
Abstract
In this study, a simple and high-throughput multiresidue pesticide analysis method based on the quick, easy, cheap, effective, rugged, safe (QuEChERS) extraction combined with gas chromatography-triple quadrupole mass spectrometry (GC-MS/MS) and ultra-high performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS) is presented for the determination of 113 pesticides in green and ripe mangoes. Different buffer systems and purification sorbents were optimized to get better recovery. Extracts from mangoes after acid acetonitrile and ammonium acetate extraction were directly injected to UHPLC-MS/MS analysis, whereas other GC-amenable compounds were treated with cleanup approaches with a mixture of sorbents [400 mg primary-secondary amine (PSA) and 400 mg graphitized carbon black (GCB)] for GC-MS/MS analysis. Recoveries for the majority of pesticides at spike levels of 10, 50, and 100 μg/kg were ranged between 70 and 120% with RSD values below 20%. Limits of detection (LODs) were below 4 μg/kg, whereas limits of quantification (LOQs) were below 10 μg/kg. Response linearity was good in the range between 5 and 500 μg/kg. The matrix effect for 56 pesticides by GC-MS/MS analysis exhibited a non-significant matrix effect (≤ 25%) in green and ripe mangoes. For 57 pesticides used for UHPLC-MS/MS analysis, the optimized method in green mango provided no significant matrix effect (≤ 25%) for 63% of the compounds, whereas only for 16 and 21% compounds, the matrix effect was medium (25–40%) and strong (> 40%); the optimized method in ripe mango provided no significant matrix effect (≤ 25%) for 61% of the compounds, whereas only for 26 and 12% compounds, the matrix effect was medium (25–40%) and strong (> 40%). Following the application of a “top-down” approach, the expanded measurement uncertainty was both 21% on average (coverage factor k = 2, confidence level 95%) in green and ripe mangoes. The application of GC-MS/MS and UHPLC-MS/MS coupled with the modified QuEChERS extraction procedures was proved to adequately quantitate these pesticides in different maturity levels of mangoes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mango is considered one of the most important tropical fruits and is widely acceptable throughout the world because of its attractive flavor, aroma, and color. Furthermore, mango is a good dietetic source of carotenoids, mineral salts, carbohydrates, ascorbic acid, and B vitamins (Srivastava et al. 2014). According to FAO’s latest statistics, China ranked second in mango production in the world during 2013 (Food and Agriculture Organization of the United Nations, Food and Agricultural Commodities Production 2013). However, the tropical agricultural environment is of high temperature and high humidity, and there are many types of pests and pathogens in mangoes. To increase its productivity and prevent economic losses, pesticides are used for pest and fungal control during mango growing and post-harvest stages. For example, a serious disease in mango is anthracnosis. The common practice is immersion of the mangoes in water containing fungicides such as thiabendazole (Filho et al. 2010). Despite the usefulness of pesticides for the control of pest and disease, agro-products are liable to contain or accumulate from agricultural practices and storage periods (Nieto-Garcia et al. 2015; Jin et al. 2012; Amvrazi and Albanis 2006; Yang et al. 2011; Stajnbaher and Zupancic-Kralj, 2003). Food intake with pesticides could be concerned to risks to a consumer’s health (Slowik-Borowiec et al. 2015; Hayward et al. 2013; Chu et al. 2005; Castillo et al. 2012; Rissato et al. 2004). Therefore, several countries have established maximum residue limits (MRLs) for pesticides in food including mangoes. In this context, the monitoring of pesticides in mangoes is of significant importance to guarantee food safety, also adhering to current good manufacturing practices.
For that reason, analytical approaches for the determination and quantification of residues in mango are indispensable. Pesticide residue analysis in mango matrices is still difficult to perform due to the texture of mango varying depending on the maturity level of the fruit. Green mango has an acid matrix which could easily degrade some pesticides. Ripe mango has a viscous matrix with high sugar and fatty acid content, which could adversely affect the extraction efficiency and instrument performances. Thus, the preparation method of removing interferents from the matrix is important for the determination of pesticides. In the recent years, the most popular cleanup approach for pesticide residue analysis is the quick, easy, cheap, effective, rugged, safe (QuEChERS) approach which was first introduced by Anastassiades et al. (2003). Compared with traditional methods, QuEChERS is considered quick, easy, cheap, effective, rugged, and safe. Because of its advantages, many studies applied this method for the multiresidue pesticide analysis in food (Prestes et al. 2009; Chamkasem et al. 2013; Zhao et al. 2014; Palenikova et al. 2015; He et al. 2015; Koesukwiwat et al. 2011; Furlani et al. 2011; Kwon et al. 2012; Cherta et al. 2013; Rejczak and Tuzimski 2015a, b, 2016). QuEChERS shows some important strengths such as being simple and quick, as well as weaknesses such as imperfect cleanup. Moreover, with the number of pesticides increasing, it is needed to apply efficient instruments to screen the largest possible pesticides at the lowest possible concentration at a short time. Gas chromatography (GC) or liquid chromatography (LC) coupled with triple quadrupole mass spectrometry showed that tandem mass spectrometry is one of the most efficient techniques in terms of low limits of detection, strong capacity of resisting disturbance, and simultaneous detection of many types of pesticides. Therefore, it is a preferred approach at present for the analysis of hundreds of pesticides using QuEChERS in combination with GC-MS/MS or LC-MS/MS (Qu et al. 2010; Wong et al. 2010; Anagnostopoulos et al. 2012; Navickiene et al. 2010; Rizzetti et al. 2016; Rejczak and Tuzimski 2015a).
