
Abstract
Rice blast is one of the most serious diseases in the world. The use of resistant cultivars is the most preferred means to control this disease. Resistance often breaks down due to emergence of new races; hence identification of novel resistance donors is indispensable. In this study, a panel of 80 released varieties from National Rice Research Institute, Cuttack was genotyped with 36 molecular markers that were linked to 36 different blast resistance genes, to investigate the varietal genetic diversity and molecular marker-trait association with blast resistance. The polymorphism information content of 36 loci varied from 0.11 to 0.37 with an average of 0.34. The cluster analysis and population structure categorized the 80 National Rice Research Institute released varieties (NRVs) into three major genetic groups. The principal co-ordinate analysis displays the distribution of resistant and moderately resistant NRVs into different groups. Analysis of molecular variance result demonstrated maximum (97%) diversity within populations and minimum (3%) diversity between populations. Among tested markers, two markers (RM7364 and pi21_79-3) corresponding to the blast resistance genes (Pi56(t) and pi21) were significantly associated and explained a phenotypic variance of 4.9 to 5.1% with the blast resistance. These associated genes could be introgressed through marker-assisted to develop durable blast resistant rice varieties. The selected resistant NRVs could be good donors for the blast resistance in rice crop improvement research.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is the staple food crop for more than half of the world’s human population and cultivated in diverse agro-climatic conditions. Rice blast caused by the fungus, Magnaporthe oryzae is one of the most devastating and one of the top 10 fungal diseases which is major threat to global food security (Dean et al. 2005). In India, it has caused a considerable yield losses over the past few decades ranging between 20–100% (Sharma et al. 2012). Majority of the plant pathogens mutated rapidly and resulted in the breakdown of resistance causing epidemics, further it is aggravated by conducive weather conditions, high disease pressure, and genome stability of the pathogen. The genome of M. oryzae is rich in retrotransposons and repetitive segments (Dean et al. 2005) which helps the fungus to change its virulence to overcome the resistance conferred by R-genes (Vasudevan et al. 2014). The rice blast system follows classical gene for gene model where a blast resistance (R) gene products prevent its infection by the races of M. oryzae carrying the corresponding avirulence (Avr) gene (Silue et al. 1992; Sahu et al. 2018). This disease can be managed by maintaining adequate flood depth, suitable planting dates, recommended dose of nitrogen fertilizer, chemical fungicides and use of resistant varieties (Bonman 1992). Among these, use of resistant varieties is the most economical and environment friendly method to manage this disease (Panda et al. 2017).
Until now, more than 100 race-specific R genes, and more than 350 QTLs for resistance to M. oryzae have been identified, and 27 have been cloned and characterized and most of them encode nucleotide-binding site-leucine-rich repeat (NBS-LRR) proteins except pi21 and Pid2, which encodes proline-containing protein and receptor kinase (Fukuoka et al. 2009; Kouzai et al. 2013; Zheng et al. 2016; Zhu et al. 2016; Yadav et al. 2019). Among them, eight genes have been located in two gene clusters; Pi2, Pi9 and Piz-t in Pi2 locus and Pik, Pik-m, Pik-p, Pi1 and Pi-ke in Pik locus (Wang et al. 2016). Most of the blast R genes are dominant except the recessive genes pi21, Pid-2 and pi66(t) (Liang et al. 2016). Most of the R-genes were identified in landraces, cultivars, or wild rice collections because of differential physiological races of M. oryzae (Tanksley et al. 1997). The existing phenotypic screening technique for blast resistance is time-consuming, laborious, and entailed specific procedures. Many PCR based molecular markers have been developed for fine mapped and cloned blast R genes for mining and identification of different R genes.
In the current scenario, molecular markers have a significant contribution in increasing the efficiency and precision to incorporate blast resistance genes in cultivars (Wang et al. 2014). Marker-assisted selection (MAS) is an advanced molecular tool in rice breeding for improvement of resistance to rice blast and with the aid of MAS many rice cultivars resistant to biotic stress have been developed and widely accepted by farmers (Xu and Crouch 2008). Association mapping (AM) is a molecular approach used for identifying target genes governing important traits in a natural population including diverse germplasm. AM can be categorized into candidate-gene association and genomewide association mapping (Zhang et al. 2017). In addition, AM utilizes a natural population as compared to biparental mapping population, thus it saves the time required to constitute the population and hasten the identification of gene in crops and hence the AM is a powerful means of genetic dissection and identification of a gene of interest.
