
Abstract
A rapid, simple, and sensitive multiresidue method for analysis of 53 pesticides in fruit and vegetables by ultra-performance liquid chromatography (UPLC) coupled to triple-quadrupole tandem mass spectrometry (MS-MS) has been developed and validated. Prior to analysis, analytes were extracted by use of buffered QuEChERS (quick, easy, cheap, effective, rugged, safe) methodology without further cleanup for non fatty matrices. Chromatographic conditions were optimised in order to achieve a fast separation in multiple reaction monitoring (MRM) mode. Indeed, more than 50 pesticides can be separated in less then 10 min. Four common representative matrices (cucumber, orange, strawberry, and olive) were selected to investigate the effect of different matrices on recovery and precision. Mean recoveries ranged from 70 to 109% with relative standard deviations lower than 20% for all the pesticides assayed in the four selected matrices. The method has been applied to the analysis of 200 vegetable samples, and imidacloprid was the pesticide most frequently found, with concentrations ranging from 0.01 to 1.00 mg kg−1. This methodology combines the advantages of both QuEChERS and UPLC-MS-MS producing a very rapid, sensitive, and reliable procedure which can be applied in routine analytical laboratories.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticides are among the most investigated priority pollutants in agricultural products due to their wide use and toxicity. The control of pesticide residues in food for commercial purposes involves many samples and pesticide residue analyses are usually costly and time-consuming, hence multiresidue methods are usually applied in regulatory pesticide monitoring because they increase the productivity of laboratories and reduce the cost of analysis.
During recent years, a number of papers have been published on the analysis of pesticides, and some of their degradation and transformation products, by gas chromatography coupled to tandem mass spectrometry (GC-MS-MS) [1–3] and liquid chromatography (LC)-MS-MS [4–7], providing good sensitivity, selectivity, and efficiency. Nowadays, LC-MS-MS is replacing GC methodologies, since pesticides are often more polar and less volatile, and it has been accepted as a routine technique for regulatory monitoring purposes in pesticide residue analysis [8].
The main drawbacks of the most commonly used procedures are that they require too much labour and time, and use large amounts of hazardous solvents [9]. For a method to be practical, it is necessary to consider cost of analysis, including reagents, equipment, labour, and environmental restrictions [10]. That is why high-throughput analyses are specially desired for rapid screening of numerous samples in routine laboratory analysis using fast and environmentally friendly methods.
Pesticide residue analysis methods involve two steps: extraction of target analytes from the matrix and chromatographic separation and determination; sample preparation is the bottleneck in the analysis of pesticide residues. Traditionally the extraction of pesticides from vegetables has been carried out by homogenization with organic solvent such as ethyl acetate [11] usually in a shaking apparatus or using a Polytron homogenizer [12]. In general, these methods are complicated, time-consuming, expensive, and require large amounts of solvents, so several approaches have been proposed to increase the performance of sample extraction, such as pressurized liquid extraction [13], microwave assisted extraction [13], and the QuEChERS (quick, easy, cheap, effective, robust, safe) method [14, 15]. This last technique minimises the number of sample-preparation steps and has become very popular in the last few years since it has been shown to be a powerful technique in analysis of pesticide residues in foodstuffs [16–18] and other compounds, for example drugs, in blood [19]. This methodology is based on the extraction of pesticides from the sample with acetonitrile. Removal of residual water and clean-up are performed simultaneously by using a rapid procedure, called dispersive solid-phase extraction [14], in which anhydrous MgSO4 and primary-secondary amine (PSA) sorbent are added before determination, reducing analysis cost, labour, waste, and glassware and increasing sample throughput.