Up to date, several researchers elaborated the QuEChERS procedure combined with triple quadrupole mass spectrometry for the extraction of pesticides in mango matrices. The first report on its application to pesticide extraction from mango matrices was published in 2009 by Banerjee et al. (2009), which determined 87 pesticides in mango by LC-MS/MS. Next, Navickiene et al. (2010) applied the QuEChERS method with GC-MS/MS for the extraction of seven pesticides in mango. Then, Deme and Upadhyayula, 2015used the QuEChERS approach for the determination of 40 pesticides in mango juices. After that, Sivaperumal et al. (2017) used the QuEChERS method for the analysis of 68 pesticide residues in mango matrices.
The main objective of the present work was to study the feasibility QuEChERS method coupled with GC-MS/MS and ultra-high performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS) to simultaneously determine 113 pesticides in different maturity levels of mangoes. The research was implemented to assess the fitness of the proposed method. An additional objective was to optimize the extraction method to improve sample cleanup capacity, reduce matrix interferents, and preparation time.
Experimental
Materials, Solvents, and Standards
The phytosanitary compounds listed in Tables 1 and 2 were purchased from Dr. Ehrenstorfer (Augsburg, Germany), with purity higher than 99%.
HPLC-grade acetonitrile, methanol, and hexane were purchased from Fisher Scientific (Fair Lawn, NJ, USA). The pesticides were chosen considering their application in mangoes in China. All of the standards were individually dissolved in methanol at a concentration of 1000 mg/L. Calibration standard (listed in Table 1) solutions were prepared in hexane and in blank mango extracts (matrix-matched calibration), whereas calibration standard (listed in Table 2) solutions were prepared in methanol and in blank mango extracts (matrix-matched calibration).
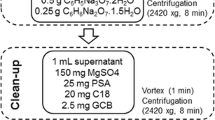
For sample preparation, commercial QuEChERS extraction salt packets containing 6 g MgSO4, 1.5 g CH3COONa or 4 g MgSO4, 1 g NaCl or 4 g MgSO4, 1 g NaCl, 1 g sodium citrate, and 0.5 g disodium hydrogencitrate sesquihydrate were used. For d-SPE cleanup, preweighed mixtures containing 400 mg of primary secondary amine (PSA), 400 mg graphitized carbon black (GCB), 1200 mg anhydrous MgSO4 or 150 mg PSA, 15 mg GCB, and 900 mg anhydrous MgSO4 were used. These materials were purchased from Agilent (Agilent Technologies, Lake Forest, CA). Analytical-grade ammonium acetate and commercial QuEChERS extraction sorbent packets containing 500 mg Zr-Sep+ or 400 mg PSA, 80 mg GCB, 1200 mg anhydrous MgSO4, and 480 mg Zr-Sep+ were acquired from Sigma-Aldrich (Supelco, Bellefonte, PA, USA).
Multiwalled carbon nanotubes (MWCNTs) (20–30 nm) were obtained from Chengdu Organic Chemicals Co. Ltd., Chinese Academy of Sciences (Chengdu, China). Reagent-grade anhydrous MgSO4, sodium chloride, and glacial acetic acid were purchased from Guangzhou Chemical Reagent Factory (Guangzhou, China).
GC-MS/MS Analysis
The analysis was carried out on a 7000 triple-quadrupole MS coupled to a 7890A GC, a 7693 autosampler, and a computer with MassHunter software (version B.05.00412) for data acquisition and processing (Agilent Technologies, Palo Alto, CA, USA). Pesticides were separated with HP-5 MS capillary columns from Agilent (0.25 mm i.d. × 30 m × 0.25 μm film thickness). The flow rate of the carrier gas (helium) was 1 mL/min. The initial oven temperature was 70 °C (hold for 2 min), and it ramped to 150 °C at a rate of 25 °C/min, then raised to 200 °C at 3 °C/min, and the temperature was increased until 280 °C at a rate of 8 °C/min, which was held for 2 min. The total run time was 33.8 min. The injector temperature was 250 °C. The injected volume was 1.0 μL with splitless mode. The temperatures of ion source and transfer line were 230 and 280 °C. The parameters of the MRM transition mode for all the test pesticides are provided in Table 1. The time of solvent delay was 4.0 min in order to prevent filament burnout. The collision gases were nitrogen and helium with flow rates at 1.5 and 2.25 mL/min for MRM mode. The calibration curve was made by solvent solution fortifying known concentration standards. Concentrations were obtained by comparing the peak area of the targets in the sample to the response of solvent solution standards in the calibration curve. Pesticide identification was confirmed by making a comparison between the expected retention time and the ratio of the two transition (primary/secondary) values to solvent solution standards, following the criteria for identification established by the FDA and European Union (EC Council Directive 2002).
UHPLC-MS/MS Analysis
The UHPLC-MS/MS system (Waters, Milford, USA) was performed using Waters Acquity UPLC™ liquid chromatography, consisting of an autosampler, a binary pump, and a column temperature controller, coupled with a Xevo TQ™ MS/MS triple quadrupole detector. An Acquity UPLC™ BEH C18 column (100 × 2.1 mm i.d., 1.7 μm particle size) from Waters (Wexford, Ireland) was used for UPLC separation at 35 °C with a sample injection volume of 5 μL. MassLynx 4.1 software (Waters, Milford, USA) was used for instrument control and data processing. A binary mobile phase was composed of (A) 1 mM ammonium acetate in water and (B) methanol. A mobile phase gradient started at 10% B at a flow rate of 0.25 mL/min and went to 70% B at 2 min (curve 6) and held for 2.5 min then 95% B at 7 min (curve 6) and held until 9 min, and was concluded by column equilibration at the initial condition for 9.1 min (curve 6) and held for 1.9 min for a total run time of 11 min. MS determination was performed in the electrospray ionization positive/negative mode (ESI+ or ESI−) with monitoring of the ion transitions (precursor/product) using MRM for each pesticide. The MS source conditions were as follows: capillary voltage, 2.8 kV; source temperature, 110 °C; desolvation temperature, 350 °C; desolvation gas (N2) flow, 900 L h−1, and cone gas (N2) flow, 50 L h-1. Collision-induced dissociation was performed using argon as collision gas with a flow rate at 0.21 mL min−1.The cone voltage, collision energy parameters, ionization mode, and MRM transition for each pesticide analyzed are shown in Table 2.