National Rice Research Institute released varieties (NRVs) can be studied as donor source of favourable genes for biotic stresses which are grown in blast endemic areas in diverse agro-ecological zones in India. During the previous study, NRVs were genotyped for 12 major blast resistant genes using 17 molecular markers (Yadav et al. 2017). In continuation of the previous study, the present study was undertaken to investigate the genetic association of 36-mapped resistance genes in 80 NRVs using linked/functional markers. The objective of the current study was to identify the candidate R genes which confer blast resistance to these NRVs and that could be used for identification of novel donor source (R genes/alleles) for blast resistance, and genomic studies.
Material and methods
Plant material and disease reaction in uniform blast nursery
A set of 80 NRVs originated from eight different agro-ecologies (table 1) was collected from the National Gene Bank, NRRI, Cuttack (table 1 in electronic supplementary material at http://www.ias.ac.in/jgenet). These NRVs were phenotyped for leaf blast under natural conditions in the uniform blast nursery (UBN). The screening was done in two replications during dry and wet seasons, 2015–2016, at the research farm of NRRI, Cuttack (\(85{^{\circ }}55'48''\)E longitudes and \(20{^{\circ }}26'35''\)N latitude). Thirty seeds of each NRVs were grown in a 50-cm long row with a 10 cm row spacing. The highly susceptible varieties HR12 and CO39 were used as a spreader row to ensure the uniform spread of the disease. The disease scoring was recorded from 25 to 40 days after sowing at 5-day intervals when the spreaders row showed more than 85% infection. Disease reaction was scored using the standard evaluation system (SES), IRRI, Philippines (2002) on a 0–9 scale as: resistant (0–3), moderately resistant (4–5), and susceptible (6–9). The higher disease score was considered for evaluation, whenever there were different blast disease score between replications as well as season.
Genomic DNA isolation
Young leaves from 3-week-old seedlings were collected and stored in –80\({^{\circ }}\)C freezer. The genomic DNA was isolated following Doyle and Doyle (1990) method with slight modification. In brief, the 200 mg leaf sample was grinded with liquid nitrogen; powder was immediately transferred to 1ml CTAB isolation buffer and incubated at 65\({^{\circ }}\)C in a recirculating water bath. After one hour, equal volume of PCI (Phenol:Chloroform:Isoamyl alcohol; 25:24:1) was added, and centrifuged at 10,000 rpm for 10 min. The aqueous phase was transferred to new 2ml tube and mixed with Chloroform:Isoamyl alcohol (24:1) and again centrifuged as above. The aqueous phase was pipetted out to a new 2ml tube and absolute alcohol was added twice the volume followed by \(1/10^{\mathrm{th}}\) of sodium acetate (3.5 M) and mixed properly. The samples were kept for 2 h in −20\({^{\circ }}\)C followed by centrifuged of 10,000 rpm for 10 min at room temperature (RT). The white pellet was wash with 70% ethanol by centrifuging at 7000 rpm for 7 min at RT followed by air dry. The completely dried pellet was dissolved in nuclease free water for further quantification and used for PCR amplification. The quantity and quality of nuclear DNA were assessed by 0.8% agarose gel electrophoresis and Nano-drop ND-1000 Spectrophotometer (Thermofisher Scientific, Waltham, USA). Nuclease-free water was used to dilute the DNA samples to the concentration of 20 ng/\(\mu \)L for PCR amplification.
PCR amplification and visualization
The polymerase chain reactions (PCR) were executed in 20 \(\mu \)L reaction volume containing 25 ng template DNA, 1xTaq buffer (10 mM Tris-HCl, 50 mM KCl, pH 8.3), 0.2 \(\mu \)M of each of dNTP, 0.2 \(\mu \)M of each forward and reverse primers, and 1 U of Taq DNA polymerase (DreamTaq, Thermo Scientific, USA). The PCR cycle was set up as follows: initial denaturation of 5 min at 94\({^{\circ }}\)C; 35 cycles at 94\({^{\circ }}\)C for 45 s, primers annealing for 45 s at varied temperature (table 2), extension at 72\({^{\circ }}\)C for 45 s, and a final extension for 10 min at 72\({^{\circ }}\)C. The PCR products were separated by electrophoresis in 3.5% agarose gels and visualized using a gel documentation system (Alpha Imager, USA). The amplified PCR products were scored as presence (1) or absence (0). The PCR reaction was repeated twice for each marker to cross-check the scoring data.
