
Abstract
The present study conducted QTL (quantitative trait locus) mapping using F2 and F2:3 generations derived from a cross between an overwintering cultivated rice Nuodao89-1 and Shuhui527, to identify potential yield component related QTLs. A total of 37 QTLs were detected across all chromosome except chromosome 7, with LOD values ranging from 3.10 to 12.67. Three QTLs including qTGW3.1, qTGW6.1 and qNEG9.1 were repeatedly detected in both generations. qTGW3.1 was from Nuodao89-1, a total of 46 functional genes involved in 229 gene ontology terms were identified within its locus. Six QTL clusters were founded and corresponding agronomic traits of those QTLs showed highly significant correlation. Three types of epistatic interaction including 47 epistatic QTL pairs for eight yield component traits were detected, but the epistatic QTLs was not so important in controlling the genetic expression of the yield-related Nuodao89-1 as the additive QTLs. Overall, this research provides a theoretical basis for the mining of yield-related genes from overwintering cultivated rice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice is one of the most important food crops in the world (Sakamoto and Matsuoka 2008; Xiao et al. 1996), and their yield traits have been receiving increased attention. Since the application of the quantitative trait locus (QTL) analysis strategy, many genes related to rice yield have been mapped and cloned, including MOC1 (Lu et al. 2009), IPA1 (Li et al. 2003), PROG1 (Jin et al. 2008), D3 (Ishikawa et al. 2005), GIF1 (Wang et al. 2008), TGW6 (Ishimaru et al. 2013), GW2 (Song et al. 2007), GS3 (Mao et al. 2010) and GS5 (Li et al. 2011). These genes are involved in various factors related to production. In rice breeding and production, continuous exploration and innovation of the use of novel rice resources is an indispensable part of the cultivation of new high-yield rice varieties, and is the driving force behind the transformation and upgradation of the rice breeding industry (Yan et al. 2019). Continuous isolation of novel QTLs/genes from different genetic resources is important for the comprehensive evaluation of the genetic mechanisms underlying yield traits and the application of such information allows improved cultivar breeding.
Overwintering cultivated rice can survive through the cold winter and germinate in spring in the following year. A rice farmer needs to sow seeds only once to harvest multiple times. This considerably reduces the workload of farmers and is of practical significance to reduce the negative impact of large-scale land abandonment that has resulted from the sharp decline in the number of rice farmers. Presently, most studies on overwintering cultivated rice have focussed on the QTL mapping of cold tolerance (Deng et al. 2018). However, studying the inheritance of yield traits in overwintering cultivated rice is equally important to allow the production of novel variants that are both high-yielding and cold-tolerant. Nuodao89-1 is a typical overwintering cultivated rice variant, which can survive cold winter owing to the dormancy of its axillary buds. These buds sprout and regenerate in the following spring. Notably, the yield of this regeneration is equivalent to that of the normal season (Zhao et al. 2012). In the present study, we conducted QTL analysis of yield component traits using the two populations, and the F2 and F2:3 populations were constructed using Nuodao89-1 and Shuhui527 as parent strains.
Materials and methods
Plant materials
In the present study two sets of populations were involved, including 201 F2 plants and 201 F2:3 lines derived from a cross between Nuodao89-1 and Shuhui 527. Nuodao89-1 can survive cold winter and germinate in the spring of the following year (Zhao et al. 2006). Shuhui 527 has high combining ability, excellent grain quality, strong resilience and resistance to rice blast, and high recovery rates for hybridization (Zhao et al. 2012).