The separation step also requires considerable time so it is mandatory to use fast analytical methodologies. In order to reduce chromatographic analysis time, several strategies have been used. Thus, different alternatives based on altering the basic geometric structure of the stationary phase (silica and polymer monoliths) [20] or exploiting the physicochemical conditions of the mobile phase (such as reducing the viscosity of the mobile phase by operating at higher temperatures) have been proposed [21]. Finally a last possibility, which has become very popular in the last few years, is the use of columns filled with particles of size below 2 μm, in what has been called “ultra-performance liquid chromatography” (UPLC) [22, 23]. This technique takes advantage of the flat nature of the van Deemter plot for stationary phases formed by particles smaller than 2 μm, generating higher chromatographic performance, and improving resolution, speed, and sensitivity [24]. The combination UPLC-MS-MS provides significant advantages concerning selectivity, sensitivity, and speed. Up to now, UPLC has been mainly applied to pharmaceutical and metabolomic fields [25–28]; few papers have been found for the determination of pesticides in food samples [29–32].
Considering these characteristics, UPLC can therefore provide high sample throughput and good sensitivity when it is combined with QuEChERS extraction procedure. Furthermore, sample pretreatment can be minimized when MS-MS detection is used, so some modifications of the QuEChERS procedure are proposed in this work, such as eliminating a clean up step and reducing sample preparation.
Thus, in this paper, a rapid multiresidue method has been developed which uses UPLC-MS-MS with an electrospray interface (ESI) and an extraction procedure based on QuEChERS methodology for the determination of 53 multiclass pesticides which are commonly used in southeast Spain. Some of these pesticides, for example thiamethoxam, fensulfothion, azaconazole, spinosad, flutalonil, and trycresyl phosphate, have not previously been investigated by use of QuEChERS. In order to evaluate the applicability of the proposed procedure, sample matrices representative of a variety of fruits and vegetables, following SANCO European guidelines [33, 34], have been selected. Thus cucumber was chosen as a representative commodity with a high water content, orange as a fruit with a high acid content, strawberry as a commodity with a high sugar content, and olive as a commodity with a high fat content. The high resolving capacity of the UPLC method combined with the fast sample preparation approach will offer significant benefits for pesticide residues analysis in food, which can be used in routine laboratories.
Experimental
Chemicals and reagents
Pesticide reference standards (purity higher than 99%) were purchased from Dr Ehrenstofer (Augsburg, Germany) and Riedel-de Haën (Seelze-Hannover, Germany). Stock standard solutions of individual compounds (with concentrations ranging from 200 and 300 mg L−1), were prepared by exact weighing of the powder and dissolution in 50 mL methanol, acetonitrile, or acetone. They were stored at 4 °C in the dark, for no more than 4 months, checking that no changes in their concentrations were detected within this period. A multicomponent working standard solution (2 mg L−1 concentration of each compound) was prepared by appropriate dilution of the stock solutions with methanol; this solution was stored under refrigeration at 4 °C. Anhydrous sodium sulphate, sodium hydroxide, acetic acid (content >97%), formic acid (content >98%), and ammonium acetate were obtained from Panreac (Barcelona, Spain). Primary-secondary amine (PSA) bonded silica (100 g, bulk) was supplied by Supelco (Bellefonte, PA, USA). Varian (Harbor City, CA, USA) 500 mg Florisil cartridges were used for clean-up of fatty matrices. Methanol, acetone, and acetonitrile (pesticide residue grade solvent) were purchased from Panreac. Highly purified water (Milli-Q, Millipore, Bedford, USA) was used throughout for the preparation of buffers, mobile phase, and other reagents.
Apparatus
Chromatographic analyses were performed with an Acquity UPLC system (Waters, Milford, MS, USA) and separations were achieved using an Acquity UPLC BEH C18 column (100 × 2.1 mm, 1.7-μm particle size) from Waters.