Statistical data analysis
A total of 36 linked/functional markers were used to score the presence or absence of the resistance genes in the 80 NRVs. Jaccard’s coefficient similarity matrix was assessed using binary data. The polymorphism information content (PIC) value, allele number and allele frequency were estimated for each marker using the PowerMarker v3.25 (Liu and Muse 2005). The genetic distance matrix among the NRVs was assessed through principal co-ordinate analysis (PCoA) in GenAlEx 6.5 software. Similarly, analysis of molecular variance (AMOVA) between and within the populations and population assignment was estimated using GenAIEx 6.5.0 (Peakall and Smouse 2012). An unweighted neighbour-joining (NJ) unrooted tree was constructed in the DARwin 5 program (Perrier and Jacquemound-Collect 2006, DARwin Software, http://darwn.cirad.fr/darwin). The dissimilarity index was estimated using NEI coefficient (Nei 1973) with a bootstrap value of 1000. The general linear model (GLM) function in TASSEL5 software was used to understand the genetic association of blast resistance genes with the disease (Bradbury et al. 2007). The GLM model of Tassel 5 software was run with permutations of 1000. The population structure analysis was performed using the Bayesian model-based approach employed in Structure v 2.3.4 software (Pritchard et al. 2000). The number of subgroups (K) in the population varied from 1 to 10. The population structure was run using the admixture model, correlated allele frequencies and five independent iterations per K with a burn-in period length of 200,000 and 200,000 Markov chain Monte Carlo (MCMC). The optimal K was determined from the peak value of \(\Delta \)K (Evanno et al. 2005) using Structure Harvester 0.6.93 (Earl 2012).

Position of marker loci used in this study.
Results
Phenotyping and genetic diversity
The NRVs panels of 80 varieties were phenotyped for resistance to leaf blast. Nineteen NRVs (24.69%) were found to be resistant, 21 (26.25%) were moderately resistant, and 40 (50%) were susceptible. The disease score ranged from 0 to 9. Interestingly, resistant NRVs were observed across all the ecologies. Highest resistant varieties proportion was observed in irrigated (5), whereas lowest proportion was in Boro, on the other hand medium deep water and coastal saline ecologies with one each (table 1). The NRVs were genotypes for 36 markers corresponding to 36 blast resistance genes (figure 1). The genetic diversity parameters of 36 marker loci measured during current study are presented in table 3. The major allele frequency varied from 0.52 to 0.93 with a mean value of 0.75. Similarly, the genetic diversity of 36 markers had a mean value of 0.34 and varied from 0.11 to 0.49. The PIC was used to measure the informativeness of a genetic marker. The PIC value for 36 markers ranged from 0.11 (RM101 and RM 11787) to 0.37 (RM72 and Pia-STS) with an average of 0.27. The PIC value of two markers, RM72 and Pia-STS, corresponding to the Pi33 and Pia genes showed the highest value of 0.37 which can be used effectively for genetic diversity study (table 3).

Unweighted–NJ tree based on molecular markers linked to blast resistance in 80 NRVs. These NRVs are represented corresponding to (a) subpopulations determined from structure analysis (SG1, red; SG2, pink; SP3, blue; admixture, green); (b) disease reaction (resistant, green; moderately resistant, pink; susceptible, red).

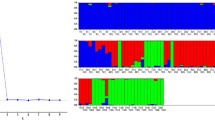
Estimated population structure of the rice NRVs which is partitioned into coloured segments that represent the estimated membership for K\(=\)3. The maximum of ad hoc measure \(\Delta \)K was observed to be K\(=\)3, which indicated that the entire population can be grouped into three subgroups.
Genetic relatedness through cluster analysis, population structure and PCoA
The cluster analysis was analysed with UPGMA and NJ methods using Darwin software based on 36 markers linked to 36 blast resistance genes. The cluster analysis categorized the NRVs into three major clusters (I, II and III) (figure 2). Cluster I consisted of 43 NRVs, was further categorized into two subclusters IA and IB. Subcluster IA included 38 NRVs, with 11 (28.94%) resistant genotypes. Subcluster IB included only five NRVs with no resistant genotype. Similarly, cluster II possessed 32 NRVs, further divided into two subclusters, IIA and IIB. Subclusters IIA consisted of 27 NRVs, having six resistant genotypes (22.22%). Conversely, subclusters IIB consisted of only five NRVs with only one resistant genotype. Cluster III is the smallest cluster comprised of five NRVs, with only one resistant variety. Interestingly, the majority of resistant genotypes were clustered together in major cluster I and few in major cluster II. Genetically similar NRVs were clustered together in the same group, on the contrary NRVs of same ecologies clustered in different groups.
