
Abstract
Non-coding RNAs (ncRNAs) are a large cluster of RNAs that do not encode proteins, but have multiple functions in diverse cellular processes. Mounting evidence indicates the involvement of ncRNAs in the physiology and pathophysiology of the central and peripheral nervous systems. It has been shown that numerous ncRNAs, especially microRNAs and long non-coding RNAs, are differentially expressed after insults such as acquired brain injury, spinal cord injury, and peripheral nerve injury. These ncRNAs affect neuronal survival, neurite regrowth, and glial phenotype primarily by targeting specific mRNAs, resulting in translation repression or degradation of the mRNAs. An increasing number of studies have investigated the regulatory roles of microRNAs and long non-coding RNAs in neural injury and regeneration, and thus a new research field is emerging. In this review, we highlight current progress in the field in an attempt to provide further insight into post-transcriptional changes occurring after neural injury, and to facilitate the potential use of ncRNAs for improving neural regeneration. We also suggest potential directions for future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In clinical practice, injuries to the central and peripheral nervous systems (CNS and PNS) are commonly encountered. Injured CNS neurons are unable to regenerate their axons spontaneously because of suppression by glial scar-associated inhibitors and myelin-derived molecules as well as loss of the capacity for developmentally regulated intrinsic growth [1, 2]. In contrast, PNS neurons show a robust intrinsic regenerative capacity after traumatic injury, but functional outcomes are often unsatisfactory [3]. Accordingly, much research has been devoted to the development of therapeutic interventions to improve neural regeneration based on an understanding of the molecular mechanisms underlying the responses to injury and regeneration.
To initiate a regenerative response, the PNS neuron must shift from a transmitting state to a regenerative state, which requires the initiation of a growth program through gene transcription and the activation of local signaling cascades that control axon assembly. Knowledge of the gene transcription responses of PNS neurons to injury has provided insight into the genes associated with regeneration. Many regeneration-associated genes have been identified by examining gene expression changes after injury. These genes include transcription factors, such as ATF3, c-Jun, C/EBPb, CREB, NFIL3, p53, SMAD1, SOX11, STAT3, and KLF family members, while non-transcription factor “terminal” genes consist of those that encode adhesion/guidance molecules (integrin subunits and CD44), neuropeptides (VIP and CGRP), structural and cytoskeleton-associated proteins (GAP43, CAP23, SCG10, and CRMP2), and metabolic enzymes (arginase 1) [4]. In addition to increasing RAG expression, another approach to enhancing regeneration is to increase the “metabolic growth state” of neurons by up-regulating anabolic processes, such as mTOR activation of protein translation or transcriptional regulation of anabolic processes [5, 6].
Clearly, the molecular approaches to neural regeneration noted above are mainly based on transcriptional regulation. Recently, many studies on non-coding RNAs (ncRNAs) have revealed an emerging layer of post-transcriptional gene regulation for the post-neural-injury process. ncRNAs are a large cluster of RNAs that are not translated into proteins. As the sequencing of the human genome reported the surprising finding that about 20,000 protein-coding genes represent <2% of the total genomic sequence, the investigation of ncRNAs has attracted increasing attention because they have multiple functions at the transcriptional and post-transcriptional levels in diverse cellular processes [7].
Recently, a number of studies have shown that ncRNAs, mainly microRNAs and long ncRNAs, are differentially expressed in injured neural tissue after various types of injury. Dysregulation of ncRNA expression affects the survival and growth of neurons and regulates the phenotypic modulation of glial cells. These intriguing results contribute to the potential use of ncRNAs as diagnostic markers and therapeutic targets for neural injury. This review aims to summarize current research progress in understanding the involvement of ncRNAs in CNS and PNS injury and the effects of ncRNAs on neural regeneration. We also suggest potential directions for future research.
Classification of Non-coding RNAs
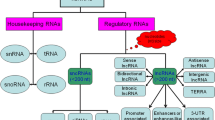
The ncRNAs have a high degree of heterogeneity in sequence, structure, and biological function. They are usually classified into subtypes according to various criteria. For instance, ncRNAs are divided into housekeeping and regulatory types according to their biological functions. The former includes ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), guide RNAs (gRNAs) and telomerase RNAs, while the latter includes microRNAs (miRNAs, miRs), small interfering RNAs (siRNAs), Piwi-interacting RNAs (piRNAs), and long chain non-coding RNAs (long non-coding RNAs, lncRNAs). Also, ncRNAs are divided into nuclear and cytoplasmic types according to subcellular localization, or divided into those with a polyA tail (polyA-plus ncRNAs) and those without (polyA-minus ncRNAs). In addition, ncRNAs are divided into short and long types according to the length of the transcript. The short type includes miRNAs, siRNAs, piRNAs, snRNAs, and snoRNAs [8], which are <200 nucleotides (nt) long, typically 20–30 nt, while the long type (lncRNAs) are >200 nt in length, and account for at least 80% of mammalian genome transcription [9].
In this review, we focus on two members of the ncRNA family to describe the regulation of neural regeneration by miRNAs at the post-transcriptional level and by lncRNAs at both the transcriptional and post-transcriptional levels [10].
miRNAs are a class of endogenous small single-strand ncRNAs of about 22 nt. They have been widely investigated since the first miRNA (lin-4) was identified. They are generated by RNA polymerase II, and their encoding sequence is often found in an intergenic region in the form of a single copy, multiple copies, or a gene cluster, while the encoding sequences of other miRNAs occur in the exon or intron regions of a gene. Mature miRNAs combine with Argonaute 1 to form an RNA-induced silencing complex (RISC) that is involved in the regulation of cellular life [11]. Put simply, the RISC influences the stability and translation of messenger RNA (mRNA) through direct effects on the 3′-untranslated region of the target mRNA, thereby resulting in translation repression or degradation of the mRNA. Importantly, miRNAs are abundant in the nervous system where they function in development [12] and maintenance of the neuronal phenotype [13], influence the maturation of dendrites and spines [14], and serve as effectors of synaptic plasticity and function [15, 16]. Lack of a specific miRNA, miRNA overexpression, or miRNA mutation may lead to abnormal cellular function and even neurological disorders [17].
lncRNAs are >200 nt in length but lack open reading frames. They can be classified on the basis of genomic location and biogenesis into (1) sense lncRNAs that are transcribed on the same strand of an exon; (2) antisense lncRNAs that are transcribed on the opposite strand of an exon; (3) bidirectional lncRNAs that are located on the opposite strand from a protein-coding gene whose transcription is initiated <1000 bp away; (4) intronic lncRNAs; (5) intergenic lncRNAs (also called long intergenic noncoding RNAs, lincRNAs) that occur between two genes; and (6) circular RNAs with exonic or intronic linear sequences that circularize after alternative splicing [18, 19]. lncRNAs were once considered to be intermediate products in the transcription process, and to have no biological functions. Nowadays, however, increasing evidence has shown that lncRNAs have complex functions, including activation or reduction of the expression of specific genes, especially adjacent protein-encoding genes, and are associated with the pathogenesis of some diseases.