Phenotypic evaluation
All materials were planted at the experimental station of Chongqing Normal University, Chongqing (N29°32, E106°320) in China. The parents, F2 and F2:3 populations were planted in the summer of 2017 and 2018, respectively, and three plants of the parents and three effective panicles of individuals of F2 and F2:3 populations were harvested randomly to evaluate the phenotype each year. The field management was similar to that under normal rice production conditions. A total of nine yield traits including panicle length (PL), number of panicles (NP), number of filled grains (NFG), number of empty grains (NEG), spikelet per panicle (SPP), grain-setting density (GSD), seed-setting rate (SSR), grain yield plant–1 (GYP) and thousand-grain weight (TGW) were measured in a single plant in the F2 population and in three plants in the middle of rows for each F2:3 line at maturity stage. Phenotypic evaluation was performed following the method described by Shen (1995). For the F2 population and the F2:3 lines, the single plant trait value and the mean trait values for three individuals in the middle of rows were used as input data to identify QTLs, respectively. Data analysis and processing were performed using Microsoft excel 2003 and R statistics software (R Core Team 2017).
Molecular markers
To evaluate the polymorphisms that developed between the parent, we used 548 pairs of SSR primers and 31 pairs of STS markers developed by Tian et al. (2010). These markers covered all the 12 rice chromosomes. Polymorphic primers uniformly distributed across the 12 rice chromosomes were used to genotype the F2 individual. All primers were synthesized by Sangon Biotech (Shanghai, China).
Genomic DNA extraction and genotyping
Total genomic DNA was extracted from young quadrifoliate leaves of parental and F2 generations using the cetyltrimethyl ammonium bromide (CTAB) method. A total volume of 20 μL reaction mixture composed of 10 μL 2×Taq MasterMix, 1 μL template DNA (50 ng μL–1), 1 μL of each primer (10 μM), and 7 μL ddH2O was applied for polymerase chain reaction (PCR). The amplification procedure was performed according to the manufacturer’s instructions (Genstar). The PCR products were separated using an 8% nondenatured polyacrylamide gel, followed by silver staining. Marker analysis was performed using the method described by Suh et al. (2009).
Construction of linkage maps and QTL analysis
Genetic linkage map construction and QTL mapping analysis were performed using the QTL ICIM Mapping v4.1 software, and all primers were added based on their physical positions on the chromosome. Using the BIP function, additive QTLs were identified by inclusive composite interval mapping (ICIM) at a threshold value of LOD ≥ 3.0, and pairwise epistatic interactions between all markers were tested at a threshold value of LOD ≥ 6.0. The mean trait values for each line in both populations were used for QTL analysis.
Results
Evaluations of yield component traits
A highly significant difference in NFG, TGW, GYP and SPP (P<0.01), and a significant difference in NEG and GSD (P<0.05), between the two parents Nuodao89-1 and Shuhui527 were observed. Each trait showed a pattern of continuous distribution and exhibited broad distribution in each population. Transgressive segregations were observed for all of the traits over the two population, studied (table 1; figure 1 in electronic supplementary material at http://www.ias.ac.in/jgenet/).

Correlation between yield component traits in the two populations. (a) F2 generation; (b) F2:3 generation. Red colour shows positive correlation and blue colour negative correlation (P<0.05).
Traits correlations
Correlation coefficient analysis was performed using regression effect values from one trait when applied to the others. Results showed that there is a significant correlation between most of the traits in these two populations (figure 1). Of the 36 possible correlations from each population, 26 (23 positive and three negative) were significant (P< 0.05) in F2, while 25 correlations (20 positive and five negative) were significant in F2:3. Most of the correlations between the traits were similar in both populations. Panicle length was positively correlated with grain number per panicle and thousand-grain weight. The spikelet per panicle was positively correlated with seed-setting rate and grain-setting density. The GYP was significantly positive correlated with other seven traits except the number of empty grains. These results indicate the significance of other traits towards yield.