The population structure of 80 NRVs were explored using the model-based population structure based on 36 markers corresponded to 36 blast resistance genes. The peak plateau of ad hoc statistic \(\Delta \)K was observed to be K=3 (figure 3), which indicated the presence of three subgroups (SG1, SG2 and SG3) in the NRVs. The threshold value of >55%, classified the entire NRVs into three subgroups with two admixture (table 4). The SG1 consisted of 24 NRVs, of which eight (33.33%) were highly resistant. The SG2 comprised of 27 NRVs, of which six (22.22%) were highly resistant. Similarly, SG3 included 27 NRVs with five (18.51%) highly resistant NRVs. Interestingly, resistant NRVs were distributed in all the three subgroups with maximum percentage observed in SG1. The moderately resistant genotypes were mostly in SG1 and SG1 whereas SG3 was dominated by susceptible genotypes. Further, high resistant variety, Sarasa belonged to SG2. Accordingly, structure analysis could not differentiate resistant genotypes but partially categorized moderately resistant and susceptible genotypes.
The molecular markers genotypic data were used to calculate the PCoA to estimate the genetic relationship among NRVs. Based on disease reaction, NRVs exhibited uniform distribution across the two axes. According to the PCoA analysis, the first two axes explained 12.71% and 8.93% of the total variance (table 5). In PCoA, resistant landraces were observed to be distributed mostly in the first quadrant, moderately resistant genotypes in third and fourth quadrants, whereas, susceptible genotypes were distributed in all the four quadrants (figure 4). The population assignment test was estimated using GenAlex which partly distinguished resistant populations from moderately resistant or susceptible populations (figure 5, a&b). Similarly, it was partly able to distinguish between moderately resistant from susceptible populations (figure 5c).

PCoA of 36 molecular markers linked to blast resistance in 80 NRVs.
AMOVA
AMOVA is a statistical method to detect molecular variation using molecular markers. In AMOVA analysis, 80 NRVs were categorized into three groups based on their disease reaction: resistant (19), moderately resistant (21) and susceptible (40). Through AMOVA analysis, greater variance (97%) was observed within the population, whereas less (3%) between population (figure 6; table 6a). The highest pairwise fixation indices \((F_{\mathrm{ST}})\) value of 0.040 was observed between the resistant and susceptible, while the lowest was observed between the resistant and moderately resistant populations. The inbreeding coefficient \(F_{\mathrm{IS}}\) and \(F_{\mathrm{IT}}\) were observed to be 1.000. This suggested that individuals from different populations are weakly isolated and genetically more closely related. The pairwise Nei’s genetic distance ranged from 0.34 (between resistant and moderately resistant population) to 0.043 (resistant and susceptible population) (table 6b).

Population assignment of NRVs signifying the log likelihood assignment of each NRVs using disease reaction: (a) resistant and susceptible populations, (b) resistant and moderately resistant populations, (c) moderately resistant and susceptible populations.
Genetic association of blast resistant genes
The genetic relatedness between disease score and molecular markers was investigated using the GLM to know any significance relatedness. Among the 36 markers corresponding to the 36 blast resistant genes, only two markers (RM7364, and pi21_79-3) corresponding to the blast resistant genes (Pi56(t) and pi21) were observed to be significantly associated with blast disease resistance. The phenotypic variance of the two markers varied from 4.9 to 5.1% (table 7). Among these markers, pi21_79-03 exhibited the highest phenotypic variance (5.1%) followed by RM7364 (4.9), whereas remaining markers did not exhibit significant association at \(P<\) 0.1.
Discussion
Genetic diversity of crop plants has been eroded due to replacement of landraces and traditional local varieties with improved and high yielding varieties (Tanksley et al. 1997). The emergence of new and virulent races imposed a constant threat to sustainable rice production and global food security. To keep pace with the pathogen, it is necessary to identify the potential donor for novel resistance genes/alleles to combat the nuisance caused by this disease. In this study, we performed the candidate gene-based screening of blast resistance donors (NRVs) in rice breeding distributed over eight ecologies using 36 known blast resistance genes.

AMOVA analysis of NRVs.
Among the NRVs screened for blast disease resistance in the uniform blast nursery under natural screening at NRRI Cuttack, 19 NRVs were found to be resistant to the leaf blast disease. Among 19 NRVs, seven were reported to be released as resistant to M. oryzae (Yadav et al. 2017). These seven NRVs belonged to three different agro-ecologies, namely Satya Krishna, Chandrama, and Abhishek (irrigated), Sahbhagidhan (upland), Sumit, Reeta and Samalei (shallow low land). Interestingly, all these NRVs were released after 2006 except Samalei. Similarly, Zhu et al. (2016) reported 40 cultivars as highly resistant in China and 20 were previously reported to be resistant. During the breeding programme, identification of the individual resistance gene is often difficult through phenotype-based screening, as it is influenced by the developmental stage and environmental conditions. Instead, DNA markers linked to R genes are the easy and quickest way to identify and select several blast resistance genes without performing phenotype-based screening (Hayashi et al. 2006).
