin and Moeller 2006). Studies on the evolution of plant defenses have focused largely on gene-for-gene resistance (Stahl and Bishop 2000) but evolution of other positively selected defense genes, including chitinases (Bishop et al. 2000; Tiffin 2004), β-1,2-endoglucanases (Bishop et al. 2005), polygalacturanase inhibitor proteins (Bishop 2005; Stotz et al. 2000), proteinases, proteinase inhibitors, and lectins (Roth and Liberles 2006), has also been investigated. These resistance mechanisms function primarily against bacteria and fungi, are inherited dominantly, and involve the modification of a gene product to fulfill a new role. In contrast, many viral resistance genes are recessively inherited (Kang et al. 2005b; Provvidenti and Hampton 1992; Robaglia and Caranta 2006) and are presumably involved in modifying a host protein to avoid interaction with viral pathogenicity factors (Diaz-Pendon et al. 2004; Fraser 1990). These genes, like eIF4E, are typically host factors involved in basic cellular processes that are used by the virus to complete its life cycle. They function as resistance genes pleiotropically once natural mutations arise that prevent viruses from using them. Because they are recessively inherited, natural selection can act on resistance alleles only when they are present in the homozygous state. In this regard it is remarkable that a signature of positive selection was detected at eIF4E given that heterozygotes would not possess a selective advantage even in the presence of viral pathogens.
Tests of Positive Selection Accurately Identify Resistance-Determining Codons
The amino acids detected as being positively selected represent one of a few amino acid changes found in resistance alleles relative to susceptible wild-type sequences. Site 76 is one of three amino acid changes in pvr1 9 responsible for providing resistance to two strains of Potato virus Y (PVY) (Charron et al. 2008). Sites 76 and 77 are two of five and two of three polymorphisms between wild-type pea eIF4E and resistance alleles sbm1 1 and sbm1 2, respectively (Gao et al. 2004). Similarly, site 77 is one of four amino acid changes in pvr1 6 and one of four amino acid changes in resistance allele pot-1 (Charron et al. 2008). For site 110, we have direct empirical evidence linking this single amino acid specifically with virus resistance. When the glycine-to-arginine mutation was induced at site 110 in susceptible pepper eIF4E, the protein–protein interaction with VPg was interrupted and, when expressed transgenically in tomato plants, conferred resistance to three strains of Tobacco etch virus (Yeam et al. 2007).
It is interesting that unique mutations in different species tend to be similar among the positively selected sites. In several cases, the specific amino acid change between wild-type and mutant alleles is the same in different species. For instance, site 76 is an alanine in wild-type alleles from both pepper and pea that mutates to aspartic acid in resistance alleles from both species. Site 77 is an alanine-to-aspartic acid mutation in resistance alleles in all three species considered. Finally, the amino acid change at site 110 is a glycine-to-arginine change in resistance alleles from both pepper and pea. This suggests that only particular amino acid changes are sufficient to confer virus resistance. Mutations at those sites that produce other amino acids would tend to be lost by genetic drift or purifying selection if they did not confer a selective advantage by providing resistance to invading viruses.
Coevolutionary History of Virus and Host Drive Positive Selection of eIF4E
Positive selection at eIF4E is particularly interesting considering that the viral protein VPg, which interacts directly with eIF4E in susceptible host–pathogen combinations (Schaad et al. 2000), is also positively selected, and the specific viral amino acids under positive selection are found in regions of the protein known to be virulence determinants (Moury et al. 2004). The data we present for positive selection acting within eIF4E as well as VPg therefore provides empirical evidence supporting the hypothesis that a coevolutionary “arms race” occurs between host and pathogen in this system (Charron et al. 2008; Dawkins and Krebs 1979). As expected under an “arms race” scenario, multiple resistance alleles with unique resistance spectra and multiple viral strains with unique infectivity are known to exist (Charron et al. 2008; Kang et al. 2005a; Kyle and Palloix 1997; Ruffel et al. 2002). This has been intensively studied in pepper, where nine distinct resistance alleles were investigated for resistance to four viral strains (Charron et al. 2008). Experimental modification of many of the amino acids involved in this protein–protein interaction proves that these sites are directly involved in virus resistance. Positively selected codons of VPg are directly involved in overcoming resistance eIF4E alleles (Ayme et al. 2007; Moury et al. 2004). In this study, we found that amino acids involved in host plant resistance are positively selected. The detailed evolutionary knowledge of eIF4E and VPg makes this coevolutionary pathosystem among the best-studiedin all of biology.