Genetic linkage maps
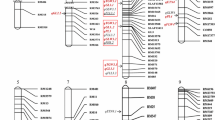
We used 579 markers (31 STS makers and 548 SSR markers) to determine the polymorphism between the two parent strains (Nuodao89-1 and Shuhui527). However, most of the markers were monomorphic. In total, 157 polymorphic markers (table 1 in electronic supplementary material) including 15 STS makers and 142 SSR markers were scored and used to construct the genetic linkage map (figure 2). Among these, chromosome 7 had the most markers (22) and chromosome 12 the least (5). The full map covered a genetic length of 2763.37 cM with a mean intermarker distance of 17.6 cM. The highest marker density was found on chromosome 8, with an average interval of 10.6 cM.

Linkage map constructed based on the F2 population of Nuodao89-1× Shuhui527.
QTL analysis
ICIM mapping was employed to identify putative QTLs and revealed a total of 37 QTLs (table 2; figure 2) were identified. The 37 QTLs for yield component traits were distributed across all chromosomes, with the exception of chromosome 7, with LOD values ranging from 3.10 to 12.67 and an explained PVE ranging from 4.83% to 43.59%. Of these, 16 QTLs were identified in F2 and 24 in F2:3. Three QTLs including qTGW3.1, qTGW6.1 and qNEG9.1 were detected in both populations. The details of the QTLs are presented in table 2. Four QTLs (qNEG12.1, qNEG6.1, qNEG2.1 and qNEG6.2) for number of empty grains mapped to chromosomes 2, 6 and 12, explained 2.55%, 1.76%, 3.37% and 2.29% of the phenotypic variation, respectively. Two SPP QTLs (qSPP1.1 and qSPP11.1) with significant LOD values (3.44 and 3.46) were detected on chromosomes 1 and 11 in F2 and F2:3, respectively. ICIM-ADD detected five significant QTLs (qSSR12.1, qSSR6.1, qSSR2.1, qSSR11.1 and qSSR9.1) with LOD values ranging from 3.19 to 11.06 for SSR on chromosomes 12, 6, 2, 11 and 9 in F2:3, which explained the PVE range from 1.4% to 9.63%. Two SSR QTLs (qSSR6.2 and qSSR6.3) with significant LOD values (3.67 and 3.86) were detected on chromosome 6 in F2, these two QTLs had a PVE of 9.24% and 9.64%, respectively. A total of nine QTLs for thousand-grain weight were detected in both populations and mapped to chromosomes 1, 2, 3, 6 and 9, with LOD values ranging from 3.18 to 9.21, PVE ranged from 3.85% to 13.95%. Of these, qTGW3.1 and qTGW6.1 were identified in both populations with an average LOD value of 7.92 and 3.55, and accounted for an average PVE of 15.35% and 7.20%, respectively. The synergistic alleles, qTGW3.1 and qTGW6.1, originated from Nuodao89-1. Two grain density QTLs were detected in F2:3 and were mapped to the positive traits from Shuhui 527. Three QTLs (qGYP1.1, qGYP6.1 and qGYP10.1) for grain yield per plant mapped to chromosomes 1, 6 and 10, explained 12.61%, 36.49% and 43.59% of the phenotypic variation, respectively.
Gene ontology annotation of the QTL- qTGW3.1
IM was conducted to confirm the validity of the three QTLs (qNEG9.1, qTGW3.1 and qTGW6.1) that were repeatedly detected in both populations during ICIM mapping. The result showed that qTGW3.1 and qNEG9.1 could also be detected by IM (figure 3, a&b). Based on the linked marker information, qTGW3.1 was located at 916.87 kb region between RM156 and RM7642 on chromosome 3. Gene ontology (GO) annotation was performed via the rice genome annotation project (http://rice.plantbiology.msu.edu) and revealed that this QTL locus contained 124 candidate genes. A total of 229 GO terms under 46 functional genes (table 2 in electronic supplementary material) were found and they were divided into three major GO types, namely, ‘cellular component’, ‘biological process’, and ‘molecular function’. Within these categories, the four most represented GO terms were ‘biological process’, ‘metabolic process’, ‘cellular process’, and ‘cellular component’ (figure 3c).