MS-MS detection was performed on a Micromass Quattro Premier tandem quadrupole mass spectrometer (Waters, Manchester, UK). The instrument was operated using an electrospray (ESI) source in positive mode. The ionisation source parameters were: capillary voltage 3.0 kV, extractor voltage 5 V, source temperature 110 °C, desolvation temperature 350 °C, cone gas flow 80 L h−1 and desolvation gas flow 600 L h−1 (both gases were nitrogen). Collision-induced dissociation was performed using argon as collision gas at a pressure of 4 × 10−3 mbar in the collision cell. The multiple reaction monitoring (MRM) transitions and the cone and collision energy voltages applied are summarized in Table 1. Data acquisition were performed using MassLynx 4.0 software with QuanLynx software (Waters).
Centrifugation was performed with a high-volume centrifuge equipped with a bucket rotor (4 × 250 mL) from Orto Alresa, Mod. Consul (Madrid, Spain). A Heidolph model Reax 2000 vortex mixer and an analytical AB204-S balance (Mettler Toledo, Greinfesee, Switzerland) were used. All pH measurements were made with a Crison Basic 20 pH-meter (Insulab, Valencia, Spain) equipped with a combined AgCl-glass electrode assembly. A Braum MX 32 kitchen blender (Barcelona, Spain) was used to process samples.
Extraction procedure
Pesticides were extracted from non-fatty fruit and vegetables using an extraction procedure based on the buffered QuEChERS procedure [16]. Briefly, 1 kg sample was reduced by half by quartering and using two opposite quarters. These quarters were homogeneously mixed with a blender until the texture of the sample was homogeneous, and ca 200 g was transferred to a screw-top container. A representative subsample (10 g) was then weighed in a polypropylene centrifuge tube (40 mL), 10 mL 1% of acetic acid in acetonitrile solution was added, and the mixture was vortex mixed for 1 min. Afterwards, 4 g anhydrous magnesium sulfate and 1 g ammonium acetate were added and the tubes were shaken immediately for 1 min. After centrifugation at 4,300 g for 5 min the supernatant was analysed by UPLC-MS-MS.
For the extraction of pesticides from fatty matrices, after the centrifugation step, the upper layer was transferred on to a Florisil cartridge, collecting the cleaned extract. Finally, 1 mL was transferred to a vial and analysed by UPLC-MS-MS.
Chromatographic analysis
The standards and sample extracts were separated by UPLC with a mobile phase gradient prepared from 0.01% formic acid in water (eluent A) and methanol (eluent B). The gradient profile started at 90% eluent A and decreased linearly to 10% in 5 min. This composition was held for a further 2 min before being returned to the initial conditions in 0.5 min, followed by a re-equilibration time of 2.5 min, giving a total run time of 10 min. The column temperature was maintained at 30 °C and the flow rate was 0.35 mL min−1. The injection volume was 5 μL.
Validation study
Four validation data sets were carried out for each type of sample, based on the European SANCO guideline [34] and European Commission Decision 2002/657 [35]. Linearity was studied using matrix-matched calibration by analysing samples of orange, strawberry, cucumber, and olive spiked at four concentrations between 10 and 500 μg kg−1 (10, 50, 150, and 500 μg kg−1). Precision and accuracy of the method were tested with spiked samples of each commodity. Thus, recovery and repeatability were evaluated at three concentrations (11.5, 50, and 150 μg kg−1), analyzing six replicates for the lowest and highest levels and five for the middle level. Interday precision was evaluated at 11.5 μg kg−1. For that purpose, five spiked samples were analysed daily for a period of 6 days. Furthermore, limits of detection (LOD) and quantification (LOQ), and uncertainty, were also evaluated in the four matrices.
Samples
For development of a multiresidue method applicable to a variety of vegetable matrices, blank samples of four types of commodity were obtained from local organic produce stores and used to prepare fortified samples and matrix-matched calibration standards for validation purposes. Thus, orange was selected as a food commodity with high acidity, strawberry as a high sugar-content commodity, cucumber as high water-content commodity, and olive as a representative matrix with a high fat content.
Samples of the different matrices were obtained from greenhouses located in the province of Almeria (southeast of Spain) and were analysed within 24 h.