Roles of miRNAs in Responses to Neural Injury
During neurogenesis, neuronal maturation, and brain development, miRNAs serve as fine regulators of genetic networks [20]. For the development and maintenance of neurons, miRNAs play roles in cell specification, axonal path-finding, and apoptosis [21, 22]. It is important to determine the dysregulation of miRNAs during neurodevelopmental abnormalities and neurodegenerative disorders such as fragile X syndrome, autism spectrum disorder, Rett syndrome, depression, drug addiction, Huntington’s disease, and schizophrenia [23–26]. Here, however, we focus on the involvement of miRNAs in the responses to various types of neural injury.
miRNAs in Acquired Brain Injury
Acquired brain injury (ABI) is defined as an injury to the brain occurring after birth. It is not a hereditary, congenital, or degenerative disease [27, 28], but is caused by stroke, hemorrhage, infection, or trauma. Several miRNAs, such as miR-9, miR-183, miR-134, miR-135, miR-124a, miR-124b, miR-153, and miR-219, are highly enriched and specifically located in the brain [29, 30]. The expression of miR-146b, miR-551b, miR-92b, and miR-384 is several-fold higher in the hippocampus than in the cortex [31]; miR-132, miR-212, miR-221, miR-222, and let-7 are predominantly expressed in forebrain regions; and miR-206 and miR-497 are mainly expressed in the cerebellum. This region-specific expression of miRNAs in the brain suggests that they play specific regulatory roles in ABI [32].
Ischemic Brain Injury
Stroke is a major cause of serious long-term disability after focal cerebral ischemic injury. Microarray data on a large scale has identified the miRNA expression profiles after middle cerebral artery occlusion (MCAO)-induced focal cerebral ischemia. It has been shown that 12 miRNAs are up-regulated and 18 are down-regulated in the infarct region after 6 h of MCAO [33]. The serum miR-126 levels appear to differ in permanent versus transient ischemia, and the changes in these levels may be used to distinguish severe permanent ischemia from transient ischemia [34].
miRNAs can regulate the ischemic brain injury caused by a thrombus, embolus, or other interruption of the arterial supply [35, 36]. Expression of the glutamate receptor subunits GluR2 and NR2B, together with N-methyl-d-aspartate receptor-mediated Ca2+ influx, is inhibited by the overexpression of miR-223, thereby protecting neurons from cell death [37]. Administration of anti-miR-320a leads to a reduction of infarct volume as well as an increase in the expression of aquaporins 1 and 4 after cerebral ischemia [38]. The expression of let-7c-5p is decreased in the plasma of patients with ischemic stroke, but its overexpression suppresses microglia activation against ischemic damage [39].
After ischemic brain injury, neuronal death is one of the most important events that influences recovery, and miRNAs regulate neuronal survival. Two key regulator of apoptosis, B cell lymphoma 2 (Bcl-2) and Bcl-w, are regulated by miR-15b [40], miR-29b [41], miR-181a [42], and miR-497 [43]. miR-181c suppresses the expression of tumor necrosis factor-α (a key pro-inflammatory cytokine) to protect neurons from cell death [44]. miR-592 decreases the expression of p75NTR, an ischemia-induced neurotrophin receptor, attenuates the activation of pro-apoptotic signaling pathways, and prevents neuronal apoptosis [45]. miR-134 down-regulates the expression of heat shock protein A12B (HSPA12B) and promotes neuronal death after ischemic injury [46].
After transient cerebral ischemia, miR-200c expression in the brain increases rapidly, contributing to cell death by inhibiting Reelin expression [47]. Down-regulation of miR-30a expression prevents neuronal ischemic injury by up-regulating the expression of HSPA5, while decreased endoplasmic reticulum stress-induced apoptosis might be one of the mechanisms underlying HSPA5-mediated neuroprotection [48, 49].
miR-424 and miR-23a-3p inhibit neuronal apoptosis after ischemia, reduce the levels of reactive oxygen species in cortex, and abrogate H2O2-induced injury by increasing cellular viability and manganese superoxide dismutase activity [50, 51]. miR-134 regulates ischemic/reperfusion injury-induced neuronal death via CREB (cAMP response element-binding protein) signaling [46, 52]. miR-22 inhibits nuclear factor-κB activity by decreasing the nuclear receptor coactivator 1 expression and caspase-3 activity and thus reduces cortical neuronal apoptosis [53]. miR-23a/b and miR-27a/b suppress Apaf-1 (apoptotic protease activating factor 1) protein and alleviate the neuronal apoptosis induced by intrauterine hypoxia [54]. miR-124 targets Ku70 to improve ischemia/reperfusion-induced brain injury and dysfunction [55].
Collectively, the above findings confirm that miRNAs are key regulators of neuronal cell death. As potential targets for promoting neuronal survival, miRNAs contribute to recovery after ischemic brain injury.
Traumatic Brain Injury
In contrast to ischemic brain injury that generally occurs in the older population, traumatic brain injury (TBI) is a leading cause of death, disability, and cognitive impairment in children and young adults.
Microarray analysis has shown that many miRNAs exhibit differential expressions after TBI. Since TBI is attenuated by hypothermia, it may be linked to the temperature-sensitive miRNAs [56–58]. Mitochondria-associated miRNAs, such as miR-155 and miR-223, both of which play roles in inflammatory processes, are significantly dysregulated in the hippocampus after TBI [59]. Rapid up-regulation of miR-711 following secondary injury after TBI stimulates neuronal death by inhibiting the serine/threonine kinase Akt and activating FoxO3, GSK3α/β, PUMA, and Bim [60]. miR-21 expression changes in response to TBI, inhibits apoptosis, and promotes angiogenesis by down-regulating the expression of apoptosis- and angiogenesis-related molecules and PTEN as well as increasing the phosphorylation of Akt [61, 62]. The expression of miR-23a and miR-27a is down-regulated after TBI, thus contributing to neuronal death by up-regulating members of the Bcl-2 family [63].