Potential Effect of Positively Selected Sites on Translation Initiation
It is interesting to consider what role positively selected amino acids from eIF4E play in host translation initiation. The region of eIF4E that binds the 5′ cap overlaps with the region involved in determining whether the plant is resistant to an invading virus. Sites required for translation initiation are strongly conserved given that recruiting mRNA to the ribosomal complex is an important function of plant cells that must be maintained (Marcotrigiano et al. 1997). When the G110R mutation was induced in wild-type pepper eIF4E, it lost its ability to bind both VPg and the 5′ cap (Yeam et al. 2007). In addition, the pvr1 allele from Capsicum chinense that contains this mutation and two others is also unable to bind the 5′ cap (Yeam et al. 2007). However, transgenic tomato plants containing pepper eIF4E with this mutation alone exhibit no obvious abnormalities (Yeam et al. 2007). In addition, homozygous pvr1 pepper plants do not appear to exhibit any negative phenotypic effects. Either the mutated form of eIF4E is still able to participate in translation initiation despite not being able to bind to the 5′ cap in vitro, or the presence of eIF4E paralogs are responsible for reducing the evolutionary constraints of this locus. Under the latter scenario, paralogs would take over the cap-binding function while the version of eIF4E involved in resistance is allowed to diverge (Robaglia and Caranta 2006). However, because nine other known resistance alleles in pepper are able to complement an eIF4E knockout mutant in yeast, it seems more likely that translation initiation function continues in resistance alleles (Charron et al. 2008).
HyPhy FEL Detects Resistance Sites More Accurately and With Higher Power
Identification of positively selected genes does not provide detailed understanding of adaptive change. We contest that several additional pieces of information are required. First, the area under positive selection must be further localized to specific amino acids. Statistical procedures have been developed for this purpose (Massingham and Goldman 2005; Nielsen and Yang 1998; Suzuki and Gojobori 1999). These techniques provide information on the physical distribution of molecular evolution that may be used to better understand selective forces acting on a gene (Anisimova et al. 2002). In addition, changes in particular amino acids must be correlated with an associated change in phenotype (Golding and Dean 1998). In this study, we attempted to fulfill these criteria to confidently connect positive selection with causal forces. We have used popular statistical methods to identify site-specific positive selection of a well-studied resistance gene in plants. Both methods identified a small number of positively selected amino acid sites associated with resistance alleles. Our findings indicate that both PAML Model 8 and HyPhy FEL accurately and precisely pinpoint single amino acids that are known to function in resistance alleles to interrupt the virus life cycle. However, HyPhy FEL does so with greater power and precision because all three amino acids predicted to be positively selected are associated with virus resistance. We conclude, therefore, that HyPhy FEL is an appropriate method for similar data sets to identify sites having biologic relevance to the fitness of the host.
Conclusion
Interaction with translation initiation factors appears to be a common infection strategy for viruses that infect plants (Schaad et al. 2000). Amino acid changes in eIF4E that result in resistance to a number of viral families have evolved independently in a variety of plant taxa (Charron et al. 2008; Gao et al. 2004; Kang et al. 2005a; Morales et al. 2005; Nicaise et al. 2003; Nieto et al. 2006; Ruffel et al. 2002, 2005; Wicker et al. 2005). The results of this study provide some insight into the driving evolutionary forces responsible for this process. In addition, our results support the use of ML and Bayesian methods for detecting site-specific positive selection, particularly in natural populations where the fitness consequences of single amino acid changes cannot be demonstrated experimentally. In conclusion, we suggest that the accurate identification of amino acids with dramatic biologic consequences in this system make these methods a powerful predictive tool for targets of genetic crop improvement for a number of agronomically important traits.