Results and discussion
Selection of MS-MS conditions
To optimise the MS-MS conditions for each analyte, experiments were carried out by infusion of a standard solution of 15 mg L−1 of each pesticide using electrospray ionisation in positive mode. Full-scan mass spectra and MS-MS spectra were acquired in order to obtain at least one precursor and two product ions for each compound for both identification and quantification purposes, selecting the most abundant product ion for quantification and the others as confirmation transitions. Table 1 indicates MS-MS transitions for identification and quantification, and cone voltages and collision energy values optimised for each of the compounds selected. Other parameters, for example desolvation and cone gas flow, source and desolvation temperature and capillary voltages were studied, indicating the optimum working conditions in the Experimental section.
UPLC-MS-MS determination
For the optimisation of a multiresidue pesticide method, special attention must be paid to the optimisation of the LC-MS system. Thus a suitable compromise between resolution and analysis time should be obtained, and the MS instrumentation should collect sufficient data across the peak to enable reliable integration.
To separate the selected pesticides and to provide and overall optimum response for MS-MS detection, gradient elution was performed, using methanol and an aqueous solution of formic acid (0.01%) as mobile phase components. With these conditions, good separation of the selected pesticides in less than 10 min and good MS sensitivity were achieved. Other parameters such as flow rate, injection volume, and column temperature were optimised in order to get a fast and reliable separation: 0.35 mL min−1 as optimum flow rate, 30 °C as column temperature, and 5 μL as injection volume. Under these conditions, retention times of the pesticides in all the matrices analysed were constant, ranging from 1.53 (pymetrozine) to 6.50 min (fenazaquin) with a relative standard deviation lower than 0.25% when the same column is used.
Using these conditions, the analytes were distributed in 17 overlapping functions, using a maximum of ten transitions (five pesticides) per function, centring the chromatographic peak in the time window, minimizing the risk of peak loss due to unexpected slight changes in retention time. Bearing in mind that peak widths were approximately 6–10 s, the number of spectral data points across the peaks was much smaller than for conventional HPLC, and this could be have a deleterious effect on the spectral quality. Different dwell times (from 5 to 50 ms) were tested to find the best detection parameters to obtain a sufficient number of data point across the peak, observing that negligible differences were found at low dwell times. The optimum value selected, in order to obtain at least 15 points per peak, was 15 ms; this provided reproducible results for determination and confirmation.
Using the conditions and parameters described in Experimental section, a peak had more than 10 points, which provided reproducible results for determination and confirmation.
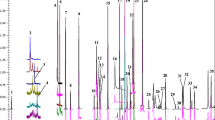
Figure 1 shows a representative chromatogram obtained from standards, using the conditions described in the Experimental section. Complete resolution between the pesticides was not achieved, but this can be resolved due to the high specificity of MS-MS detection.

Combined UPLC-MS-MS chromatogram from a standard mixture of pesticides (50 μg L−1) based on quantifying MS-MS transitions. The peak numbering is shown in Table 1. Conditions are as described in the Experimental section
Selection of the extraction and clean up procedure
As already indicated before, sample extraction was carried out following the buffered QuEChERS procedure [16]. However several modifications were included. Thus, it was observed that when ammonium acetate was used instead of sodium acetate before the centrifugation step, better sensitivities and recoveries were obtained.
Acetonitrile extraction was also evaluated for different clean up approaches for the four food commodities evaluated in this work. Thus, for non fatty matrices, (orange, strawberry, and cucumber) no clean up, dilution of the extract with water, and dispersive solid phase extraction with 50 mg PSA were necessary. When no clean up or 50 mg of PSA were applied, recoveries ranged between 70 and 110% for all the selected compounds. Considering that no clean up reduces the extraction time, and good chromatograms without interferences were obtained, no PSA was added for clean up. Finally when the extract was diluted with water before injection into the chromatographic system, irregular peak shapes were obtained, so no further clean up or dilution was carried out after the centrifugation step.
