
Abstract
The NMDA receptor, which is heavily involved in several human brain diseases, is a heteromeric ligand-gated ion channel that interacts with multiple intracellular proteins through the C-termini of different subunits. GluN2A and GluN2B are the two primary types of GluN2 subunits in the forebrain. During the developmental period, there is a switch from GluN2B- to GluN2A-containing NMDA receptors in synapses. In the adult brain, GluN2A exists at synaptic sites more abundantly than GluN2B. GluN2A plays important roles not only in synaptic plasticity but also in mediating physiological functions, such as learning and memory. GluN2A has also been involved in many common human diseases, such as cerebral ischemia, seizure disorder, Alzheimer’s disease, and systemic lupus erythematosus. The following review investigates the functional and molecular properties, physiological functions, and pathophysiological roles of the GluN2A subunit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The N-methyl-d-aspartate receptor (NMDAR) is a heteromeric protein containing two obligate GluN1 subunits and a variety of GluN2 and GluN3 subunits [1]. GluN2A is one of the primary types of GluN2 subunits in the forebrain. The predicted amino acid sequence for GluN2A exhibits a sequence of 1464 amino acids [2]. The 1464-aa predicted sequence encoded by human GluN2A cDNA exhibits a 95.2 % identity with those of its mouse and rat homologues. GluN2A contains four putative transmembrane domains (M1–M4) and a C-terminal extension of greater than 600 residues that provides additional target sites for cellular constituents [3]. The expression of the GluN2A gene is under developmental control. There is an increasing trend for GluN2A expression as development progresses. Synaptic activity and sensory experiences cause this developmental switch from GluN2B- to GluN2A-containing NMDARs [4]. Over the last decade, an increasing number of reports have demonstrated important roles for GluN2A in physiological and pathophysiological processes. The following review provides a summary of recent findings regarding the functional and molecular properties, physiological functions, and pathophysiological roles of GluN2A.
The Functional and Molecular Properties of GluN2A
Electrophysiological Properties
In neurons of the cerebral cortex and regardless of postnatal age, cells expressing GluN2A subunit messenger RNA (mRNA) have faster NMDAR excitatory postsynaptic currents than cells that do not express the GluN2A subunit [5]. Chen et al. reported that GluN1/GluN2A-mediated peak current densities are ∼4 times larger than GluN1/GluN2B, and the peak channel open probability is significantly higher for GluN1/GluN2A than for GluN1/GluN2B [6]. The transition of NMDARs from the open to the closed state is also regulated by the GluN2 subunit. Based on calcium-dependent inactivation, GluN2A-containing NMDARs show a reversible inactivation that is highly similar to native NMDARs in cultured hippocampal neurons; however, those containing GluN2B exhibit no significant inactivation [7]. Moreover, recovery from desensitization was faster for GluN1/GluN2A- than for GluN1/GluN2B-containing channels [8]. The above results suggest that GluN2A-containing NMDARs desensitize more and take less time to recover than GluN2B-containing receptors [9]. These electrophysiological properties could enable GluN2A subunits to more flexibly regulate synapse activity.
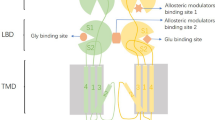
Domains in the Extracellular Regions of GluN2A
From the N- to C-terminal, the GluN2A subunit contains an N-terminal domain containing a modulatory site that binds Zn2+ followed by an agonist-binding domain, a channel-forming domain comprised of four transmembrane segments (M1–M4) and an intracellular C-terminal domain [10]. The extracellular regions of GluN2A include an N-terminal region and an extracellular loop between M3 and M4 [11]. The agonist-binding domain and the extracellular loop form the glutamate-binding site. Although the subunit-specific gating of NMDARs is controlled by the region that is formed by the GluN2 N-terminal domain [12], the interface of the agonist-binding domain dimer between GluN1 and GluN2A could be a major structural determinant that controls the allosteric modulation of GluN2A [10]. Moreover, the N-terminal domains of both GluN1 and GluN2 subunits determine allosteric Zn2+ inhibition and the glycine affinity of NMDARs [13]. The extracellular N-terminal domain of GluN2A also contains an endoplasmic reticulum retention signal that can be specifically masked by the N-terminal domain of GluN1α [14].