After injury, the axons of retinal ganglion cells in adult mammals rapidly degenerate and the cell bodies may die, while glial cells at the injury site undergo scar formation. However, miR-30b decreases the sema3A levels to promote axon outgrowth [64]. In zebrafish retina, the miRNA and mRNA expression profiles indicate that miR-29b and miR-223 promote regeneration by regulating key biological processes, including cell survival/apoptosis, extracellular matrix-cytoskeleton signaling, and heparan sulfate proteoglycan binding [65].
miRNAs in Spinal Cord Injury
Spinal cord injury (SCI) is followed by excitotoxicity, edema, inflammation, ischemia, and chronic demyelination as secondary injuries, leading to additional damage [66]. Numerous miRNAs are highly expressed and localized in the spinal cord as well as in the brain. In adult rats, >77% of the identified mature miRNAs are expressed in the spinal cord [67]. Several, such as miR-1, miR-10a, miR-338, miR-451, miR-34a, miR-133a, miR-133b, miR-142-3p, miR-199, miR-10b, and miR-219 are highly enriched in the spinal cord [21], and their expression changes dynamically after SCI [67, 68]. For example, increased miR-223 expression regulates the expression of early-phase genes after SCI [68]. The expression of miR-124, which controls neurogenesis and neurite outgrowth during differentiation, is down-regulated after SCI [69, 70], and it affects inflammatory nociception by regulating methyl-CpG-binding protein 2 (MeCP2) and inflammation-related genes [71]. Further, miR-124 also targets the transcription factor CEBPα, holding promise as a target for treating neuroinflammation [72]. miR-486 down-regulates neurogenic differentiation 6 (Neurod 6) expression, thus enhancing apoptosis and functional deficits in neurons after SCI [73]. miR-126 targets such genes as SPRED1, PIK3R2, and VCAM1 to rescue tissue damage and to improve the functional deficit; its expression is down-regulated after SCI [74].
Astrocytes, specialized glial cells, perform supportive, metabolic, and homeostatic functions in the CNS [75]. miR-181 is a negative regulator of astrocyte activation, and its expression is down-regulated by inflammatory stimuli. Accordingly, miR-181 affects inflammatory cytokine secretion in astrocytes and modulates astrocyte activation and differentiation by targeting MeCP2 (methyl-CpG-binding protein 2) [44]. Similarly, miR-146a regulates the release of cytokines from astrocytes [76]. miR-17-5p targets the cell-cycle inhibitors P21 and RB1 to promote the proliferation of reactive astrocytes and facilitate functional recovery after SCI [77]. Transfection with miR-124 can improve the outcome of neural stem-cell transplantation in SCI rats by increasing the numbers of neurons and reducing the numbers of GFAP-positive astrocytes [78].
In the spinal cord, motor neuron subtypes are organized into columns that project axons to specific target muscles. For instance, the medial motor columns innervate axial muscles, while the lateral motor columns innervate limb muscles [79, 80]. Specification of the motor columns requires extrinsic signaling pathways to induce sequential Hox transcription-factor-mediated responses [79, 81]. miRNAs participate in the process of motor neuron gene regulation, including development, motor neuron disease, axon regeneration, and synaptic connection. miR-20a causes continuing motor neuron degeneration by down-regulating neurogenin 1 while up-regulation of neurogenin may protect motor neurons from aggressive secondary injury [82]. miR-29b reduces the expression of Bad, Bim, Noxa, and Puma, and plays a role in neuronal apoptosis in SCI. Down-regulation of miR-20a and miR-29b expression may cooperatively protect motor neurons from cell death by down-regulating the Mcl-1 (myeloid cell leukemia 1) and up-regulating BH3-only proteins after SCI [83]. miRNAs are also important for axonal regeneration in spinal motor neurons. After SCI, the elevated expression of miR-133b represses mRNA translation of RhoA, promoting the functional recovery of motor neuron axons [84].
Taken together, miRNAs play regulatory roles in neuronal-subtype specification, functional maintenance, and motor neuron regeneration after SCI.
miRNAs in Peripheral Nerve Injury
After peripheral nerve injury, the regenerating axons in the proximal nerve stump can grow across the lesion due to activation of the intrinsic growth capacity of neurons and the formation of a regenerative microenvironment. De-differentiated Schwann cells replenish lost or damaged tissues by proliferation, and produce a favorable environment for axonal outgrowth by helping to clear myelin debris and forming cellular conduits or corridors that guide axons through the degenerated nerve stump and back to their targets [85]. A recent study has suggested that knockout of Dicer impedes regenerative axon growth as well as anatomical, physiological, and functional recovery. The data suggest that an intact Dicer-dependent miRNA pathway is critical for successful peripheral nerve regeneration after injury [86].
Neuronal Survival
The survival of injured neurons is a necessary prerequisite for axonal regrowth. The expression of miR-21 and miR-222 increases continuously in dorsal root ganglia (DRG, L4-L6) during the initial 7-day period after sciatic nerve transection, and tissue inhibitor of metalloproteinase 3 (TIMP3) has been identified as a common target of miR-21 and miR-222. Overexpression of miR-21 and miR-222 reduces apoptosis and enhances the viability of cultured DRG neurons. Interleukin 6 (IL-6) up-regulates the miR-21 expression in these neurons [87]. miR-146a mediates apoptosis in DRG neurons under hyperglycemic conditions, which down-regulate miR-146a expression, improving the protein level of both IL-1 receptor-activated kinase and tumor necrosis factor receptor-associated factor 6 in DRGs [88].