For samples with high fat content, such as olives, a clean up procedure after extraction with acetonitrile was necessary, because a small amount of fat can be coextracted during acetonitrile extraction, even though lipids are not very soluble in this solvent [36]. Thus, conventional solid-phase extraction (SPE) was used, using Florisil as sorbent [37]. In this case, a clean solution was obtained after passing the acetonitrile extract through the Florisil cartridge, and this was analysed by UPLC-MS-MS. The methodology was evaluated by spiking blank samples with 50 μg kg−1 pesticides; the results obtained are listed in Table 2. Good recoveries were obtained for the four matrices evaluated. It can be noted that pesticides such as thiamethoxam, fensulfothion, azaconazole, spinosad, and flutalonil, which have not previously been evaluated by QuEChERS, provided good recoveries, indicating that this extraction procedure can also be applied for these pesticides. A representative UPLC-MS-MS chromatogram of orange fortified at the 25 μg kg−1 level is shown in Fig. 2.

UPLC-MS-MS chromatograms obtained from a blank orange sample spiked at 25 μg kg−1. Selected pesticides were: pymetrozine (m/z 218.2 > 104.9), carbendazim (m/z 192.0 > 160.0), imidacloprid (m/z 256.2 > 209.3), thiacloprid (m/z 253.3 > 126.0) imazalil (m/z 297.2 > 159.0), flutalonil (m/z 324.4 > 242.3), flusilazol (m/z 316.1 > 247.3), diclobutrazol (m/z 328.2 > 70.2), and fenazaquin (m/z 307.3 > 161.2). Conditions are as described in the Experimental section
Finally it can be emphasized that considering the extraction time using the approach proposed in this paper (approx. 10 min) and the separation-detection time provide by UPLC-MS-MS (10 min), it is possible to determine more than 50 pesticides in one sample in less than 20 min. UPLC combined with QuEChERS provides a valuable tool for routine analyses, reducing analysis time and the volume of solvent used as mobile phase.
Validation
As already indicated, a validation procedure in accordance with the SANCO guideline [34] was carried out to evaluate response linearity, repeatability, interday precision, accuracy (expressed as recovery), limits of detection and quantification, and uncertainty for the four matrices studied in this work-cucumber, orange, strawberry and olive.
First, the compounds were identified by searching the appropriate retention time windows (RTWs), which were given by the mean retention time ± three standard deviations of the retention time of ten blank samples spiked with a mid-level calibration standard for each compound (Table 1). The identity was then confirmed by acquisition of two MS-MS transitions and comparing the intensity ratios of both (quantification and confirmation). Confirmation was considered reliable if the ratio was within the criteria laid down in the European Commission Decision 2002/657 [35]. Selectivity of the method was evaluated by analysing unspiked samples. The absence of any signal at the same elution time as the target pesticide suggested there were no matrix interferences that may give a false positive signal.
When electrospray ionisation is used, it is well known that the presence of matrix components can affect the ionisation of the target compounds, reducing or enhancing the response compared with standards in solvents. Therefore, quantification was carried out by use of matrix-matched calibration standards prepared as described in the Experimental section for every type of food commodity, to counteract this matrix effect. Peak area was used as response and good linearity was obtained for all pesticides at concentrations within the tested interval (from 10 to 500 μg kg−1), with coefficients of determination higher than 0.98. Deviation of the residuals of each calibration point was also studied, checking it was always equal or lower than 20%.