The average gene diversity was found to be 0.34, ranging from 0.11 to 0.49, whereas the major allele frequency had a mean value of 0.75 and varied from 0.52 to 0.93. The gene diversity was observed to be 0.67 in 107 NE collections, 0.25 in 80 NRVs, 0.227 in 288 landraces, and 0.32 in 167 landraces (Roy et al. 2016; Yadav et al. 2017; Susan et al. 2019; Yadav et al. 2019). The degree of polymorphism was detected by calculating PIC values that varied from 0.11 (RM101 and RM11787) to 0.37 (RM72 and Pia-STS) with an average of 0.27. It was higher than that observed by Yadav et al. (2017) in 80 NRVs (0.18) and Susan et al. (2019) and slightly lower than Roy et al. (2016) with 0.62 in NE Himalayan landraces. In the present study, low gene diversity and PIC values were observed as compared to landraces. These NRVs are developed as a result of strong artificial selection pressure, whereas it does not strongly operate in case of landraces.
Based on marker genotype data, distance based clustering categorized the entire NRVs into three major clusters. Most of the resistant NRVs were clustered in major cluster I followed by major cluster II. Our results are in accordance with the previous studies where resistant accessions were clustered in one group and susceptible in another group (Yadav et al. 2017; Susan et al. 2019). Interestingly, NRVs of same ecologies were not grouped together whereas genetically similar NRVs were clustered together.
The genetic architecture of NRVs was investigated using the model-based structure software based on 36 molecular markers. Population structure differentiated 80 NRVs into three subgroups (SG1, SG2 and SG3) with two admixtures. Most of the resistant genotypes (eight) belonged to SG1 whereas, SG2 and SG3 included six (22.22%) and five (18.51%) resistant NRVs, respectively. Consequently, a weak association for blast reaction was observed through structure analysis. Yadav et al. (2017) categorized 80 NRVs into three subgroups through structure analysis using 17 markers linked to blast resistance. Similarly, Roy et al. (2016) and Susan et al. (2019) divided the NE landraces into three and two subpopulation, respectively. The result of cluster analysis is in accordance with the structure analysis of the NRVs. The corresponding NRVs in SG1 and SG2 were found concurrent with the cluster I, NRVs of the cluster III is harmonized with SG3 whereas most of the NRVs of subcluster III belonged to SG3.
The PCoA through the scatter plots partitioned the resistant and susceptible NRVs into different quadrant. Likewise, previous study also showed the portioning of resistant and susceptible genotypes into different groups (Yadav et al. 2017; Susan et al. 2019). However, population assignment partially differentiated resistant and moderately resistant populations.
AMOVA is a method of estimating molecular variance within the species. Based on AMOVA, variation within the population was higher (97%) as compared with between populations (3%). The highest \(F_{\mathrm{ST}}\) was observed between the resistant and susceptible populations (0.040) while, the minimum was observed between the resistant and moderately resistant populations. The inbreeding coefficient \(F_{\mathrm{IS}}\) and \(F_{\mathrm{IT}}\) were observed to be 1.0. The lower \(F_{\mathrm{ST}}\) value indicated that the lower divergence between subgroups. Yadav et al. (2017) observed more variance within the population (96%), whereas less between populations (4%) and \(F_{\mathrm{ST}}\) was in accordance with our result. Similarly, Susan et al. (2019) reported higher (96%) variance within the population and lower (4%) between the populations in the NE landraces. The pairwise Nei’s genetic distance ranged from 0.034 (between resistant and moderately resistant population) to 0.043 (resistant and susceptible population).
Candidate gene-based association mapping is an approach to dissect trait of interest that investigate individual genes for genetic association with a phenotype (Neale and Savolainen 2004). Through GLM, among 36 markers tested, two markers corresponding to two blast resistance genes, Pi56(t) and pi21 were identified to be significantly associated with phenotypic variance varied from 4.9 to 5.1%. Interestingly, Pi56(t) and pi21 genes were reported to be a broad spectrum in nature and individual markers can be used for identification of rice blast resistant genes in the diverse rice germplasm. The genetic association of blast resistance in 80 NRVs, 167 landraces and 288 NE landraces against rice blast varied from 6.5 to 7.7% and explained its implication in markers identification associated with the blast resistance (Yadav et al. 2017; Susan et al. 2019; Yadav et al. 2019). However, the present study did not completely explain the genetic differences between the resistance gene(s) and the disease reaction. Similarly, Yadav et al. (2017) was not able to explain the resistance spectrum using 17 markers corresponding to the 12 blast resistance genes. The resistance spectrum of these NRVs could be explained through the identification of new blast resistance genes, their allelic variant or QTLs. In addition, these varieties had been released with multiple stress tolerance for different ecologies, which could be tested for other biotic and abiotic stresses. These resistant NRVs identified in the present study could be used as a potential donor for the breeding of blast resistance as well as genetic material for identification cloning and characterization of new blast resistant genes.