Neurite Outgrowth
Microarray analysis and deep sequencing have revealed that many miRNAs regulate the expression of transcription factors and signaling mediators that are important for peripheral nerve regeneration [86]. In particular, the influences of miRNAs on neurite outgrowth from DRG neurons have been extensively investigated. For example, miR-21 promotes axonal growth from adult DRG neurons by targeting Sprouty2 (a specific inhibitor of the Ras/Raf/ERK pathway) [89], and miR-222 also promotes neurite outgrowth from these neurons by targeting PTEN (phosphatase and tensin homolog deleted on chromosome 10, a negative regulator of Akt) [90]. Several other miRNAs, including miR-8, miR-431, miR-145, and miR-138, have been shown to play regulatory roles in neurite outgrowth. Their targets are the cell-adhesion molecules Fasciclin III (Fas III) and Neuroglian (Nrg), Kremen1 (an antagonist of Wnt/β-catenin signaling), Robo2 (a transmembrane receptor), and Sirtuin type 1 (an NAD-dependent histone deacetylase), respectively [91–94].
Multiple targets of miR-21 have been validated, two-thirds of which are linked to intrinsic and/or extrinsic pathways of apoptosis [95]. miR-21 promotes neurite outgrowth by down-regulating the expression of its target gene, SPRY2142. Moreover, miR-21 expression is up-regulated in DRG neurons after sciatic nerve injury [87] (Fig. 1). After this injury, miR-21 and miR-222 promote neurite outgrowth and inhibit apoptosis by repressing TIMP3 in DRGs, suggesting that the two miRNAs are candidate hub molecules for triggering intrinsic neurite growth in injured DRG neurons [87, 89] (Fig. 1).

Schematic diagram illustrating (1) the joint inhibitory effects of miR-21 and miR-222 on neuronal apoptosis through suppressing TIMP3 after peripheral nerve injury, and (2) the promoting effects of miR-21 and miR-222 on neurite regrowth through suppressing sprouty2 and PTEN, respectively, after peripheral nerve injury. T-shaped lines indicate an inhibitory effect or negative regulation while arrows indicate a promoting effect.
miR-132 plays roles in dendrite morphology and synaptic function [96]. Its knockdown reduces axonal extension in cultured DRG neurons while overexpression increases axonal extension. Moreover, miR-132 regulates the mRNA level of RAS p21 protein activator 1 gene, serving as a positive regulator of developing axon extension [97]. miR-26a specifically targets glycogen synthase kinase 3β (GSK3β) to rescue axon regeneration, and the miR-26a-GSK3β pathway regulates axon regeneration at the neuronal soma by controlling the expression of Smad1, a regeneration-associated transcription factor [98]. let-7 inhibits the lin-41 expression in older neurons while lin-41 inhibits the let-7 expression in younger neurons. A let-7-lin-41 regulatory circuit can ensure that axon regeneration is inhibited only in older neurons [99].
Schwann Cell Phenotype Modulation
Evidence has identified a specific cohort of miRNAs as epigenetic regulators of the transition between the differentiation and de-differentiation of Schwann cells during the acute phase of PNS injury. miR-138 and miR-709 show the highest affinity for regulating the expression of Egr2, Sox-2, and c-Jun after PNS injury [100]. miR-204 negatively regulates Nrn1 protein expression and activates cleaved caspase-3, stimulating the apoptosis of Schwann cells after exposure to H2O2-induced oxidative stress [101]. miR-182 inhibits the proliferation and migration of Schwann cells by targeting fibroblast growth factor 9 and neurotrimin, respectively, at an early stage following sciatic nerve injury [102]. miR-221 and -222 promote the proliferation and migration of Schwann cells by targeting longevity assurance homologue 2, a suppressor of cell growth and metastasis, which can increase the intracellular H+ concentration by interacting with V-ATPase [103].
miR-9 is an important functional regulator of Schwann cell migration by directly targeting collagen triple-helix repeat-containing protein 1, which in turn inactivates downstream Rac1 GTPase [104]. let-7 miRNA significantly reduces the proliferation and migration of primary Schwann cells by suppressing the protein translation of nerve growth factor (NGF). The detailed mechanism seems to be that the NGF expression inhibited by let-7 miRNA can regulate the miR-221/222 expression to affect the Schwann cell phenotype [105]. Increased miR-132 expression induced by hypoxia enhances Schwann cell migration and down-regulates the target, PRKAG3, to facilitate peripheral nerve regeneration [106].
miR-34a is highly expressed in the adult nervous system, and Notch1 and cyclin D1 are its targets in cancer cells [107]. These two targets are also important mediators of Schwann cell dedifferentiation and proliferation after peripheral nerve injury [108, 109]. miR-140 targets the transcription factor Egr2, a master regulator of myelination, and modulates axonal myelination in co-cultures of DRG neurons and Schwann cells [110]. miR-29a inhibits peripheral myelin protein, which is a dose-sensitive, disease-associated protein primarily expressed in myelinating Schwann cells [111]. The effects of several miRNAs on Schwann cells after peripheral nerve injury are illustrated in Fig. 2.

After peripheral nerve injury, the expression of miR-204, let-7, mir-27a, and miR-29 is constantly down-regulated (downward arrows), while that of of miR-182, miR-221/222, miR-27a, and miR-132, and miR-29a is constantly up-regulated (upward arrows), and that of of miR-34a and miR-140 is dysregulated (curved arrows). After peripheral nerve injury, these miRNAs regulate the behavior of Schwann cells, such as apoptosis, proliferation, migration, and myelination, as indicated by arrows or T-shaped lines (positive and negative regulation, respectively). Also shown (middle) is a schematic showing the process of axonal regrowth from an injured peripheral neuron while reaching the target organ for re-innervation, coupled with myelination of the re-growing axon by Schwann cells.
For the sake of convenience to the reader, we summarize the above description by listing many miRNAs that have been reported to be associated with various injuries to the nervous system and highlighting their functional significance in neural regeneration (Table 1).
Roles of lncRNAs in Responses to Nerve Injury
lncRNAs are specifically expressed in the CNS and PNS, and may be involved in regeneration. To date, several reports have described the roles of lncRNAs in CNS development and neurogenesis [112, 113]. It has been shown that a total of 322 lncRNAs are differentially expressed in the brain with hypoxic-ischemic damage, and silencing of the lncRNA BC088414 decreases apoptosis and increases cell proliferation [114]. These findings suggests the roles of lncRNAs in CNS injury.
In investigations of the impact of lncRNAs on the intrinsic regenerative capacity of peripheral neurons, a total of 105 lncRNAs have been found to show significant differential expression in DRGs after sciatic nerve injury. Among these, BC089918 and uc.217 have been specifically investigated and the results showed that down-regulation of BC089918 expression [115], and silencing of uc.217 expression by siRNA both enhance neurite outgrowth of DRG neurons [116].