Table 2 shows the results obtained from the recovery study; it can be observed that recoveries ranged from 70 to 109%, in agreement with previously reported results [15, 16]. Some differences can be observed between the three levels assayed. RSD values range from 5 to 19% for the low level (11.5 μg kg−1), from 5 to 15% for the middle level (50 μg kg−1), and from 2 to 13% when 150 μg kg−1 was evaluated. It can also be observed that good recoveries were obtained for the four matrices evaluated, that recoveries did not depend on the matrix evaluated, and no significant differences were observed. Furthermore, it can be observed that higher RSD values were obtained when olive was used as matrix, because a clean up step has to be introduced to remove fat from the samples. In summary, despite the well-established RSD threshold of 20%, repeatability was below 10% for 17 pesticides in cucumber, 11 in strawberry, 15 in orange, and 13 in olive for the low level studied, whereas for the high level evaluated, RSD values were lower than 10% for 45 pesticides in cucumber, 47 in strawberry, 51 in orange, and 44 in olive.
Interday precision was evaluated at 11.5 μg kg−1; the results obtained are shown in Table 4. It can be noted that these values were similar or slightly higher than repeatability values, ranging from 7 to 20%, indicating the stability of the developed method.
LOD was evaluated by injecting pesticides (3, 6, and 8 μg kg−1) in blank matrices; the LOD was the amount for which signal-to-noise ratios (S/N) were equal to or slightly higher than 3. LOQ were evaluated as the lowest concentrations tested for which recoveries ranged from 70 to 110% and precision was lower than 20% [38]. The results obtained are shown in Table 3, and it can be observed that LODs were always lower than 3 μg kg−1, whereas LOQs were lower than 10 μg kg−1. Furthermore, it can be observed that LODs were lower in olives than in the other matrices, because of the additional clean up step included in the extraction procedure, which eliminates some interferences and increases S/N. It can be emphasized that for all of the pesticides in the fruit and vegetable matrices investigated the quantification limit was equal to or lower than the maximum residue limits established by European Union [39] and it is considered as the level at which the laboratory guarantees that residues have been quantified satisfactorily.
Uncertainty was estimated applying the bottom-up approach [40], using the data obtained during the validation process at two concentrations (15% over the first calibration point, 11.5 μg kg−1). Finally expanded uncertainty (U) was obtained by multiplying the relative combined uncertainty by a coverage factor of 2, which is related to a confidence level of 95%. The values obtained (Table 4) ranged from 11 to 24% for cucumber, from 12 to 23% for strawberry, from 9 to 23% for orange, and from 14 to 24% for olive. Considering 15–20% as the commonly accepted valued for method precision, most uncertainties estimated in this work are in accordance with established requirements for analytical results. In fact, for cucumber, uncertainty was higher than 20% for 11 pesticides only. For strawberry, orange, and olive, respectively, 9, 6 and 18 pesticides have uncertainties higher than 20%.
Application to real samples
The proposed method was used for routine analysis of more than 200 vegetable samples with different matrices, such as those evaluated (olives, cucumbers, oranges, and strawberries) and tomato, eggplants, and peppers, in less than 2 months. The laboratory is accredited by UNE-EN-ISO/IEC 17025 for pesticide residue analysis, so internal quality control was applied for every batch of samples to check if the system is under control. This quality control implies a matrix-matched calibration, a reagent blank, a matrix blank, and a spiked blank sample at 11.5 μg kg−1 in order to evaluate the stability of the proposed method with time.
The results obtained are summarised in Table 5, and it can be noted that 21% of the samples gave positive values (higher than the LOQ) and 9% exceeded the levels established by the European Union. Although the low number of samples rejected for consumption is indicative of correct use of pesticides in agriculture in the studied area, it is important to highlight that according to good agricultural practice no sample should contain residues above the MRLs, so more work should be carried out in order to achieve samples with pesticide contents below the established MRLs. The most frequently found pesticide was imidacloprid, with concentrations ranging from 0.01 to 1.00 mg kg−1. This insecticide was found in 45 samples of the commodities pepper, tomato, eggplant, and cucumber, indicating that it is often used in the studied area (southeast Spain). Other pesticides detected were nitenpyram, methomyl, carbendazim, acetamiprid, and spinosad, which were found in several samples. For instance, methomyl residues exceeded the MRLs in three samples, whereas it was not found in other samples. Traces of other compounds such as indoxacarb, promecarb, and pymetrozine were detected, whereas other pesticides such as cymoxanil and aldicarb were not found in any samples.