Sample and Sample Preparation
Mangoes were obtained from different local markets in Haikou, Hainan Province, China. The fruits were skinned, cored, and homogenized in a food processor, with pulsed action until contents were uniform and had the consistency of smooth paste. Then, the samples were transferred to polypropylene (PP) bottles, labeled, and if necessary, stored at −20 °C until used.
Several steps of the QuEChERS method were optimized in order to obtain the best extraction. Ten grams of pesticide-free mango sample was transferred to a 50-mL polypropylene centrifuge tube; the matrices were spiked with adequate concentrations of standards, according to the spiking level at 0.05 mg/kg. After 30 min, four different buffer systems were added respectively. Four different buffer systems are listed as follows: (A) citrate buffer system (EN method), 10.0 mL acetonitrile and the preweights of 4.0 g anhydrous MgSO4, 1.0 g NaCl, 1.0 g sodium citrate, and 0.5 g disodium hydrogen citrate sesquihydrate; (B) none buffer system, 10.0 mL acetonitrile and the preweights of 4.0 g anhydrous MgSO4 and 1.0 g sodium chloride; (C) sodium acetate buffer system (AOAC method), 10.0 mL of 1% (v/v) acetic acid in acetonitrile, and the preweights of 6.0 g anhydrous MgSO4 and 1.5 g CH3COONa; and (D) ammonium acetate buffer system, 10.0 mL of 1% (v/v) acetic acid in acetonitrile, and the preweights of 4.0 g anhydrous MgSO4 and 1.7 g CH3COONH4.
Next, the tubes were immediately shaken for 1 min to avoid crystallization and were centrifuged at 4500 rpm for 5 min. Then, 1 mL of the supernatant was filtered through a 0.22-μm PTFE filter for UHPLC-MS/MS analysis. Six milliliters of the supernatant was pipetted into a 15-mL dispersive tube containing sorbents.
The optimization of QuEChERS sorbents was carried out by the ammonium acetate buffer system. Briefly, after 10.0 mL acidified acetonitrile and ammonium acetate extraction as mentioned above, five different types of dispersive tubes containing different sorbents were optimized: (i) AOAC method, 400 mg primary secondary amine (PSA), 400 mg GCB, and 1200 mg anhydrous MgSO4; (ii) EN method, 150 mg PSA, 15 mg GCB, and 900 mg anhydrous MgSO4; (iii) 60 mg multiwalled carbon nanotubes; (iv) 500 mg Zr-Sep+; and (v) 400 mg primary secondary amine (PSA), 80 mg GCB, 1200 mg anhydrous MgSO4, and 480 mg Zr-Sep+.
Then the tubes were shaken by hand for 1 min and centrifuged at 4500 rpm for 5 min. For the analysis by GC-MS/MS, a 2-mL aliquot of the upper layer was evaporated using a vacuum rotary evaporator and reconstituted with 1 mL of n-hexane.
Method Validation
The developed method was validated following the European Union SANTE/11945/2015 guidelines, which include recovery, precision, linearity, limits of detection (LODs), and limits of quantification (LOQs). Accuracy and precision analysis was conducted in terms of intra-day, inter-day, and intra-laboratory reproducibility studies. Intra-day accuracy and precision were studied by spiked mango blank samples with a mixture of the investigated pesticides for three levels at 10, 50, and 100 μg/kg. Each concentration level comprised five replicates. Spiked samples were left for 30 min prior to preliminary treatment. The experiments were repeated in three following days, producing results (n = 15) for inter-day accuracy and precision evaluation. Intra-laboratory reproducibility was studied by two different analysts using the same instrumentation, but using a self-produced solution for spiking procedures and calibration data. In total, 25 replicates (n = 25) were analyzed for each spiking level, and overall accuracy (recovery %) and precision (RSD%) are presented in Tables 3 and 4.
The calibration curves were obtained by injecting five levels of reference standard solutions. The matrix-matched calibration curves were constructed by a fortified appropriate volume mixed standard to the blank sample extract. Linearity was acceptable when the regression coefficient (r2) was > 0.99 with mass accuracy ≤ 5 ppm for all the analytes (SANTE 2015).
The matrix-matched standard was also used to evaluate the matrix effect (ME) for each analyte in green and ripe mango samples. ME values were calculated as the difference between the signal from the pesticide in the matrix and the signal in the injection solvent, divided by the signal in the injection solvent; the derived value was then expressed as a percentage: %ME = [(peak area of matrix standard − peak area of solvent standard) / peak area of solvent standard] × 100.
For the limit of detection (LOD) and the limit of quantification (LOQ) study, blank mango samples were fortified with lower concentration levels of pesticides (0.1, 0.2, 0.5, 1, 2, 5, 10 μg/kg). LOD was determined by the characteristic ion of the lowest spiked concentration pesticide which shows a chromatographic signal that is at least three times its respective peak to peak noise. LOQ was estimated based on the lowest fortified concentration, which provides suitable recovery and precision (SANTE 2015).