References
Barman S., Gouda M., Venu R. and Chatto B. 2004 Identification of a major blast resistance gene in the rice cultivar “Tetep”. Plant Breed. 123, 300–302.
Berruyer R., Adreit H., Milazzo J., Gaillard S, Berger A, Dioh W. et al. 2003 Identification and fine mapping of Pi33, the rice resistance gene corresponding to the Magnaporthe grisea avirulence gene ACE1. Theor. Appl. Genet. 107, 1139–1147.
Bonman J. 1992 Durable resistance to rice blast disease–environmental influences. Euphytica 63, 115–123.
Bradbury P. J., Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y. and Bucker E. S. 2007 TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635.
Chen S., Wang L., Que Z., Pan R. and Pan Q. 2005 Genetic and physical mapping of Pi37(t), a new gene conferring resistance to rice blast in the famous cultivar St. No. 1. Theor. Appl. Genet. 111, 1563–1570.
Chen X. W., Li S. G., Xu J. C., Zhai W. X ., Ling Z. Z, Ma B. T. et al. 2004 Identification of two blast resistance genes in a rice variety, Digu. J. Phytopathol. 152, 77–85.
Dean R. A, Tabot N. J., Ebbole D. J., Farman M. L., Mitchell T. K., Orbach M. J. et al. 2005 The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434, 980–986.
Doyle J. J. and Doyle J. L. 1990 Isolation of plant DNA from fresh tissue. Focus 12, 39–40.
Earl D. A. 2012 STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361.
Evanno G., Regnaut S. and Goudet J. 2005 Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620.
Fjellstrom R., Conawaybormans C. A., Mcclung A. M., Marchetti M. A., Shank A. R. and Park W. D. 2004 Development of DNA markers suitable for marker assisted selection of three Pi genes conferring resistance to multiple Pyricularia grisea pathotypes. Crop Sci. 44, 1790–1798.
Fukuoka S., Saka N., Koga H., Ono K., Shimizu T., Ebana K. et al. 2009 Loss of function of a proline–containing protein confers durable disease resistance in rice. Science 325, 998–1001.
Gowda M., Roy-Barman S. and Chattoo B. B. 2006 Molecular mapping of a novel blast resistance gene Pi38 in rice using SSLP and AFLP markers. Plant Breed. 125, 596–599.
Hayashi K., Yoshida H. and Ashikawa I. 2006 Development of PCR based allele specific and InDel marker sets for nine rice blast resistance genes. Theor. Appl. Genet. 113, 251–260.
Hayashi K., Yasuda N., Fujita Y., Koizumi S. and Yoshida H. 2010 Identification of the blast resistance gene Pit in rice cultivars using functional markers. Theor. Appl. Genet. 121, 1357–1367.
He X., Liu X., Wang L., Wang L., Lin F., Cheng Y. et al. 2012 Identification of the novel recessive gene pi55(t) conferring resistance to Magnaporthe oryzae. Sci. China Life Sci. 55, 141–149.
Huang H., Huang L., Feng G., Wang S., Wang Y., Liu J. et al. 2011 Molecular mapping of the new blast resistance genes Pi47 and Pi48 in the durably resistant local rice cultivar Xiangzi–3150. Phytopathology 101, 620–626.
Jiang H., Feng Y., Bao L., Li X., Gao G., Zhang Q. et al. 2012 Improving blast resistance of Jin 23B and its hybrid rice by marker–assisted gene pyramiding. Mol. Breed. 30, 1679–1688.
Kim D. M., Ju H. G., Kang J. W., Han S. S. and Ahn S. N. 2013 A new rice variety ‘Hwaweon 5’ with durable resistance to rice blast. Korean J. Breed Sci. 45, 142–147.
Koide Y., Kawasaki A., Telebanco-Yanoria M. J., Hairmansis A., Nguyet N. T., Bigirimana J. et al. 2010 Development of pyramided lines with two resistance genes, Pish and Pib, for blast disease (Magnaporthe oryzae B. Couch) in rice (Oryza sativa L.). Plant Breed. 129, 670–675.
Koide Y., Telebanco–Yanoria M. J., Fukuta Y. and Kobayashi N. 2013 Detection of novel blast resistance genes, Pi58(t) and Pi59(t), in a Myanmar rice landrace based on a standard differential system. Mol. Breed. 32, 241–252.