GO annotation of QTL-qTGW3.1. (a) IM of qTGW3.1 on rice chromosome 3, (b) ICIM of qTGW3.1 on rice chromosome 3, and (c) distribution of GO terms.
Genomic regions of QTL clusters
Six regions with several significant QTLs for yield-related traits were detected on chromosome 2 (RM262-RM166), 6 (RM217-RM2615 and SSII-3M1-RM528), 9 (RM444-AGPsmaM1), 11 (RM254-RM224), 12 (RM19-RM270), and four of six genomic regions were detected in the F2:3 population. The QTL (qPL11.1 and qSPP11.1) genomic region was flanked repeatedly by RM254 and RM224 on chromosome 11 affecting panicle length and spikelet per panicle. Two QTLs including thousand-grain weight and grain yield per plant were detected at the region flanking SSII-3M1 and RM528 on chromosome 6. The other four regions contained a QTL for number of empty grains and a QTL for seed setting rate, respectively. These QTL clusters with large effects on yield traits lay good foundation for the future genetic mechanism elucidation.
Epistatic interactions
Epistasis is a phenomenon where the function of one gene is influenced by one or more other genes (Mo et al. 2018). In the present study, 47 epistatic interactions (table 3) were detected for yield component traits, distributed across all chromosomes. The proportions of PVE explained by these epistatic interactions ranged from 2.53% to 16.94% in F2 and 1.07% to 18.54% in F2:3. Of the 47 digenic interactions, three types of digenic interactions were identified: (i) four interactions between QTLs, including marker interval RM2615-RM276 for SSR on chromosome 6 and marker interval ISAM1-RM281 for TGW on chromosome 8. (ii) 14 interactions between QTLs and background loci including marker interval RM119-RM273 for TGW and background loci RM332-RM287 with LOD of 6.21 for SSR. (iii) 29 interactions between complementary loci, including marker interval RM153-RM13 on chromosome 5 with marker interval RM445-RM6767 or RM206-RM254. There were four digenic interactions with large effects (>10%) for GSD, SSR, TGW and PL. For PL, the interactions of loci on chromosomes 2 and 9 accounted for 18.54% PVE while for SSR, the interactions of loci on chromosomes 4 and 11 accounted for 16.94% PVE. But the contribution rate and effect values for most of the epistasis interactions were limited.
Discussion
QTL mapping analysis is a useful tool to identify genomic regions related to yield component traits (Jia et al. 2019). A total of 37 yield component trait QTLs were detected in this study. Among them, 22 had a contribution rate greater than 10%, and thus were classified as major QTLs. However, only three QTLs could be repeatedly detected in both populations, and the locations of the remaining QTLs in the F2 population significantly differed in the F2:3 population. This could be due to agronomical trait in QTL mapping population that were easily affected by multi environmental factors including water, temperature, gas, light and fertilizer (Liang et al. 2019). The synergistic genes of multiple QTLs were from Shuhui527, which were consistent with the results of multiple traits which are significantly better in Shuhui527.