References
Anisimova M, Bielawski JP, Yang Z (2001) Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol 18:1585–1592
Anisimova M, Bielawski JP, Yang Z (2002) Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol 19:950–958
Athey-Pollard AL, Kirby M, Potter S, Stringer C, Mills PR, Foster GD (2002) Comparison of partial sequence of the cap binding protein (eIF4E) isolated from Agaricus bisporus and its pathogen Verticillium fungicola. Mycopathologia 156:19–23
Ayme V, Petit-Pierre J, Souche S, Palloix A, Moury B (2007) Molecular dissection of the potato virus Y VPg virulence factor reveals complex adaptations to the pvr2 resistance allelic series in pepper. J Gen Virol 88:1594–1601
Bishop JG (2005) Directed mutagenesis confirms the functional importance of positively selected sites in polygalacturonase inhibitor protein. Mol Biol Evol 22:1531–1534
Bishop JG, Dean AM, Mitchell-Olds T (2000) Rapid evolution in plant chitinases: molecular targets of selection in plant-pathogen coevolution. Proc Natl Acad Sci USA 97:5322–5327
Bishop JG, Ripoll DR, Bashir S, Damasceno CM, Seeds JD, Rose JK (2005) Selection on Glycine beta-1, 3-endoglucanase genes differentially inhibited by a Phytophthora glucanase inhibitor protein. Genetics 169:1009–1019
Charron C, Nicolai M, Gallois JL, Robaglia C, Moury B, Palloix A, Caranta C (2008) Natural variation and functional analyses provide evidence for co-evolution between plant eIF4E and potyviral VPg. Plant J 54:56–68
Dawkins R, Krebs JR (1979) Arms races between and within species. Proc R Soc Lond B Biol Sci 205:489–511
Diaz-Pendon JA, Truniger V, Nieto C, Garcia-Mas J, Bendahmane A, Aranda MA (2004) Advances in understanding recessive resistance to plant viruses. Mol Plant Pathol 5:223–233
Ford MJ (2002) Applications of selective neutrality tests to molecular ecology. Mol Ecol 11:1245–1262
Fraser RSS (1990) The genetics of resistance to plant viruses. Annu Rev Phytopathol 28:179–200
Gao Z, Johansen E, Eyers S, Thomas CL, Noel Ellis TH, Maule AJ (2004) The potyvirus recessive resistance gene, sbm1, identifies a novel role for translation initiation factor eIF4E in cell-to-cell trafficking. Plant J 40:376–385
Gingras AC, Raught B, Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68:913–963
Golding GB, Dean AM (1998) The structural basis of molecular adaptation. Mol Biol Evol 15:355–369
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hughes AL, Nei M (1988) Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335:167–170
Kang BC, Yeam I, Frantz JD, Murphy JF, Jahn MM (2005a) The pvr1 locus in Capsicum encodes a translation initiation factor eIF4E that interacts with Tobacco etch virus VPg. Plant J 42:392–405
Kang BC, Yeam I, Jahn MM (2005b) Genetics of plant virus resistance. Annu Rev Phytopathol 43:581–621
Kosakovsky Pond SL, Frost SDW (2005) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Kyle MM, Palloix A (1997) Proposed revision of nomenclature for potyvirusresistance genes in Capsicum. Euphytica 97:183–188
Leonard S, Plante D, Wittmann S, Daigneault N, Fortin MG, Laliberte JF (2000) Complex formation between potyvirus VPg and translation eukaryotic initiation factor 4E correlates with virus infectivity. J Virol 74:7730–7737
Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK (1997) Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89:951–961
Massingham T, Goldman N (2005) Detecting amino acid sites under positive selection and purifying selection. Genetics 169:1753–1762
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654
Meyers BC, Shen KA, Rohani P, Gaut BS, Michelmore RW (1998) Receptor-like genes in the major resistance locus of lettuce are subject to divergent selection. Plant Cell 10:1833–1846
Morales M, Orjeda G, Nieto C, van Leeuwen H, Monfort A, Charpentier M, Caboche M, Arus P, Puigdomenech P, Aranda MA et al (2005) A physical map covering the nsv locus that confers resistance to melon necrotic spot virus in melon (Cucumis melo L.). Theor Appl Genet 111:914–922
Moury B, Morel C, Johansen E, Guilbaud L, Souche S, Ayme V, Caranta C, Palloix A, Jacquemond M (2004) Mutations in potato virus Y genome-linked protein determine virulence toward recessive resistances in Capsicum annuum and Lycopersicon hirsutum. Mol Plant Microbe Interact 17:322–329
Nicaise V, German-Retana S, Sanjuan R, Dubrana MP, Mazier M, Maisonneuve B, Candresse T, Caranta C, LeGall O (2003) The eukaryotic translation initiation factor 4E controls lettuce susceptibility to the potyvirus lettuce mosaic virus. Plant Physiol 132:1272–1282
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Nieto C, Morales M, Orjeda G, Clepet C, Monfort A, Sturbois B, Puigdomenech P, Pitrat M, Caboche M, Dogimont C et al (2006) An eIF4E allele confers resistance to an uncapped and non-polyadenylated RNA virus in melon. Plant J 48:452–462