The measurement uncertainty was estimated according to the “top-down” approach using the data obtained in the validation study (Walorczyk 2014; Medina-Pastor et al. 2011). The major uncertainty sources were from precision and accuracy/bias; see Eq. (1). Combined uncertainty was calculated from the measurement uncertainty of the lowest spiking level and the high spiking level, by using Eq. (2). Finally, the relative expanded uncertainty was determined by using the coverage factor k = 2 at the confidence level of 95%.
where u(%) represents measurement uncertainty, RSD represents relative standard deviation (%), R represents recovery(%), uc(%) represents combined measurement uncertainty, u0.01(%) represents measurement uncertainty at 0.01 mg/kg, and u0.1(%) represents measurement uncertainty at 0.1 mg/kg.
Results and Discussion
In the study, 113 pesticides were analyzed by using the QuEChERS procedure. The optimized parameters of GC-MS/MS and UHPLC-MS/MS for all the pesticides are shown in Tables 1 and 2, which presented retention time, precursor ions, product ions, and collision energy. Due to the complexity of the green and mature mango matrices and the large number of the pesticides with different physical and chemical properties, the extraction and cleanup procedure must be paid more attention in the study.
Sample Extraction Solvents and Buffer Systems
Solvent extraction is a critical step for the analytical method to reduce the interferents in matrices and get acceptable sensitivity. According to the literature (Nannan et al. 2016), acetonitrile has good recoveries in QuEChERS extraction and the extracts could easily be separated from the aqueous phase through a salting-out effect. Thus, acetonitrile was chosen for the sample extraction. However, acetonitrile was seldom used in GC analysis for its large solvent expansion volume, high toxicity, and low volatility (Li et al. 2008). Therefore, acetonitrile extracts were evaporated using a vacuum rotary evaporation apparatus and redissolved with n-hexane for GC-MS/MS analysis.
The buffer systems were composed of different extraction solvents and salts. The different pH values of buffer systems would result in different recoveries for some specific pesticides, which undergo ionization and/or degradation during extraction. Besides, through a hydration pattern and salting out, the salt could increase the solubility of the polar pesticides in the organic phase (Frenich et al. 2008). Therefore, different buffer systems were investigated to improve the sensitivity.
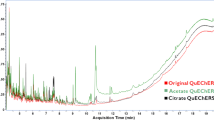
Figures 1 and 2 show the obtained results of the selected pesticides of green mango using different buffer systems. It was demonstrated that the extraction efficiency of pesticides can be enhanced by using acetate buffers (method C and method D). The response areas of carbosulfan is almost zero using method A and B buffer systems. On the other hand, when method C and method D were used for the extraction of the target compound, the signal of carbosulfan dramatically increased. With the acetate buffers, the extraction efficiency of fenthion, bifenthrin, and others were also improved. This was possible because the pH of the green mango matrix was close to 2.0. In the presence of sodium acetate (method C) and ammonium acetate (method D) buffer systems, the pH of the extract gets close to 5.2–5.5, then the responses of some unstable pesticides in acid/neutral condition were improved. Moreover, with the addition of 1% acetic acid-acetonitrile, better extraction of instable pesticides was observed. Although sodium acetate (method C) and ammonium acetate (method D) generally gave preferable results, the best buffer system was with ammonium acetate. Thus, ammonium acetate was selected for QuEChERS salt package for green mango pretreatment.

Response of the selected pesticides extracted from 10 g of green mango using different buffer systems and analyzed by GC-MS/MS

Response of the selected pesticides extracted from 10 g of green mango using different buffer systems and analyzed by UHPLC-MS/MS
Figures 3 and 4 show the mean responses of the chosen pesticides extracted from 10 g of ripe mango using different buffer systems. It demonstrated no significant signal enhancement for most of the pesticides, except for dichlorvos and carbosulfan. The higher signal of dichlorvos and carbosulfan was observed using sodium acetate (method C) and ammonium acetate (method D). For the responses of pesticides, method C was comparable to method D. Therefore, the ammonium acetate buffer system (method D) was added in the ripe mango pretreatment to improve the extraction efficiency in the study.

Response of the selected pesticides extracted from 10 g of ripe mango using different buffer systems and analyzed by GC-MS/MS

Response of the selected pesticides extracted from 10 g of ripe mango using different buffer systems and analyzed by UHPLC-MS/MS
Evaluation of Purification Sorbents
The cleanup step was important for target analysis because the matrices of mangoes were complex. Besides, due to a few drawbacks in maintaining GC-MS/MS compared with UHPLC-MS/MS, the cleanup steps were carried out only for solution which was injected into GC-MS/MS in this study. Various sorbents including GCB, PSA, Zr-Sep+, and multiwalled carbon nanotubes were tested; these sorbents were considered useful for removing special coextractive substances. PSA was a sorbent exhibiting retaining organic acids, sugars, and pigments (Nieto-Garcia et al. 2015). GCB could be useful to remove sterols and pigments such as carotenoid, which is the main component in mango. Zr-Sep+ is a sorbent which enables Lewis acid/base interactions for efficient interference removal (Rejczak and Tuzimski, 2017a, b). Multiwalled carbon nanotubes exhibited hydrophobicity and high surface area, which could remove lots of interferents (Deme and Upadhyayula 2015). Therefore, in order to find an efficient cleanup method to increase the lifetime of GC components from the column and inlet liner, we evaluated the five approaches by employing the following: (i) AOAC method, 400 mg PSA + 400 mg GCB; (ii) EN method, 150 mg PSA + 15 mg GCB; (iii) 60 mg multiwalled carbon nanotubes; (iv) 500 mg Zr-Sep+; and (v) 400 mg PSA + 80 mg GCB + 480 mg Zr-Sep+. The experiments were carried out at the concentration level of 0.05 mg/kg by adding the blank mango samples with the tested pesticides. The samples were extracted with 1% acetic acid-acetonitrile and salting out by anhydrous MgSO4 and CH3COONH4, and then underwent dispersive SPE cleanup by the five methods. A total of 56 compounds for GC-MS/MS were analyzed by different purification sorbents. It was a qualified result when the recoveries were between 70 and 120% and RSD < 20%, as stipulated in the EU method validation criteria of SANTE, 2015. The number of compounds with qualified result can be observed in Fig. 5.