Kouzai Y., Kaku H., Shibuya N., Minami E. and Nishizawa Y. 2013 Expression of the chimeric receptor between the chitin elicitor receptor CEBiP and the receptor–like protein kinase Pi–d2 leads to enhanced responses to the chitin elicitor and disease resistance against Magnaporthe oryzae in rice. Plant Mol. Biol. 81, 287–295.
Lei C., Hao K., Yang Y., Ma J., Wang S. Wang J. et al. 2013 Identification and fine mapping of two blast resistance genes in rice cultivar 93–11. Crop J. 1, 2–14.
Liang Z., Wang L. and Pan Q. 2016 A new recessive gene conferring resistance against rice blast. Rice 9, 47.
Liu K. and Muse S. V. 2005 PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129.
Liu X., Yang Q., Lin F., Hua L., Wang C. Wang L. et al. 2007 Identification and fine mapping of Pi39(t), a major gene conferring the broad–spectrum resistance to Magnaporthe oryzae. Mol. Gen. Genomics 278, 403–410.
Liu X. Q., Wang L., Chen S., Lin F. and Pan Q. H. 2005 Genetic and physical mapping of Pi36(t), a novel rice blast resistance gene located on rice chromosome 8. Mol. Gen. Genomics 274, 394–401.
Liu Y., Liu B., Zhu X., Yang J., Bordeos A. Wang G. et al. 2013 Fine–mapping and molecular marker development for Pi56(t), a NBS–LRR gene conferring broad– spectrum resistance to Magnaporthe oryzae in rice. Theor. Appl. Genet. 126, 985–998.
Ma J., Lei C., Xu X., Hao K., Wang J. Cheng Z. et al. 2015 Pi64, encoding a novel CC–NBS–LRR protein, confers resistance to leaf and neck blast in rice. Mol. Plant Microbe Interact. 28, 558–568.
Matsushita K., Yasuda N., Koizumi S., Ashizawa T., Sunohara Y. Iida S. et al. 2011 A novel blast resistance locus in a rice (Oryza sativa L.) cultivar, Chumroo, of Bhutan. Euphytica 180, 273–280.
Neale D. B. and Savolainen O. 2004 Association genetics of complex traits in conifers. Trends Plant Sci. 9. 325–330.
Nei M. 1973 Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70, 3321–3323.
Nguyen T. T., Koizumi S., La T. N., Zenbayashi K. S., Ashizawa T. Yasuda N. et al. 2006 Pi35(t), a new gene conferring partial resistance to leaf blast in the rice cultivar Hokkai 188. Theor. Appl. Genet. 113, 697–704.
Okuyama Y., Kanzaki H., Abe A., Yoshida K., Tamiru M. Saitoh H. et al. 2011 A multifaceted genomics approach allows the isolation of the rice Pia-blast resistance gene consisting of two adjacent NBS-LRR protein genes. Plant J. 66, 467–479.
Panda G., Sahu C., Yadav M. K., Aravindan S., Umakanta N. Raghu S. et al. 2017 Morphological and molecular characterization of Magnaporthe oryzae from Chhattisgarh. Oryza 54, 330–336.
Peakall R. and Smouse P. E. 2012 GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research an update. Bioinformatics 28, 2537–2539.
Pritchard J. X., Stephens M. and Donnelly P. 2000 Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Roy S., Marndi B. C., Mawkhlieng B., Banerjee A., Yadav R. M., Misra A. K. et al. 2016 Genetic diversity and structure in hill rice (Oryza sativa L.) landraces collected from the north–eastern Himalayas of India. BMC Genet. 17, 107.
Sahu, C., Yadav M. K., Panda G., Aravindan S., Umakanta N., Raghu S. et al. 2018 Morphological and molecular characterization of Magnaporthe oryzae causing rice blast disease in Odisha. Oryza 55, 467–472.
Sharma T. R., Rai A. K., Gupta S. K., Vijayan J., Devanna B. N. and Ray S. 2012 Rice blast management through host plant resistance: retrospect and prospects. Agric. Res. 1, 37– 52.
Shi B. H., Zhang J. H., Zheng Y. M., Liu Y. Q., Cruz C. V. Zheng T. Q. et al. 2012 Identification of a new resistance gene Pi–Da(t) from Dacca6 against rice blast fungus(Magnaporthe oryzae) in Jin23B background. Mol. Breed. 30, 1089–1096.
Silue D., Notteghem J. L. and Tharreau D. 1992 Evidence of a gene or gene relationship in the Oryza sativa–Magnaporthe grisea pathosystem. Phytopathology 82, 577–580.