Conclusions
The tissue-specific expression and functional roles of ncRNAs in the nervous system under physiological conditions determine their putative involvement in the pathophysiological processes of neural injury, which include immune/inflammatory responses, glial scar formation, neuronal apoptosis, cell proliferation and migration, axonal regrowth, and target organ re-innervation. After different types of CNS and PNS injury, such as ABI, SCI, and peripheral nerve injury, diverse ncRNAs, mainly miRNAs and lncRNAs, are differentially expressed in the injured neural tissue, and play unique regulatory roles through binding to the 3′-untranslated regions of target mRNAs, leading to translation repression or degradation of the mRNAs. The critical regulation of neural injury and regeneration is reflected in the promoting or suppressing effect on neuronal survival, neurite outgrowth, and glial phenotype.
The involvement of ncRNAs in numerous cellular processes and human diseases predicts that the two types of ncRNAs, miRNAs and lncRNAs, may be used as potential diagnostic biomarkers and therapeutic targets in the clinic. More importantly, both miRNAs and lncRNAs are readily detectable in bodily fluids, thus enabling them to be useful for therapeutic applications, including for neural injury [117]. However, neural injury is a complex process integrating multiple signaling pathways in the nervous, immune, and vascular systems, accompanied by various cellular and molecular mechanisms. Hence, single-target therapies are usually inadequate for treating neural injury. The further identification of ncRNAs whose expression is likely to be changed after neural injury will contribute to a global understanding of the molecular regulation of injury responses and regeneration, and will also facilitate the development of clinical applications of ncRNAs.
Another challenge to the potential use of ncRNAs in the clinic is the preparation of ncRNA amplifiers and inhibitors and improving the relevant performance, including delivery, bioavailability, function, and adverse side-effects of both amplifiers and inhibitors. Currently, several preclinical animal studies have been reported. For example, implantation of a silicone tube injected with a 1:1 mixture of Matrigel and steroid-conjugated miR-9 agomir for bridging the rat sciatic nerve gap reduces Schwann cell migration within the tube due to increased expression of miR-9 [104]. Another example is that a silicone tube injected with a 1:1 mixture of Matrigel and let-7d antagomir enhances Schwann cell migration and axon outgrowth after implantation of the tube [105].
Importantly, a systems-level analysis of transcriptional changes in neural injury has been attracting increasing attention in that this new methodology can advance our understanding of ncRNA regulation. To conduct a systems-level analysis, massive data sets have been processed using Ingenuity Pathway Analysis [118], a web-based functional analysis tool, to generate gene networks, which may be used to search the signaling pathways and provide profound insights into the regulation of neural injury responses and regeneration at the transcriptional and post-transcriptional levels. Overall, further studies are needed to fully understand the functional roles of ncRNAs in neural injury and to actively develop ncRNA-based therapies for improving neural regeneration.
References
Sun F, He Z. Neuronal intrinsic barriers for axon regeneration in the adult CNS. Curr Opin Neurobiol 2010, 20: 510–518.
Shen D, Wang X, Gu X. Scar-modulating treatments for central nervous system injury. Neurosci Bull 2014, 30: 967–984.
Battiston B, Papalia I, Tos P, Geuna S. Chapter 1: Peripheral nerve repair and regeneration research: a historical note. Int Rev Neurobiol 2009, 87: 1–7.
Ma TC, Willis DE. What makes a RAG regeneration associated? Front Mol Neurosci 2015, 8: 43.
Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322: 963–966.
Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, et al. Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 2015, 86: 1000–1014.
Liu Q, Paroo Z. Biochemical principles of small RNA pathways. Annu Rev Biochem 2010, 79: 295–319.
Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol 2010, 220: 126–139.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316: 1484–1488.
Kaur P, Liu F, Tan JR, Lim KY, Sepramaniam S, Karolina DS, et al. Non-Coding RNAs as Potential Neuroprotectants against Ischemic Brain Injury. Brain Sci 2013, 3: 360–395.
Ghildiyal M, Xu J, Seitz H, Weng Z, Zamore PD. Sorting of Drosophila small silencing RNAs partitions microRNA* strands into the RNA interference pathway. RNA 2010, 16: 43–56.
Sun E, Shi Y. MicroRNAs: Small molecules with big roles in neurodevelopment and diseases. Exp Neurol 2015, 268: 46–53.
Jovicic A, Roshan R, Moisoi N, Pradervand S, Moser R, Pillai B, et al. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci 2013, 33: 5127–5137.
Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A 2010, 107: 20382–20387.
Aksoy-Aksel A, Zampa F, Schratt G. MicroRNAs and synaptic plasticity—a mutual relationship. Philos Trans R Soc Lond B Biol Sci 2014, 369(1652). doi:10.1098/rstb.2013.0515.
Olde Loohuis NF, Kos A, Martens GJ, Van Bokhoven H, Nadif Kasri N, Aschrafi A. MicroRNA networks direct neuronal development and plasticity. Cell Mol Life Sci 2012, 69: 89–102.
Lee HJ. Exceptional stories of microRNAs. Exp Biol Med (Maywood) 2013, 238: 339–343.
Arthanari Y, Heintzen C, Griffiths-Jones S, Crosthwaite SK. Natural antisense transcripts and long non-coding RNA in Neurospora crassa. PLoS One 2014, 9: e91353.
Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res 2012, 40: 6391–6400.
Kapsimali M, Kloosterman WP, de Bruijn E, Rosa F, Plasterk RH, Wilson SW. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol 2007, 8: R173.
Kosik KS. The neuronal microRNA system. Nat Rev Neurosci 2006, 7: 911–920.
He L, Lu QR. Coordinated control of oligodendrocyte development by extrinsic and intrinsic signaling cues. Neurosci Bull 2013, 29: 129–143.
Beveridge NJ, Cairns MJ. MicroRNA dysregulation in schizophrenia. Neurobiol Dis 2012, 46: 263–271.
Lee ST, Chu K, Im WS, Yoon HJ, Im JY, Park JE, et al. Altered microRNA regulation in Huntington’s disease models. Exp Neurol 2011, 227: 172–179.
Merico D, Costain G, Butcher NJ, Warnica W, Ogura L, Alfred SE, et al. MicroRNA Dysregulation, Gene Networks, and Risk for Schizophrenia in 22q11.2 Deletion Syndrome. Front Neurol 2014, 5: 238.