We found 2060 records related to yield-related QTLs on the gramene website (http://www.gramene.org). Among the 37 QTLs for nine yield-related traits, 19 QTLs have been previously located and, in some cases, cloned. The region where qPL1.1 is located contains cloned gene FLR2, which is involved in tillering of rice, and regulate the yield of rice (Li et al. 2016). The OsMCA1 (Liu et al. 2015) and PAD (Kurusu et al. 2012) gene were cloned at the qPL3.1 locus. The cloned gene of UAP1 (Wang et al. 2015) was located at genetic locus of qPL8.2 and regulated panicle length, seed setting rate, spikelet per panicle and thousand-grain weight. For GSD, two intervals (RM259-RM306 and RM401-RM119) containing seven cloned genes, including IPI1 (Wang et al. 2003) and Gnp4 (LAX2) (Zhang et al. 2011; Hiroaki et al. 2011). The cloned gene of TGW6 was located at genetic locus of qTGW6.1, the loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield (Ishimaru et al. 2013). qNEG2.1 and qSSR2.1 were located at the same genomic region of qSPP2 reported by Zhang et al. (2009) on chromosome 2. The region where qTGW3.3 was located contains the cloned gene GL3.1, which controls rice grain size and yield by regulating Cyclin-T1;3 (Qi et al. 2012). qGSD1.1, qNEG6.1 and qNEG6.2 were in the same interval as qGSD1, qNEG6a, and qNEG6b reported by Yan et al. (2019) separately, and they may even refer to the same site. The cloned gene of OsCNGC13 was located at genetic locus of qSSR6.1 and regulated seed setting rate by facilitating pollen tube growth in stylar tissues (Xu et al. 2017). These QTLs have shown large and consistent effects under varying environmental and across a wide range of genetic backgrounds, may be genetically independent and could be used for MAS (Swamy et al. 2016). Eighteen QTLs including 11 major QTLs are reported for the first time and are considered novel. Among them, six QTLs were from overwintering rice, which may be a unique genetic pattern for overwintering cultivated rice. qTGW3.1, explained 43.59% of the phenotypic variation, is a positive QTL for yield improvement, could be repeatedly detected in two populations, and originated from Nuodao89-1. Further, IM also detected qTGW3.1 at the interval of RM156 (17715030 bp) and RM7642 (17789026 bp). A total of 46 functional genes involved in 229 GO terms were found at this location, and these candidate genes might affect thousand-grain weight in Nuodao89-1. But none of them were previously reported genes related to grain weight directly. It was still difficult to accurately predict a single candidate gene from this large region. The synergistic allele for qTGW3.1 was from Nuodao89-1, fine mapping and gene cloning of qTGW3.1 may be important for increasing yields of overwintering cultivated rice in molecular breeding. Thus, fine mapping at the qTGW3.1 region and the related near-isogenic lines should be developed to identify candidate genes in future research.
Some of the genomic regions can be associated with more than one trait based on multi-trait QTL mapping (Zuo et al. 2019). The cluster distribution was generally founded in yield-related traits. For instance, Brondani et al. (2002) detected that specific marker regions strongly associated with multiple traits including panicle number, 100-grain weight, spikelets per panicle, percentage of filled grains per panicle, grain yield per panicle, filled grain number per panicle and grain yield per plant. Zhao et al. (2013) found that the same chromosome segment is often associated with the expression of several traits, which may be the result of the pleiotropic effects of a single gene or the effect of tightly linked genes. The present study detected six regions associated with more than one trait. The QTLs (qPL11.1 and qSPP11.1) were flanked repeatedly by RM3484 and RM6776 on chromosome 11, simultaneously affecting the genetic expression of panicle length and spikelet per panicle in the F2 population. QTLs (qNEG6.1, qNEG12.1, qNEG2.1 and qNEG9.1) for NEG always shared the same location with the SSR QTLs (qSSR6.1, qSSR12.1, qSSR2.1 and qSSR9.1). qGYP6.1 and qTGW6.1 were detected at the region flanking SSII-3M1 and RM528 on chromosome 6. Notably, there was a significant correlation between PL and SPP, NEG and SSR, GYP and TGW. It is consistent with the findings that QTL for significantly correlated traits usually had same chromosome location reported by Hittalmani et al. (2003) and Tian et al. (2006). In addition, the cloning and functional studies of QTL also indicate the pleiotropic effects of various QTLs. For instance, Ghd7 (Xue et al. 2008) simultaneously controls the number of grains per panicle, heading date and plant height. DEP1 (Huang et al. 2009) affects many traits including panicle length, grain-setting density, grain number per panicle, grain length and grain weight. These results provide an explanation for the correlation between traits.