Sun P., Liu J., Wang Y., Jiang N., Wang S. Dai Y. et al. 2013 Molecular mapping of the blast resistance gene Pi49 in the durably resistant rice cultivar Mowanggu. Euphytica 192, 45–54.
Susan A., Yadav M. K., Kar S., Aravindan S., Ngangkham U., Raghu S. et al. 2019 Molecular identification of blast resistance genes in rice landraces from northeastern India. Plant Pathol. 68, 537–546.
Tanksley S. D and Mc Couch S. R. 1997 Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277, 1063–1066.
Vasudevan K., Casiana M., Vera C., Gruissem W. and Navreet K. B. 2014 Large scale germplasm screening for identification of novel rice blast resistance sources. Front. Plant Sci. 505, 1–9.
Wang C., Yang Y., Yuan X., Xu Q., Feng Y., Yu H. et al. 2014 Genome–wide association study of blast resistance in indica rice. BMC Plant Biol. 14, 311.
Wang R., Fang N., Guan C., He W., Bao Y. and Zhang H. 2016 Characterization and fine mapping of a blast resistant gene Pi–jnw1 from the japonica rice landrace Jiangnanwan. PLoS One 11, e0169417.
Wang Y., Wang D., Deng X., Liu J., Sun P., Liu Y. et al. 2012 Molecular mapping of the blast resistance genes Pi2–1 and Pi51(t) in the durably resistant rice ‘Tianjingyeshengdao’. Phytopathology 102, 779–786.
Xu X., Chen H., Fujimura T. and Kawasaki S. 2008 Fine mapping of a strong QTL of field resistance against rice blast, Pikahei–1(t), from upland rice Kahei, utilizing a novel resistance evaluation system in the greenhouse. Theor. Appl. Genet. 117, 997–1008.
Xu X., Hayashi N., Wang C. T., Fukuoka S., Kawasaki S., Takatsuji H. et al. 2014 Rice blast resistance gene Pikahei–1(t), a member of a resistance gene cluster on chromosome 4, encodes a nucleotide–binding site and leucine–rich repeat protein. Mol. Breed. 34, 691–700.
Xu Y. and Crouch J. H. 2008 Marker–assisted selection in plant breed: from publications to practice. Crop Sci. 48, 391–407.
Yadav M. K., Aravindan S., Ngangkham U., Shubudhi H. N., Bag M. K., Adak T. et al. 2017 Use of molecular markers in identification and characterization of resistance to rice blast in India. PLoS One 12, e0176236.
Yadav M. K, Aravindan S., Ngangkham U., Raghu S., Prabhukarthikeyan S. R., Keerthana U. et al. 2019 Blast resistance in Indian rice landraces: genetic dissection by gene specific markers. PLoS One 14, e0211061.
Yang Q., Lin F., Wang L. and Pan Q. 2009 Identification and mapping of Pi41, a major gene conferring resistance to rice blast in the Oryza sativa subsp. indica reference cultivar, 93–11. Theor. Appl. Genet. 118, 1027–1034.
Zhang W., Sun J., Zhao G., Wang J., Liu H., Zheng H. et al. 2017 Association analysis of the glutelin synthesis genes GluA and GluB1 in a Japonica rice collection. Mol. Breed. 37, 129.
Zheng W., Wang Y., Wang L., Ma Z., Zhao J., Wang P. et al. 2016 Genetic mapping and molecular marker development for Pi65(t), a novel broad–spectrum resistance gene to rice blast using next–generation sequencing. Theor. Appl. Genet. 129, 1035–1044.
Zhou J. H., Wang J. L., Xu J. C., Lei C. L. and Ling Z. Z. 2004 Identification and mapping of a rice blast resistance gene Pi-g(t) in the cultivar Guangchangzhan. Plant Pathol. 53, 191–196.
Zhu M., Wang L. and Pan Q. 2004 Identification and characterization of a new blast resistance gene located on rice chromosome 1 through linkage and differential analyses. Phytopathology 94, 515– 519.
Zhu D., Kang H., Li Z., Liu M., Zhu X., Wang Y. et al. 2016 A genome–wide association study of field resistance to Magnaporthe oryzae in rice. Rice 9, 44.
Acknowledgements
The authors are extremely grateful to the Director, ICAR-National Rice Research Institute, Cuttack, India, for his support and facilitation in carrying out the research work successfully. This study was funded by the ICAR-National Rice Research Institute, Cuttack.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding Editor: H. A. Ranganath
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yadav, M.K., Aravindan, S., Ngangkham, U. et al. Candidate screening of blast resistance donors for rice breeding. J Genet 98, 73 (2019). https://doi.org/10.1007/s12041-019-1116-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-019-1116-z