Xu B, Hsu PK, Karayiorgou M, Gogos JA. MicroRNA dysregulation in neuropsychiatric disorders and cognitive dysfunction. Neurobiol Dis 2012, 46: 291–301.
O’Reilly K, Pryor J. Young people with brain injury in nursing homes: not the best option! Aust Health Rev 2002, 25: 46–51.
Chen A, Bushmeneva K, Zagorski B, Colantonio A, Parsons D, Wodchis WP. Direct cost associated with acquired brain injury in Ontario. BMC Neurol 2012, 12: 76.
Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439: 283–289.
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 2004, 5: R13.
He X, Zhang Q, Liu Y, Pan X. Cloning and identification of novel microRNAs from rat hippocampus. Acta Biochim Biophys Sin 2007, 39: 708–714.
Brosnan CA, Voinnet O. The long and the short of noncoding RNAs. Curr Opin Cell Biol 2009, 21: 416–425.
Liu C, Zhao L, Han S, Li J, Li D. Identification and Functional Analysis of MicroRNAs in Mice following Focal Cerebral Ischemia Injury. Int J Mol Sci 2015, 16: 24302–24318.
Chen F, Du Y, Esposito E, Liu Y, Guo S, Wang X, et al. Effects of Focal Cerebral Ischemia on Exosomal Versus Serum miR126. Transl Stroke Res 2015, 6: 478–484.
Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab 2009, 29: 675–687.
Jeyaseelan K, Lim KY, Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 2008, 39: 959–966.
Harraz MM, Eacker SM, Wang X, Dawson TM, Dawson VL. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc Natl Acad Sci U S A 2012, 109: 18962–18967.
Sepramaniam S, Armugam A, Lim KY, Karolina DS, Swaminathan P, Tan JR, et al. MicroRNA 320a functions as a novel endogenous modulator of aquaporins 1 and 4 as well as a potential therapeutic target in cerebral ischemia. J Biol Chem 2010, 285: 29223–29230.
Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X, et al. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun 2015, 49: 75–85.
Shi H, Sun BL, Zhang J, Lu S, Zhang P, Wang H, et al. miR-15b suppression of Bcl-2 contributes to cerebral ischemic injury and is reversed by sevoflurane preconditioning. CNS Neurol Disord Drug Targets 2013, 12: 381–391.
Shi G, Liu Y, Liu T, Yan W, Liu X, Wang Y, et al. Up-regulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Exp Brain Res 2012, 216: 225–230.
Moon JM, Xu L, Giffard RG. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab 2013, 33: 1976–1982.
Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, et al. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis 2010, 38: 17–26.
Hutchison ER, Kawamoto EM, Taub DD, Lal A, Abdelmohsen K, Zhang Y, et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia 2013, 61: 1018–1028.
Irmady K, Jackman KA, Padow VA, Shahani N, Martin LA, Cerchietti L, et al. Mir-592 regulates the induction and cell death-promoting activity of p75NTR in neuronal ischemic injury. J Neurosci 2014, 34: 3419–3428.
Chi W, Meng F, Li Y, Wang Q, Wang G, Han S, et al. Down-regulation of miRNA-134 protects neural cells against ischemic injury in N2A cells and mouse brain with ischemic stroke by targeting HSPA12B. Neuroscience 2014, 277: 111–122.
Stary CM, Xu L, Sun X, Ouyang YB, White RE, Leong J, et al. MicroRNA-200c contributes to injury from transient focal cerebral ischemia by targeting Reelin. Stroke 2015, 46: 551–556.
Wang P, Zhang N, Liang J, Li J, Han S, Li J. Micro-RNA-30a regulates ischemia-induced cell death by targeting heat shock protein HSPA5 in primary cultured cortical neurons and mouse brain after stroke. J Neurosci Res 2015, 93: 1756–1768.
Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, et al. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem Res 2014, 39: 1279–1291.
Liu P, Zhao H, Wang R, Wang P, Tao Z, Gao L, et al. MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke 2015, 46: 513–519.
Zhao H, Tao Z, Wang R, Liu P, Yan F, Li J, et al. MicroRNA-23a-3p attenuates oxidative stress injury in a mouse model of focal cerebral ischemia-reperfusion. Brain Res 2014, 1592: 65–72.
Chi W, Meng F, Li Y, Li P, Wang G, Cheng H, et al. Impact of microRNA-134 on neural cell survival against ischemic injury in primary cultured neuronal cells and mouse brain with ischemic stroke by targeting HSPA12B. Brain Res 2014, 1592: 22–33.
Yu H, Wu M, Zhao P, Huang Y, Wang W, Yin W. Neuroprotective effects of viral overexpression of microRNA-22 in rat and cell models of cerebral ischemia-reperfusion injury. J Cell Biochem 2015, 116: 233–241.
Chen Q, Xu J, Li L, Li H, Mao S, Zhang F, et al. MicroRNA-23a/b and microRNA-27a/b suppress Apaf-1 protein and alleviate hypoxia-induced neuronal apoptosis. Cell Death Dis 2014, 5: e1132.
Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci 2014, 52: 148–155.
Meissner L, Gallozzi M, Balbi M, Schwarzmaier SM, Tiedt S, Terpolilli NA, et al. Temporal profile of microRNA expression in contused cortex following traumatic brain injury in mice. J Neurotrauma 2015. doi:10.1089/neu.2015.4077.
Miao W, Bao TH, Han JH, Yin M, Yan Y, Wang WW, et al. Voluntary exercise prior to traumatic brain injury alters miRNA expression in the injured mouse cerebral cortex. Braz J Med Biol Res 2015, 48: 433–439.
Truettner JS, Alonso OF, Bramlett HM, Dietrich WD. Therapeutic hypothermia alters microRNA responses to traumatic brain injury in rats. J Cereb Blood Flow Metab 2011, 31: 1897–1907.
Wang WX, Visavadiya NP, Pandya JD, Nelson PT, Sullivan PG, Springer JE. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Exp Neurol 2015, 265: 84–93.
Sabirzhanov B, Stoica BA, Zhao Z, Loane DJ, Wu J, Dorsey SG, et al. miR-711 up-regulation induces neuronal cell death after traumatic brain injury. Cell Death Differ 2016, 23: 654–668.
Ge XT, Lei P, Wang HC, Zhang AL, Han ZL, Chen X, et al. miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci Rep 2014, 4: 6718.