Numbers of pesticide with recovery values between 70 and 120% analyzed by GC-MS/MS
Maximum recoveries of the pesticides were achieved when 400 mg PSA and 400 mg GCB were used as sorbents. But when using 400 mg PSA and 400 mg GCB as sorbent, the recoveries of profenofos, triazophos, chlorpyrifos, and phosalone were between 60 and 70%. Their low recoveries could be caused by absorption of planar molecule pesticides by GCB (Anastassiades et al. 2003; Slowik-Borowiec et al. 2015). Besides, in comparison with the EN method, efficiency of the purification using the AOAC method could be confirmed through visible decoloration of the extracts. Hence, we selected 400 mg PSA and 400 mg GCB as cleanup sorbent for further experiments.
Matrix Effect
The matrix effect should be calculated during method development, since specific co-extracted interfering components with analytes result in enhancement or suppression of the analytic signal, leading to overestimation or underestimation of the analyte concentrations (Rejczak and Tuzimski 2017b). There are several factors that affect the matrix effect, especially the nature of the matrix and the efficiency of sample preparation. In this study, the matrix effect (suppression or enhancement of exceeding 25%) is believed to significantly affect quantitative analytical results (Zhu et al. 2014). An assessment of matrix effects in raw and ripe mangoes matrices was carried out by comparing the detector response of the analyte in the specified matrices to the signal in pure solvent. The values of the matrix effect (%ME) are presented in Tables 3 and 4. For GC-MS/MS analysis, %ME values ranged from 2 to 25% in green mango and %ME values ranged from − 10 to 25% in ripe mango. No pesticide exhibited a significant matrix effect in green and ripe mango for GC-MS/MS analysis. It is possible that the cleanup pretreatment for GC-MS/MS is relatively complete. For UHPLC-MS/MS analysis, the strongest suppression of 85% was shown for abamectin and the strongest signal enhancement of 186% was determined for carbendazim in green mango. In the case of ripe mango, the strongest signal suppression (59%) for aldicarb sulfoxide and enhancement (210%) for carbendazim are observed. For a better understanding of ME% by UHPLC-MS/MS analysis, the results were classified into three groups: no significant matrix effect (± 0–25%), medium matrix effect (± 26–40%), and high matrix effect (higher than ± 41%) (Fig. 6a, b). No significant matrix effect was shown for 63 and 61% of the analytes for raw and ripe mangoes by UHPLC-MS/MS analysis. Medium matrix effect was shown for 16 and 26% of the analytes for raw and ripe mangoes by UHPLC-MS/MS analysis. A high matrix effect was shown for 21 and 12% of the analytes for raw and ripe mangoes by UHPLC-MS/MS analysis. In general, the matrix effect for UHPLC-MS/MS analysis is a little stronger than that for GC-MS/MS analysis. The most effective way to avoid matrix effects is to apply matrix-matched calibration standards. The results indicated that it is necessary to introduce matrix-matched calibration for UHPLC-MS/MS analysis to reduce the matrix effect, and directly employ solvent-matched calibration for GC-MS/MS analysis to save time, labor, and solvent.

a Percentage values of matrix effect (%ME) observed in green mango by UHPLC-MS/MS analysis. b Percentage values of matrix effect (%ME) observed in ripe mango by UHPLC-MS/MS analysis
Method Validation
The proposed QuEChERS method was evaluated for 113 pesticides in raw and ripe mango matrices. A validation study was carried out by accuracy (Recovery%) and precision (RSD%) including intra-day, inter-day (three following days), and intra-laboratory reproducibility. Overall accuracy and precision (n = 25) are presented in Tables 3 and 4.
Overall recoveries for most of pesticides were between 70 and 120% with RSD below 20% as shown in Tables 3 and 4. Some of the pesticides (chlorpyrifos, profenofos, triazophos, phosalone) had recoveries higher than 68%, but lower than 76% with RSD below 13%. The recoveries of carbosulfan were between 56 and 65% in ripe and raw mango matrices with RSD below 8%. Two of the pesticides of iprodione and azoxystrobin had recoveries higher than 120%, but lower than 130%, with RSD below 11%.
Linearity was evaluated in all matrices, and the linear range was studied at five concentration levels, between 5 and 500 μg/kg. Good linearity guaranteed accurate quantification for all target pesticides, as the coefficients of determination (R2) equal or higher than 0.99.
The LODs and LOQs were also investigated to evaluate the performance of the present method. LOD values were calculated at a signal to noise ratio greater than or equal to 3 (S/N ≥ 3), and LOQ was estimated based on the minimum fortified concentration, which has been demonstrated to be accurately quantified by the method (SANTE 2015). In the raw mango matrix, LODs and LOQs ranged from 0.01 to 4 μg/kg and from 0.05 to 10 μg/kg, respectively. In ripe mango, 113 pesticides had LODs that ranged from 0.02 to 3.5 μg/kg, and LOQs from 0.07 to 10 μg/kg, respectively. The values are presented in Tables 3 and 4.