Epistasis is an important factor underlying quantitative traits (Caicedo et al. 2004). A considerable body of classical evidence has strongly suggested the prevalence of epistatic effects on quantitative traits in genetic populations (Malmberg et al. 2005; Fei et al. 2011; Ku et al. 2012). In the present study, 47 epistatic interactions were detected for yield component traits, however, the contribution rate and effect value of most of these interactions were limited, which may show that the epistatic interactions have some impact on the inheritance of yield component traits of Nuodao89-1, but that this is relatively small. It should be noted that mean distance of markers is quite wide in the population under study, thus we cannot exclude the small epistatic effects were also could be due to low resolution of linkage map or low polymorphic markers. Seven intervals were involved in more than one distinct interaction. This might indicate that there are multi-locus associations in the development of yield component traits. Many main-effect QTLs were involved in the epistatic interactions. This indicates that epistasis, in the form of additive by additive interactions, plays an important role in controlling the expression of yield-component traits. The usual estimates of main effect of a QTL can be confounded by interactions, which may change according to genetic backgrounds, environments, and other factors (Li et al. 1997). Three types of digenic interactions were identified in this study. Corresponding to the genetic mode of action of phenotypic traits are the regulatory network of genes, the three patterns of epistatic interactions between the QTLs may reflect the positive or negative feedback inhibition mechanism between the QTL loci.
References
Brondani C., Rangel N., Brondani V. and Ferreira E. 2002 QTL mapping and introgression of yield-related traits from Oryza glumaepatula to cultivated rice (Oryza sativa L.) using microsatellite markers. Theor. Appl. Genet. 104, 1192–1203.
Caicedo A. L., Stinchcombe J. R., Olsen K. M., Schmitt J. and Purugganan M. D. 2004 Epistatic interaction between Arabidopsis FRI and FLC flowering time genes generates a latitudinal cline in a life history trait. Proc. Natl. Acad. Sci. USA 101, 15670–15675.
Deng X. X., Gan L., Liu Y., Luo A. C., Jin L., Chen J. et al. 2018 Locating QTLs controlling overwintering seedling rate in perennial glutinous rice 89–1 (Oryza sativa L.). Genes Genom. 40, 1351–1361.
Fei Z., Fei W. J., Mei W. Y., Yu B. Y. and S W. H. 2011 Quantitative trait loci controlling rice seed germination under salt stress. Euphytica 178, 297–306.
Hiroaki T., Zhang Y., Susumu H., Minami O., Sae S. S., Tetsuo O. et al. 2011 Lax panicle 2 of rice encodes a novel nuclear protein and regulates the fomation of axillary meristems. Plant Cell 23, 3276–3287.
Hittalmani H., Huang N., Courtois B., Venuprasad R., Shashidhar H. E., Zhuang J. Y. et al. 2003 Identification of QTL for growth- and grain yield-related traits in rice across nine locations of Asia. Theor. Appl. Genet. 107, 679–690.
Huang X., Qian Q., Liu Z., Sun H., He S., Luo D. et al. 2009 Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 41, 494–497.
Ishikawa S., Maekawa M., Arite T., Onishi K., Takamure I. and Kyozuka J. 2005 Suppression of tiller bud activity in tillering dwarf mutants of rice. Plant Cell Physiol. 46, 79–86.
Ishimaru K., Hirotsu N., Madoka Y., Murakami N., Hara N., Onodera H. et al. 2013 Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 45, 707–711.
Jin J., Huang W., Gao J. P., Yang J., Shi M., Zhu M. Z. et al. 2008 Genetic control of rice plant architecture under domestication. Nat. Genet. 40, 1365–1369.
Ku L. X., Sun Z. H., Wang C. L., Zhang J., Zhao R. F., Liu H. Y. et al. 2012 QTL mapping and epistasis analysis of brace root traits in maize. Mol. Breed. 30, 697–708.