Page RD (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Pond SL, Frost SD (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Pond SL, Frost SD, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Provvidenti R, Hampton RO (1992) Sources of resistance to viruses in the Potyviridae. Arch Virol Suppl 5:189–211
Robaglia C, Caranta C (2006) Translation initiation factors: a weak link in plant RNA virus infection. Trends Plant Sci 11:40–45
Roth C, Liberles DA (2006) A systematic search for positive selection in higher plants (Embryophytes). BMC Plant Biol 6:12
Rozas J, Rozas R (1995) DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data. Comput Appl Biosci 11:621–625
Ruffel S, Dussault MH, Palloix A, Moury B, Bendahmane A, Robaglia C, Caranta C (2002) A natural recessive resistance gene against potato virus Y in pepper corresponds to the eukaryotic initiation factor 4E (eIF4E). Plant J 32:1067–1075
Ruffel S, Gallois JL, Lesage ML, Caranta C (2005) The recessive potyvirus resistance gene pot-1 is the tomato orthologue of the pepper pvr2-eIF4E gene. Mol Genet Genomics 274:346–353
Savage AE, Miller JS (2006) Gametophytic self-incompatibility in Lycium parishii (Solanaceae): allelic diversity, genealogical structure, and patterns of molecular evolution at the S-RNase locus. Heredity 96:434–444
Schaad MC, Anderberg RJ, Carrington JC (2000) Strain-specific interaction of the tobacco etch virus NIa protein with the translation initiation factor eIF4E in the yeast two-hybrid system. Virology 273:300–306
Shahabuddin M, Shaw JG, Rhoads RE (1988) Mapping of the tobacco vein mottling virus VPg cistron. Virology 163:635–637
Sorhannus U (2003) The Effect of positive selection on a sexual reproduction gene in Thalassiosira weissflogii (Bacillariophyta): results obtained from maximum-likelihood and parsimony-based methods. Mol Biol Evol 20:1326–1328
Stahl EA, Bishop JG (2000) Plant-pathogen arms races at the molecular level. Curr Opin Plant Biol 3:299–304
Stotz HU, Bishop JG, Bergmann CW, Koch M, Albersheim P, Darvill AG, Labavitch JM (2000) Identification of target amino acids that affect interactions of fungal polygalacturonases and their plant inhibitors. Physiol Mol Plant Pathol 56:117–130
Suzuki Y (2004) New methods for detecting positive selection at single amino acid sites. J Mol Evol 59:11–19
Suzuki Y, Gojobori T (1999) A method for detecting positive selection at single amino acid sites. Mol Biol Evol 16:1315–1328
Suzuki Y, Nei M (2004) False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Mol Biol Evol 21:914–921
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (and other methods), version 4. Sinauer Associates, Sunderland, MA
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tiffin P (2004) Comparative evolutionary histories of chitinase genes in the Genus zea and Family poaceae. Genetics 167:1331–1340
Tiffin P, Moeller DA (2006) Molecular evolution of plant immune system genes. Trends Genet 22:662–670
Wicker T, Zimmermann W, Perovic D, Paterson AH, Ganal M, Graner A, Stein N (2005) A detailed look at 7 million years of genome evolution in a 439 kb contiguous sequence at the barley Hv-eIF4E locus: recombination, rearrangements and repeats. Plant J 41:184–194
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z (2002) Inference of selection from multiple species alignments. Curr Opin Genet Dev 12:688–694
Yang Z, Bielawski JP (2000) Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15:496–503
Yang Z, Nielsen R, Goldman N, Pedersen A-MK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Yeam I, Cavatorta JR, Ripoll DR, Kang BC, Jahn MM (2007) Functional dissection of naturally occurring amino acid substitutions in eIF4E that confers recessive potyvirus resistance in plants. Plant Cell 9:2913–2928
Acknowledgments
We thank G. Stellari, K. Zamudio, and A. Agrawal for comments. This work was supported by grants from the United States Department of Agriculture, the Kwanjeoung Educational Foundation, and the National Science Foundation Graduate Research Fellowship Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. R. Cavatorta and A. E. Savage have contributed equally to this work.
Electronic Supplementary Material
239_2008_9172_MOESM1_ESM.doc
MOESM1 [Table 1: Sequences used for constructing phylogenetic tree and running site-specific test of positive selection] (DOC 40 kb)
Rights and permissions
About this article
Cite this article
Cavatorta, J.R., Savage, A.E., Yeam, I. et al. Positive Darwinian Selection at Single Amino Acid Sites Conferring Plant Virus Resistance. J Mol Evol 67, 551–559 (2008). https://doi.org/10.1007/s00239-008-9172-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-008-9172-7