There were many sources of uncertainties derived from steps of the presented method. The “top-down” approach could simplify the calculation procedure of uncertainties and evaluate the measurement uncertainties of all analytes using the data from the validation study including recovery and RSD; see Eqs. (1) and (2). The expanded measurement uncertainties were obtained by multiplying the combined standard uncertainty by 2 (coverage factor k = 2, i.e., 95% confidence level). The results are given in Tables 3 and 4. As seen, in the case of raw mango, the pesticides had uncertainties between 6% (alpha-HCH) and 38% (phosalone) with the overall average uncertainty of 21%. In the case of ripe mango, the pesticides had uncertainties between 5% (alpha-HCH) and 41% (phosalone) with the overall average uncertainty of 21%. In the green and ripe mango matrix, the majority of the compounds had uncertainties < 30%. The highest uncertainty value was obtained for phosalone in consequence of poor recovery and high RSD of absorption of planar molecule pesticides by GCB. The expanded measurement uncertainty values below 50% for all the analytes proved that the method is suitable for the determination of the target pesticides in green and ripe mango samples.
Comparison of Proposed Method with Other Analytical Methods
It was the first time to propose the QuEChERS method for different maturity levels of mangoes. The method was comparable with other published multiresidue methods for the mango matrix in terms of the method validation parameters (Sivaperumal et al. 2017). Different cleanup sorbents were evaluated in terms of GCB, PSA, and relatively new sorbents such as Zr-Sep+ and multiwalled carbon nanotubes. In addition, the proposed method has advantages in determining the highest number of pesticides (n = 113). Remarkably, the proposed method showed better accuracy, precision, and measurement uncertainty for green and ripe mango. Thus, it was confirmed that the proposed method is efficient for screening and quantitation of residues in different maturity level of mango matrices.
Real Sample Analysis
Forty-five samples from local markets were determined to validate the application of the established method. One blank sample fortified with 100 μg/kg was used for quality control. It was demonstrated that a few of mango samples contain pesticide residues, including chlorpyrifos, imidacloprid, cypermethrin, difenoconazole, cyhalothrin, thiabendazole, azoxystrobin, and carbendazim. Chlorpyrifos was found in sample no. 7 (68 μg/kg), no. 10 (110 μg/kg), no. 43 (110 μg/kg), and no. (120 μg/kg), at concentrations higher than the MRLs established by EU (50 μg/kg) in mangoes. Imidacloprid was detected in 20% of the samples at values lower than 200 μg/kg. But China did not set MRLs (maximum residue limits) for chlorpyrifos and imidacloprid in mangoes at present (China National Food Safety Standard 2016). The two insecticides were widely used in crop growth. Cypermethrin was detected in 17 samples at a trace level (< 200 μg/kg), which was lower than MRLs set by the China National Food Safety Standard (700 μg/kg) in mangoes. This is an insecticide widely used in fruits and vegetables. Sample no. 8 showed a positive result of difenoconazole (86 μg/kg), which was fungicide and exceeded the MRL of the China National Standard (70 μg/kg). Cyhalothrin was found in no. 37 (140 μg/kg) and no. 43 (50 μg/kg), but the concentrations were lower than MRLs of mangoes (200 μg/kg). Six samples were detected to contain thiabendazole and azoxystrobin, but the concentration level was very low (< 50 μg/kg). The MRLs of thiabendazole and azoxystrobin are 5 and 1 mg/kg in mangoes, respectively. Finally, carbendazim was detected in 40% of samples and the no. 23 sample (0.8 mg/kg) above the MRL (0.5 mg/kg). Carbendazim was the main degradation product of benomyl and thiophanate methyl. Thiabendazole, azoxystrobin, and carbendazim are usually applied in post-harvest on mangoes to protect them from decay caused by various fungal pathogens.
Conclusion
The proposed QuEChERS extraction method using acidified acetonitrile and ammonium acetate proved to be suitable for the extraction of 113 pesticides in green/ripe mangoes. The optimization conditions of pretreatment revealed that the use of ammonium acetate and 400 mg PSA + 400 mg GCB is sufficient for extraction and cleanup of 56 pesticides, which were analyzed by GC-MS/MS. At the same time, the use of ammonium acetate without cleanup is effective for 57 pesticides, which were analyzed by UHPLC-MS/MS. With the advantages of high sensitivity, simple maintenance, and strongly withstanding matrix interference, UHPLC-MS/MS enabled an unnecessary cleanup, which prevents from being more time-consuming and from susceptibility to analytical errors.
From the study, pesticides for GC-MS/MS analysis did not perform a significant matrix effect and the values of the matrix effect were in the range of ± 25%. However, for pesticides used in UHPLC-MS/MS analysis, the values of the matrix effect observed in the range − 85 to 186% and − 59 to 210% in raw and ripe mangoes samples, respectively. Therefore, the use of solvent solution standard is enough to quantify those pesticides for GC-MS/MS. In order to compensate the matrix effect, matrix-matched standards were used to calculate the concentration of pesticides for UHPLC-MS/MS analysis.
The optimized QuEChERS extraction method combined with GC-MS/MS and UHPLC-MS/MS for the determination of 113 pesticides in raw/ripe mangoes has considerable superiorities in respect of being high-throughput, simple, fast, and cheap. This method gave very good results including recovery, precision, linearity, LOD, and LOQ. In conclusion, the modified method meets the China criteria, and thus is useful for routine analyses to trace level quantification of the pesticide residues from different maturity degrees of mango.