Han Z, Chen F, Ge X, Tan J, Lei P, Zhang J. miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-Akt signaling pathway in vitro after experimental traumatic brain injury. Brain Res 2014, 1582: 12–20.
Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, et al. Down-regulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci 2014, 34: 10055–10071.
Han F, Huo Y, Huang CJ, Chen CL, Ye J. MicroRNA-30b promotes axon outgrowth of retinal ganglion cells by inhibiting Semaphorin3A expression. Brain Res 2015, 1611: 65–73.
Fuller-Carter PI, Carter KW, Anderson D, Harvey AR, Giles KM, Rodger J. Integrated analyses of zebrafish miRNA and mRNA expression profiles identify miR-29b and miR-223 as potential regulators of optic nerve regeneration. BMC Genomics 2015, 16: 591.
Yilmaz T, Kaptanoglu E. Current and future medical therapeutic strategies for the functional repair of spinal cord injury. World J Orthop 2015, 6: 42–55.
Liu NK, Wang XF, Lu QB, Xu XM. Altered microRNA expression following traumatic spinal cord injury. Exp Neurol 2009, 219: 424–429.
Nakanishi K, Nakasa T, Tanaka N, Ishikawa M, Yamada K, Yamasaki K, et al. Responses of microRNAs 124a and 223 following spinal cord injury in mice. Spinal Cord 2010, 48: 192–196.
Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 2009, 12: 399–408.
Yu JY, Chung KH, Deo M, Thompson RC, Turner DL. MicroRNA miR-124 regulates neurite outgrowth during neuronal differentiation. Exp Cell Res 2008, 314: 2618–2633.
Kynast KL, Russe OQ, Moser CV, Geisslinger G, Niederberger E. Modulation of central nervous system-specific microRNA-124a alters the inflammatory response in the formalin test in mice. Pain 2013, 154: 368–376.
Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med 2011, 17: 64–70.
Jee MK, Jung JS, Choi JI, Jang JA, Kang KS, Im YB, et al. MicroRNA 486 is a potentially novel target for the treatment of spinal cord injury. Brain 2012, 135: 1237–1252.
Hu J, Zeng L, Huang J, Wang G, Lu H. miR-126 promotes angiogenesis and attenuates inflammation after contusion spinal cord injury in rats. Brain Res 2015, 1608: 191–202.
Bhalala OG, Srikanth M, Kessler JA. The emerging roles of microRNAs in CNS injuries. Nat Rev Neurol 2013, 9: 328–339.
Iyer A, Zurolo E, Prabowo A, Fluiter K, Spliet WG, van Rijen PC, et al. MicroRNA-146a: a key regulator of astrocyte-mediated inflammatory response. PLoS One 2012, 7: e44789.
Hong P, Jiang M, Li H. Functional requirement of dicer1 and miR-17-5p in reactive astrocyte proliferation after spinal cord injury in the mouse. Glia 2014, 62: 2044–2060.
Xu W, Li P, Qin K, Wang X, Jiang X. miR-124 regulates neural stem cells in the treatment of spinal cord injury. Neurosci Lett 2012, 529: 12–17.
Dasen JS, Jessell TM. Hox networks and the origins of motor neuron diversity. Curr Top Dev Biol 2009, 88: 169–200.
Jung H, Lacombe J, Mazzoni EO, Liem KF, Jr., Grinstein J, Mahony S, et al. Global control of motor neuron topography mediated by the repressive actions of a single hox gene. Neuron 2010, 67: 781–796.
Dalla Torre di Sanguinetto SA, Dasen JS, Arber S. Transcriptional mechanisms controlling motor neuron diversity and connectivity. Curr Opin Neurobiol 2008, 18: 36–43.
Jee MK, Jung JS, Im YB, Jung SJ, Kang SK. Silencing of miR20a is crucial for Ngn1-mediated neuroprotection in injured spinal cord. Hum Gene Ther 2012, 23: 508–520.
Liu XJ, Zheng XP, Zhang R, Guo YL, Wang JH. Combinatorial effects of miR-20a and miR-29b on neuronal apoptosis induced by spinal cord injury. Int J Clin Exp Pathol 2015, 8: 3811–3818.
Yu YM, Gibbs KM, Davila J, Campbell N, Sung S, Todorova TI, et al. MicroRNA miR-133b is essential for functional recovery after spinal cord injury in adult zebrafish. Eur J Neurosci 2011, 33: 1587–1597.
Parrinello S, Napoli I, Ribeiro S, Wingfield Digby P, Fedorova M, Parkinson DB, et al. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 2010, 143: 145–155.
Wu D, Raafat A, Pak E, Clemens S, Murashov AK. Dicer-microRNA pathway is critical for peripheral nerve regeneration and functional recovery in vivo and regenerative axonogenesis in vitro. Exp Neurol 2012, 233: 555–565.
Zhou S, Zhang S, Wang Y, Yi S, Zhao L, Tang X, et al. MiR-21 and miR-222 inhibit apoptosis of adult dorsal root ganglion neurons by repressing TIMP3 following sciatic nerve injury. Neurosci Lett 2015, 586: 43–49.
Wang L, Chopp M, Szalad A, Zhang Y, Wang X, Zhang RL, et al. The role of miR-146a in dorsal root ganglia neurons of experimental diabetic peripheral neuropathy. Neuroscience 2014, 259: 155–163.
Strickland IT, Richards L, Holmes FE, Wynick D, Uney JB, Wong LF. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS One 2011, 6: e23423.
Zhou S, Shen D, Wang Y, Gong L, Tang X, Yu B, et al. microRNA-222 targeting PTEN promotes neurite outgrowth from adult dorsal root ganglion neurons following sciatic nerve transection. PLoS One 2012, 7: e44768.
Liu CM, Wang RY, Saijilafu, Jiao ZX, Zhang BY, Zhou FQ. MicroRNA-138 and SIRT1 form a mutual negative feedback loop to regulate mammalian axon regeneration. Genes Dev 2013, 27: 1473–1483.
Lu CS, Zhai B, Mauss A, Landgraf M, Gygi S, Van Vactor D. MicroRNA-8 promotes robust motor axon targeting by coordinate regulation of cell adhesion molecules during synapse development. Philos Trans R Soc Lond B Biol Sci 2014, 369(1652). doi:10.1098/rstb.2013.0517.