Kurusu T., Nishikawa D., Yamazaki Y., Gotoh M., Nakano M., Hamada H. et al. 2012 Plasma membrane protein OsMCA1 is involved in regulation of hypo-osmotic shock-induced Ca2+ influx and modulates generation of reactive oxygen species in cultured rice cells. BMC Plant Biol. 12, 11.
Liang Y. S., Yan C., Zheng J., Nan W. B., Qin X. J. and Zhang H. M. 2019 Locating QTL associated with spike traits of Dongxiang wild rice (Oryza rufipogon Griff). Euphytica 215, 26.
Li C. Y., Wang L., Cui Y. C., He L. M., Qi Y. Y., Zhang J. X. et al. 2016 Two FERONIA-like receptor (FLR) genes are required to maintain architecture, fertility, and seed yield in rice. Mol. Breed. 36, 151.
Li X., Qian Q., Fu Z., Wang Y., Xiong G., Zeng D. et al. 2003 Control of tillering in rice. Nature 422, 618–621.
Li Y., Fan C., Xing Y., Jiang Y., Luo L., Sun L. et al. 2011 Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 43, 1266–1269.
Li Z. K., Pinson S. R. M., Park W. D., Andrew H. P. and James W. S. 1997 Epistasis for three grain yield components in rice (Oryza Sativa L.). Genetics 145, 453–465.
Liu Z. W., Cheng Q., Sun Y. F., Dai H. X., Song G. Y., Guo Z. B. et al. 2015 A SNP in OsMCA1 responding for a plant architecture defect by deactivation of bioactive GA in rice. Plant Mol. Biol. 87, 17–30.
Lu F., Ammiraju J. S., Sanyal A., Zhang S., Song R., Chen J. et al. 2009 Comparative sequence analysis of MONOCULM1-orthologous regions in 14 Oryza genomes. Proc. Natl. Acad. Sci. USA 106, 2071–2076.
Malmberg R. L., Held S., Waits A. and Mauricio R. 2005 Epistasis for fitness-related quantitative traits in Arabidopsis thaliana grown in the field and in the greenhouse. Genetics 171, 2013–2027.
Mao H., Sun S., Yao J., Wang C., Yu S., Xu C. et al. 2010 Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. USA 107, 19579–19584.
Mo H. Q., Li Z. and Zhu C. M. 2018 A kind of epistasis-tunable test functions for genetic algorithms. Concurrency and Computation: Practice and Experience.
Qi P., Lin Y. S., Song X. J., Shen J. B., Huang W., Shan J. X. et al. 2012 The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1;3. Cell Res. 22, 1666–1680.
R Core Team 2017 R: A language and environment for statistical computing. R foundation for statistical computing. https://www.R-projects.org/.
Sakamoto T. and Matsuoka M. 2008 Identifying and exploiting grain yield genes in rice. Curr. Opin. Plant Biol. 11, 209–214.
Shen Z. D. 1995 Crop breeding experiment, Chinese Agricultural Press, Beijing.
Song X. J., Huang W., Shi M., Zhu M. Z. and Lin H. X. 2007 A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 39, 623–630.
Suh J. P., Roh J. H., Cho Y. C., Han S. S., Kim Y. G. and Jena K. K. 2009 The pi40 gene for durable resistance to rice blast and molecular analysis of pi40-advanced backcross breeding lines. Phytopathology 99, 243–250.
Swamy B. P. M., Rahman M. A., Inabangan-Asilo M. A., Amparado A., Manito C. P., Chadha-Mohanty R. et al. 2016 Advances in breeding for high grain Zinc in rice. Rice 9, 49.
Tian F., Zhu Z. F., Zhang B. S., Tan L. B., Fu Y. C., Wang X. K. et al. 2006 Fine mapping of a quantitative trait locus for grain number per panicle from wild rice (Oryza rufipogon Griff.). Theor. Appl. Genet. 113, 619–629.
Tian Z. X., Yan C., Qian Q., Yan S., Xie H., Wang F. et al. 2010 Development of gene-tagged molecular markers for starch synthesis-related genes in rice. Chin. Sci. Bull. 55, 3768–3777.