Finally, it was found that some samples contained more than one pesticide. In fact, more than 10% of samples contained multiple residues. However, only three different pesticides were detected in one sample.
Figure 3 shows positive results for carbendazim in tomato and imidacloprid in pepper as examples of real samples analysed.

UPLC-MS-MS chromatograms obtained from (a) tomato containing carbendazim at 0.10 mg kg−1 and (b) pepper containing imidacloprid at 0.36 mg kg−1
Conclusions
UPLC coupled to tandem mass spectrometry has been shown to be a valuable technique in pesticide residue analysis of complex mixtures and matrices, providing faster analytical answers with greater sensitivity compared with traditional liquid-chromatographic methods. The extraction method was based on the QuEChERS procedure, using the extraction step without any additional “clean-up” step, which has been shown to be unnecessary for the matrices evaluated except for commodities with a high fat content, such as olives. For this type of matrix, Florisil was used as sorbent. The extraction procedure minimizes the use of organic solvents and handling, providing a simple and fast extraction method. No significant differences could be found between the relative response of different matrices. The method was validated for four representative matrices-cucumber, strawberry, orange, and olive-with good sensitivity and selectivity, providing satisfactory recoveries (70–110%) for most of the pesticides. The limits of quantification (10 μg kg−1) were always lower than the MRLs established by the European Union.
The high resolving power of the UPLC method, minimizing the run time, combined with QuEChERS, an extremely easy and fast extraction step for the determination of pesticides in food, results in a method suitable for rapid determination of pesticides in food commodities, thus increasing sample throughput. Quality-control systems applied during the assays have demonstrated very good performance and stability with time. The proposed analytical method is fast, easy to perform, and could be used for regular monitoring of pesticide residues in fruit and vegetable matrices by routine laboratories.
References
Sandra P, Tienpont B, David F (2003) J Chromatogr A 1000:299–309
Berrada H, Font G, Moltó JC (2004) J Chromatogr A 1042:9–14
Garrido Frenich A, González Rodríguez MJ, Arrebola FJ, Martínez Vidal JL (2005) Anal Chem 77:4640–4648
Garrido Frenich A, Martínez Salvador I, Martínez Vidal JL, López López T (2005) Anal Bioanal Chem 383:1106–1118
Sannino A, Bandini M (2005) Rapid Commun Mass Spectrom 19:2729–2733
Hernández F, Pozo OJ, Sancho JV, Biljsma L, Barreda M, Pitarch E (2006) J Chromatogr A 1109:242–252
Botitsi H, Economou A, Tsipi D (2007) Anal Bioanal Chem, DOI 10.1007/s00216-007-1452-3
Picó Y, Blasco C, Font G (2004) Mass Spectrom Rev 23:45–85
Cacho C, Turiel E, Martín-Esteban A, Pérez-Conde C, Cámara C (2003) Anal Bioanal Chem 376:491–496
Obana H, Akutsu K, Okihashi M, Kakimoto S, Hori S (1999) Analyst 124:1159–1165
Mol HGJ, van Dam RCJ, Steijger OM (2003) J Chromatogr A 1015:119–127
Garrido Frenich A, Martínez Vidal JL, López López T, Cortés Aguado S, Martínez Salvador I (2004) J Chromatogr A 1048:199–206
Barriada-Pereira M, González-Castro M, Muniategui-Loreno S, López-Mahía P, Prada-Rodríguez D, Fernández-Fernández E (2007) Talanta 71:1345–1351