Wu D, Murashov AK. MicroRNA-431 regulates axon regeneration in mature sensory neurons by targeting the Wnt antagonist Kremen1. Front Mol Neurosci 2013, 6: 35.
Zhang HY, Zheng SJ, Zhao JH, Zhao W, Zheng LF, Zhao D, et al. MicroRNAs 144, 145, and 214 are down-regulated in primary neurons responding to sciatic nerve transection. Brain Res 2011, 1383: 62–70.
Buscaglia LE, Li Y. Apoptosis and the target genes of microRNA-21. Chin J Cancer 2011, 30: 371–380.
Siegel G, Saba R, Schratt G. microRNAs in neurons: manifold regulatory roles at the synapse. Curr Opin Genet Dev 2011, 21: 491–497.
Hancock ML, Preitner N, Quan J, Flanagan JG. MicroRNA-132 is enriched in developing axons, locally regulates Rasa1 mRNA, and promotes axon extension. J Neurosci 2014, 34: 66–78.
Jiang JJ, Liu CM, Zhang BY, Wang XW, Zhang M, Saijilafu, et al. MicroRNA-26a supports mammalian axon regeneration in vivo by suppressing GSK3beta expression. Cell Death Dis 2015, 6: e1865.
Zou Y, Chiu H, Zinovyeva A, Ambros V, Chuang CF, Chang C. Developmental decline in neuronal regeneration by the progressive change of two intrinsic timers. Science 2013, 340: 372–376.
Adilakshmi T, Sudol I, Tapinos N. Combinatorial action of miRNAs regulates transcriptional and post-transcriptional gene silencing following in vivo PNS injury. PLoS One 2012, 7: e39674.
Gao R, Wang L, Sun J, Nie K, Jian H, Gao L, et al. MiR-204 promotes apoptosis in oxidative stress-induced rat Schwann cells by suppressing neuritin expression. FEBS Lett 2014, 588: 3225–3232.
Yu B, Qian TM, Wang YJ, Zhou SL, Ding GH, Ding F, et al. miR-182 inhibits Schwann cell proliferation and migration by targeting FGF9 and NTM, respectively at an early stage following sciatic nerve injury. Nucleic Acids Res 2012, 40: 10356–10365.
Yu B, Zhou S, Wang Y, Qian T, Ding G, Ding F, et al. miR-221 and miR-222 promote Schwann cell proliferation and migration by targeting LASS2 after sciatic nerve injury. J Cell Sci 2012, 125: 2675–2683.
Zhou S, Gao R, Hu W, Qian T, Wang N, Ding G, et al. MiR-9 inhibits Schwann cell migration by targeting Cthrc1 following sciatic nerve injury. J Cell Sci 2014, 127: 967–976.
Li S, Wang X, Gu Y, Chen C, Wang Y, Liu J, et al. Let-7 microRNAs regenerate peripheral nerve regeneration by targeting nerve growth factor. Mol Ther 2015, 23: 423–433.
Yao C, Shi X, Zhang Z, Zhou S, Qian T, Wang Y, et al. Hypoxia-Induced Up-regulation of miR-132 Promotes Schwann Cell Migration After Sciatic Nerve Injury by Targeting PRKAG3. Mol Neurobiol 2015. doi:10.1007/s12035-015-9449-y.
Pang RT, Leung CO, Ye TM, Liu W, Chiu PC, Lam KK, et al. MicroRNA-34a suppresses invasion through down-regulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis 2010, 31: 1037–1044.
Kim HA, Ratner N, Roberts TM, Stiles CD. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J Neurosci 2001, 21: 1110–1116.
Woodhoo A, Alonso MB, Droggiti A, Turmaine M, D’Antonio M, Parkinson DB, et al. Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat Neurosci 2009, 12: 839–847.
Viader A, Chang LW, Fahrner T, Nagarajan R, Milbrandt J. MicroRNAs modulate Schwann cell response to nerve injury by reinforcing transcriptional silencing of dedifferentiation-related genes. J Neurosci 2011, 31: 17358–17369.
Verrier JD, Lau P, Hudson L, Murashov AK, Renne R, Notterpek L. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia 2009, 57: 1265–1279.
Goff LA, Groff AF, Sauvageau M, Trayes-Gibson Z, Sanchez-Gomez DB, Morse M, et al. Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A 2015, 112: 6855–6862.
Kour S, Rath PC. Age-dependent differential expression profile of a novel intergenic long noncoding RNA in rat brain. Int J Dev Neurosci 2015, 46: 55–66.
Zhao F, Qu Y, Liu J, Liu H, Zhang L, Feng Y, et al. Microarray Profiling and Co-Expression Network Analysis of LncRNAs and mRNAs in Neonatal Rats Following Hypoxic-ischemic Brain Damage. Sci Rep 2015, 5: 13850.
Yu B, Zhou S, Hu W, Qian T, Gao R, Ding G, et al. Altered long noncoding RNA expressions in dorsal root ganglion after rat sciatic nerve injury. Neurosci Lett 2013, 534: 117–122.
Yao C, Wang J, Zhang H, Zhou S, Qian T, Ding F, et al. Long non-coding RNA uc.217 regulates neurite outgrowth in dorsal root ganglion neurons following peripheral nerve injury. Eur J Neurosci 2015, 42: 1718–1725.
Delay C, Mandemakers W, Hebert SS. MicroRNAs in Alzheimer’s disease. Neurobiol. Dis. 2012, 46: 285–290.
Li S, Xue C, Yuan Y, Zhang R, Wang Y, Yu B, et al. The transcriptional landscape of dorsal root ganglia after sciatic nerve transection. Sci Rep 2015, 5: 16888.
Acknowledgments
We thank Professor Jie Liu for help in manuscript preparation. Researches from the corresponding author’s laboratory were supported by the National Basic Research Development Program (973 Program) of China (2014CB542202), the National High-Technology Research Development Program (863 Program) of China (2012AA020502), the Natural Science Foundation of Jiangsu Province, China (BK20151270), the National Natural Science Foundation of China (31200799 and 81571198), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhou, S., Ding, F. & Gu, X. Non-coding RNAs as Emerging Regulators of Neural Injury Responses and Regeneration. Neurosci. Bull. 32, 253–264 (2016). https://doi.org/10.1007/s12264-016-0028-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-016-0028-7