Wang E., Wang J., Zhu X., Hao W., Wang L., Li Q. et al. 2008 Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 40, 1370–1374.
Wang J., Yu H., Xiong G. S., Lu Z. F., Jiao Y. Q., Meng X. B. et al. 2003 Tissue-specific ubiquitination by IPA1 interacting protein1 modulates IPA1 protein levels to regulate plant architecture in rice. Plant Cell. 29, 697–707.
Wang Z. H., Wang Y., Hong X., Hu D. H., Liu C. X., Yang J. et al. 2015 Functional inactivation of UDP-N-acetylglucosamine pyrophosphorylase 1 (UAP1) induces early leaf senescence and defence responses in rice. J. Exp. Bot. 66, 973–987.
Xiao J., Li J., Yuan L. and Tanksley S. D. 1996 Identification of QTLs affecting traits of agronomic importance in a recombinant inbred population derived from a subspecific rice cross. Theor. Appl. Genet. 92, 230–244.
Xue W., Xing Y., Weng X., Zhao Y., Tang W., Wang L. et al. 2008 Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 40, 761–767.
Xu Y., Yang J., Wang Y. H., Wang J. C., Yu Y., Long Y. et al. 2017 OsCNGC13 promotes seed-setting rate by facilitating pollen tube growth in stylar tissues. PLoS Genet. 13, e1006906.
Yan C., Zheng J., Duan W. J., Nan W. B., Qin X. J., Zhang H. M. et al. 2019 Locating QTL controlling yield traits in overwintering cultivated rice. Crop. Sci. 45, 522–536.
Zhang Y., Luo L., Liu T., Xu C., Xing Y. and TAG. 2009 Four rice QTL controlling number of spikelets per panicle expressed the characteristics of single Mendelian gene in near isogenic backgrounds. Theor. Appl. Genet. 118, 1035–1044.
Zhang Z. Y., Li J. J., Yao G. X., Zhang H. L., Dou H. J., Shi H. L. et al. 2011 Fine mapping and cloning of the grain number per-panicle gene (Gnp4) on chromosome 4 in rice (Oryza sativa L.). Agr. Sci. China. 10, 1825–1833.
Zhao J. G., Jiang K. F., Yang L., Yang Q. H., Wan X. Q., Cao Y. J. et al. 2013 QTL mapping for yield related components in a RIL population of rice. Chin. J. Rice Sci. 27, 344–352 (in Chinese with English abstract).
Zhao Z. W., Li S. G. and Lei S. F. 2006 Genetic variation of main agronomic traits in a new germplasm resource of perennial rice. Sci. Agr. Sinica. 20, 481–487 (in Chinese with English abstract).
Zhao Z. W., Luo A. C., Le T. and Li S. G. 2012 Identification of quantitative trait locus for overwintering germinability in rice (Oryza sativa L.). J. Integr. Agr. 11, 1767–1774.
Zuo J., Lin C. T., Cao H., Chen F., Liu Y. and Liu J. 2019 Genome-wide association study and quantitative trait loci mapping of seed dormancy in common wheat (Triticum aestivum L.). Planta 250, 187–198.
Acknowledgements
This research was supported by the National Natural Sciences Foundation (31670326), Technology Innovation and Application Development Program in Chongqing (cstc2019jscx-msxmX0353), the Program for Innovative Research Team in University, Chongqing (CXTDX201601018), Achievement Transfer Program of Institutions of Higher Education in Chongqing (KJZH17114), the key Natural Science Foundation of Chongqing (CSTC2015jcyjBX0091).
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: R. S. Sangwan
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
CHEN, J., TAI, L., LUO, L. et al. Mapping QTLs for yield component traits using overwintering cultivated rice. J Genet 100, 31 (2021). https://doi.org/10.1007/s12041-021-01279-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-021-01279-1