References
Amvrazi EG, Albanis TA (2006) Multiresidue method for determination of 35 pesticides in virgin olive oil by using liquid-liquid extraction techniques coupled with solid-phase extraction clean up and gas chromatography with nitrogen phosphorus detection and electron capture detection. J Agric Food Chem 54(26):9642–9651
Anagnostopoulos CJ, Sarli PA, Liapis K, Haroutounian SA, Miliadis GE (2012) Validation of two variations of the QuEChERS method for the determination of multiclass pesticide residues in cereal-based infant foods by LC-MS/MS. Food Anal Methods 5(2):664–683
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in produce. J AOAC Int 86(2):412–431
Banerjee K, Oulkar DP, Patil SB, Jadhav MR, Dasgupta S, Patil SH, Bal S, Adsule PG (2009) Multiresidue determination and uncertainty analysis of 87 pesticides in mango by liquid chromatography-tandem mass spectrometry. J Agric Food Chem 57(10):4068–4078
Castillo MLR, Rodriguez-Valenciano M, Moreno FP, Blanch GP (2012) Evaluation of pesticide residue contents in fruit juice by solid-phase microextraction and multidimensional gas chromatography coupled with mass spectrometry. Talanta 89:77–83
China National food safety standard (2016) Maximum residue limits for pesticides in food. GB:2763–2016
Chamkasem N, Ollis LW, Harmon T, Lee S, Mercer G (2013) Analysis of 136 pesticides in avocado using a modified QuEChERS method with LC-MS/MS and GC-MS/MS. J Agric Food Chem 61:2315–2329
Cherta L, Beltran J, Pitarch E, Hernandez F (2013) Comparison of simple and rapid extraction procedures for the determination of pesticide residues in fruit juices by fast gas chromatography-mass spectrometry. Food Anal Methods 6(6):1671–1684
Chu XG, Hu XZ, Yao HY (2005) Determination of 266 pesticide residues in apple juice by matrix solid-phase dispersion and gas chromatography-mass selective detection. J Chromatogr A 1063(1-2):201–210
Deme P, Upadhyayula VVR (2015) Ultra performance liquid chromatography atmospheric pressure photoionization high resolution mass spectrometric method for determination of multiclass pesticide residuesin grape and mango juices. Food Chem 173:1142–1149
EC Council Directive 2002/657/EC (2002) Implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results.
Filho AM, Santos FN, Pereira PAP (2010) Development, validation and application of a methodology based on solid-phase microextraction followed by gas chromatography coupled to mass spectrometry (SPME/GC-MS) for the determination of pesticide residues in mangoes. Talanta 81:346–354
Food and Agriculture Organization of the United Nations (2013) Food and agricultural commodities production. < http://faostat.fao.org/site/339/default.aspx> 16.05.14.
Frenich AG, Vidal JLM, Pastor-Montoro E, Romero-Gonzalez R (2008) High-throughput determination of pesticide residues in food commodities by use of ultra-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 390:947–959
Furlani RPZ, Marcilio KM, Leme FM, Tfouni SAV (2011) Analysis of pesticide residues in sugarcane juice using QuEChERS sample preparation and gas chromatography with electron capture detection. Food Chem 126(3):35–1287
Hayward DG, Wong JW, Shi F, Zhang K, Lee NS, Dibenedetto AL, Hengel MJ (2013) Multiresidue pesticide analysis of botanical dietary supplements using salt-out acetonitrile extraction, solid-phase extraction cleanup column, and gas chromatography-triple quadrupole mass spectrometry. Anal Chem 85:4686–4693
He ZY, Wang L, Peng Y, Luo M, Wang WW, Liu XW (2015) Multiresidue analysis of over 200 pesticides in cereals using a QuEChERS and gas chromatography-tandem mass spectrometry-based method. Food Chem 41:372–380
Jin BH, Xie LQ, Guo YF, Pang GF (2012) Multi-residue detection of pesticides in juice and fruit wine: a review of extraction and detection methods. Food Res Int 46(1):399–409
Koesukwiwat U, Lehotay SJ, Leepipatpiboon N (2011) Fast, low-pressure gas chromatography triple quadrupole tandem mass spectrometry for analysis of 150 pesticide residues in fruits and vegetables. J Chromatogr A 1218(39):7039–7050
Kwon H, Lehotay SJ, Geis-Asteggiante L (2012) Variability of matrix effects in liquid and gas chromatography-mass spectrometry analysis of pesticide residues after QuEChERS sample preparation of different food crops. J Chromatogr A 1270:235–245
Li L, Li W, Ge J, Wu YJ, Jiang SR, Liu FM (2008) Use of graphitic carbon black and primary secondary amine for determination of 17 organophosphorus pesticide residues in spinach. J Sep Sci 31:3588–3594
Medina-Pastor P, Valverde A, Pihlstrom T, Masselter S, Gamon M, Mezcua M, Rodriguez-Torreblanca C, Fernandez-Alba AR (2011) Comparative study of the main top-down approaches for the estimation of measurement uncertainty in multiresidue analysis of pesticides in fruits and vegetables. J Agric Food Chem 55:7609–7619
Nannan P, Tielong W, Jieye H (2016) Method validation and dissipation kinetics of four herbicides in maize and soil using QuEChERS sample preparation and liquid chromatography tandem mass spectrometry. Food Chem 58:793–800
Navickiene S, Aquino A, Bezerra DSS (2010) A matrix solid-phase dispersion method for the extraction of seven pesticides from mango and papaya. J Chromatogr Sci 48(9):750–754
Nieto-Garcia AJ, Romero-Gonzalez R, Frenich AG (2015) Multi-pesticide residue analysis in nutraceuticals from grape seed extracts by gas chromatography coupled to triple quadrupole mass spectrometry. Food Control 47:63–380