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenk FJ (2003) J AOAC Int 86:412–431
Lehotay SJ, de Kok A, Hiemstra M, van Bodegraven P (2005) J AOAC Int 88:595–614
Lehotay SJ, Mastovska K, Lightfield AR (2005) J AOAC Int 88:615–629
Diez C, Traga WA, Zommer P, Marinero P, Atienza J (2006) J Chromatogr A 1131:11–23
Hercegova A, Domotrova M, Kruzlicova D, Matisova E (2006) J Sep Sci 29:1102–1109
Plössl F, Giera M, Bracher F (2006) J Chromatogr A 1135:19–26
Nguyen DTT, Guillarme D, Rudaz S, Veuthey JL (2006) J Chromatogr A 1128:105–113
Nguyen DTT, Guillarme D, Rudaz S, Veuthey JL (2006) J Sep Sci 29:1836–1848
MacNair JE, Lewis KC, Jorgenson JW (1997) Anal Chem 69:983–989
Yu K, Little D, Plumb R, Smith B (2006) Rapid Commun Mass Spectrom 20:544–552
Plumb R, Castro-Pérez J, Granger J, Beattie I, Joncour K, Wright A (2004) Rapid Commun Mass Spectrom 18:2331–2337
Johnson KA, Plumb R (2005) J Pharm Biomed Anal 39:805–810
Chan ECY, Yap SL, Lau AJ, Leow PC, Toh DF, Koh HL (2007) Rapid Commun Mass Spectrom 21:519–528
Chesnut SM, Salisbury JJ (2007) J Sep Sci 30:1183–1190
Kasprzyk-Hordern B, Dinsdale RM, Guwy AJ (2007) J Chromatogr A 1161:132–14528
Leandro CC, Hancock P, Fussell RJ, Keely BJ (2006) J Chromatogr A 1103:94–101
Kovalczuk T, Jech M, Poustka J, Hajslova J (2006) Anal Chim Acta 577:8–17
Leandro CC, Hancock P, Fussell RJ, Keely BJ (2007) J Chromatogr A 1144:161–169
Pozo OJ, Barreda M, Sancho JV, Hernández F, LLiberia JLl, Cortés MA, Bagó B (2007) Anal Bioanal Chem DOI 10.1007/s00216-007-1407-8
Guidance document on residue analytical methods, Directorate General Health and Consumer Protection, SANCO/825/00rev7, 17/03/2004, European Commission
Quality control procedures for pesticide residue analysis, Directorate General Health and Consumer Protection, SANCO/10232/ 2006, 24/03/2006, European Commission
Cunha SC, Lehotay SJ, Mastovska K, Fernandes JO, Beatriz M, Oliveira PP (2007) J Sep Sci 30:620–632
Lehotay SJ, Mastovska K, Yun SJ (2005) J AOAC Int 88:630–638
Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Official Journal of the European Union, L 221, 17th August 2002, p. 16
Hamilton DJ, Ambrus A, Dieterle RM, Felsot AS, Harris CA, Holland PT, Katayama A, Kurihara N, Linders J, Unsworth J, Wong SS (2003) Pure Appl Chem 75:1123–1155
European Council Directives 76/895/EEC, 83/362/EEC, 86/363/EEC and 90/642/EEC, updated on 04/11/2004, European Union, Brussels. Available at http://ec.europa.eu/comm/food/plant/protection/pesticides/index_en.htm
Cuadros-Rodríguez L, Hernández Torres ME, Almansa López E, Egea González FJ, Arrebola Liébanas FJ, Martínez Vidal JL (2002) Anal Chim Acta 454:297–314
Acknowledgments
The authors gratefully acknowledge the Spanish Ministry of Education and Science (MEC-FEDER) (Project Ref. AGL2006-12127-C02-01 and Project Ref. CTQ2005-03654/BQU) for financial support. RRG is also gratefully for personal funding through the Juan de la Cierva program (Spanish Ministry of Education and Science).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Garrido Frenich, A., Martínez Vidal, J.L., Pastor-Montoro, E. et al. High-throughput determination of pesticide residues in food commodities by use of ultra-performance liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 390, 947–959 (2008). https://doi.org/10.1007/s00216-007-1746-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1746-5