Palenikova A, Martinez-Dominguez G, Arrebola FJ, Romero-Gonzalez R, Hrouzkova S, Frenich AG (2015) Multifamily determination of pesticide residues in soya-based nutraceutical products by GC/MS-MS. Food Chem 173:796–807
Prestes OD, Friggi CA, Adaime MB, Zanella R (2009) QuEChERS—a modern sample preparation method for pesticide multiresidue determination in food by chromatographic methods coupled to mass spectrometry. Quimica Nova 32:1620–1634
Qu LJ, Zhang H, Zhu JH, Yang GS, Aboul-Enein HY (2010) Rapid determination of organophosphorous pesticides in leeks by gas chromatography-triple quadrupole mass spectrometry. Food Chem 122(1):327–332
Rejczak T, Tuzimski T (2015a) A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem 13:980–1010
Rejczak T, Tuzimski T (2015b) Recent trends in sample preparation and liquid chromatography/mass spectrometry for pesticide residue analysis in food and related matrixes. J AOAC Int 98(5):1143–1162
Rejczak T, Tuzimski T (2016) Simple, cost-effective and sensitive liquid chromatography diode array detector method for simultaneous determination of eight sulfonylurea herbicides in soya milk samples. J Chromatogr A 78:56–65
Rejczak T, Tuzimski T (2017a) Method development for sulfonylurea herbicides analysis in rapeseed oil Samples by HPLC-DAD: comparison of zirconium-based sorbents and EMR-lipid for clean-up of QuEChERS Extract. Food Anal Methods 10:3666-3679
Rejczak T, Tuzimski T (2017b) QuEChERS-based extraction with dispersive solid phase extraction clean-up using PSA and ZrO2-based sorbents for determination of pesticides in bovine milk samples by HPLC-DAD. Food Chem 84:225–233
Rissato SR, Galhiane MS, Knoll FRN, Apon BM (2004) Supercritical fluid extraction for pesticide multiresidue analysis in honey: determination by gas chromatography with electron-capture and mass spectrometry detection. J Chromatogr A 1048(2):153–159
Rizzetti TM, Kemmerich M, Martins ML, Prestes OD, Adaime MB, Zanella R (2016) Optimization of a QuEChERS based method by means of central composite design for pesticide multiresidue determination in orange juice by UHPLC-MS/MS. Food Chem 196:25–33
SANTE (2015) Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. SANTE/11945/2015 30 November-1 December 2015 rev. 0
Sivaperumal P, Salauddin A, Kumar AR, Santhosh K, Rupal T (2017) Determination of pesticide residues in mango matrices by ultra high-performance liquid chromatography coupled with quadrupole time-of-flight massspectrometry. Food Anal Methods 10:2346–2357
Slowik-Borowiec M, Szpyrka E, Walorczyk S (2015) Gas chromatographic determination of pesticide residues in white mustard. Food Chem 173:997–1005
Srivastava AK, Rai S, Srivastava MK, Lohani M, Mudiam MKR, Srivastava LP (2014) Determination of 17 organophosphate pesticide residues in mango by modified QuEChERS extraction method using GC-NPD/GC-MS and hazard index estimation in Lucknow, India. PLOS ONE 9(5):e96493. https://doi.org/10.1371/journal.pone.0096493
Stajnbaher D, Zupancic-Kralj L (2003) Multiresidue method for determination of 90 pesticides in fresh fruits and vegetables using solid-phase extraction and gas chromatography-mass spectrometry. J Chromatogr A 1015(1-2):185–198
Wong JW, Zhang K, Tech K, Hayward DG, Makovi CM, Krynitsky AJ, Schenck FJ, Banerjee K, Dasgupta S, Brown D (2010) Multiresidue pesticide analysis in fresh produce by capillary gas chromatography –mass spectrometry/selective ion monitoring (GC-MS/SIM) and –tandem mass spectrometry (GC-MS/MS). J Agric Food Chem 58(10):5868–5883
Walorczyk S (2014) Validation and use of a QuEChERS-based gas chromatographic-tandem mass spectrometric method for multiresidue pesticide analysis in blackcurrants including studies of matrix effects and estimation of measurement uncertainty. Talanta 120:106–113
Yang X, Zhang H, Liu Y, Wang J, Zhang YC, Dong AJ, Zhao HT, Sun CH, Cui J (2011) Multiresidue method for determination of 88 pesticides in berry fruits using solid-phase extraction and gas chromatography-massspectrometry: determination of 88 pesticides in berry using SPE and GC-MS. Food Chem 127(2):855–865
Zhao MA, Feng YN, Zhu YZ, Kim JH (2014) Multi-residue method for determination of 238 pesticides in Chinese cabbage and cucumber by liquid chromatography-tandem mass spectrometry: comparison of different purification procedures. J Agric Food Chem 62:11449–11456
Zhu YZ, Zhao MA, Feng YN, Kim JH (2014) Multiresidue method for the determination of 227 pesticides in hot pepper (Capsicum annuum L.) by liquid chromatography with tandem mass spectrometry. J Sep Sci 37(20):119–2954
Acknowledgements
This work was financially supported by the special fund of the Supervision Bureau of Quality and Safety on Agro-Products in the Ministry of Agriculture and Rural Affairs of China (GJFP201700402).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Pingping Li declares that she has no conflict of interest. Yun Duan declares that he has no conflict of interest. Huilin Ge declares that he has no conflict of interest. Yue Zhang declares that she has no conflict of interest. Xiaofang Wu declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent is not applicable for this study.
Rights and permissions
About this article

Cite this article
Li, P., Duan, Y., Ge, H. et al. Multiresidue Analysis of 113 Pesticides in Different Maturity Levels of Mangoes Using an Optimized QuEChERS Method with GC-MS/MS and UHPLC-MS/MS. Food Anal. Methods 11, 2742–2757 (2018). https://doi.org/10.1007/s12161-018-1263-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1263-5