Signaling Proteins Associated to the Intracellular C-Terminal of GluN2A
The C-terminal of GluN2A provides additional target sites for downstream signaling molecules (Fig. 1). The 1349–1464 amino acid sequence in the C-terminal of GluN2A is responsible for the binding of Ca2+/calmodulin-dependent protein kinase II (CaMKII) [15]. The last three amino acid sequences in the C-terminal of GluN2A binds directly to several neuronal scaffolding proteins, including postsynaptic density protein-95 (PSD-95), postsynaptic density protein-93 (PSD-93), synapse-associated protein 102 (SAP102), and synapse-associated protein 97 (SAP97). Based on these scaffolding proteins, GluN2A could transduce many signal types to its downstream proteins.

Signaling molecules associated to the C-terminal of GluN2A. The amino acid sequence in the C-terminal of GluN2A is responsible for the binding of CaMKII and PSD-95. Based on the scaffolding protein PSD-95, GluN2A could transduce many signal types to its downstream proteins. PSD-95, p35, and cdk5 can form a complex in synaptosomes. The N-terminal region of PSD-95 is essential for the interaction between PSD-95 and Src. PKCα, PTEN, nNOS, and Fyn are the PDZ ligands of PSD-95. Pyk2 can bind to the SH3 domain of PSD-95
Among the scaffolding proteins mentioned above, PSD-95 is the most widely studied. PSD-95, p35, and cyclin dependent kinase-5 (cdk5) can form a complex in synaptosomes [16]. Phosphatase and tensin homolog located on chromosome 10 (PTEN) contains a PDZ-binding motif at its C-terminus, and NMDAR activation triggers a PDZ-dependent association between PTEN and PSD-95 [17]. PSD-95, GluN2A, and PTEN interact with each other at the synapse [18]. The PDZ3 domain of PSD-95 and the SH2 domain of Fyn are responsible for the association between the two proteins [19]. The 43–57 amino acid sequence in the N-terminal region of PSD-95 is essential for the interaction between PSD-95 and Src [20]. Proline-rich tyrosine kinase 2 (Pyk2) can bind to the SH3 domain of PSD-95, and this binding is facilitated by the Src-induced phosphorylation of PSD-95Y523 [21]. PKCα has a classic T/SXV motif (QSAV) and can interact with all three PDZ domains of PSD-95 and SAP102 [22, 23].
Additional scaffolding proteins can also couple the signaling of several proteins. PSD-93 also interacts with GluN2A and with Fyn in the mouse cerebral cortex [24]. A ternary complex assembled by PSD-93 with GluN2A and nNOS also exists in cultured cortical neurons [25]. SAP97 directly associates with GluN2A through its PDZ1 domain, and the CaMKII-dependent phosphorylation of SAP97-Ser-232 disrupts its interaction with GluN2A [26].
Physiological Functions
Synaptic Plasticity
Many studies have indicated that GluN2A is a critical factor that determines the polarity of synaptic plasticity. However, the exact role of GluN2A in long-term potentiation (LTP) and long-term depression (LTD) is still a matter of controversy (Table 1).
With regard to the role for GluN2A in LTP, several research groups have observed opposing results. Following the targeted disruption of the mouse NR2A gene, the LTPs induced by high-frequency stimulation in the hippocampal CA1 and juvenile superior colliculus were significantly reduced [27] and blocked [28], respectively. Furthermore, there was a considerable reduction in LTP in CA1 slices from 3-month-old GluN2A C-terminal-truncated mice, and this reduction could be due to impairments in cellular signal transduction events involved in LTP induction [29]. However, using RNA interference (RNAi) and overexpression, Foster et al. found that GluN2A is not essential for LTP that is induced by pairing postsynaptic depolarization to 0 mV with presynaptic stimulation consisting of 200 pulses at 2 Hz [30]. In addition, these researchers determined that the cytoplasmic tail of GluN2A appears to carry inhibitory factors for LTP. However, in these genetic experiments, indirect effects on synaptic plasticity resulting from compensatory mechanisms were difficult to describe. In addition, the stimulations used were also different from other experiments.
Others studies have used pharmacological approaches to study the direct effect of GluN2A on LTP. Several have indicated that the inhibition of GluN2A by NVP-AAM077 blocked or reduced LTP in the hippocampal CA1 [31, 32], dentate gyrus [33], lateral amygdala [34], and visual Cortical [35]. Although these results are highly consistent, there are two issues worth addressing. One is that the selectivity of NVP-AAM077 drops to tenfold for rodent GluN2A over GluN2B; at the concentrations used, NVP-AAM077 also partially blocks GluN2B. The second is how to interpret the effects of NVP-AAM077 on GluN1/GluN2A/GluN2B NMDARs.
Most studies concluded that GluN2A plays a critical role in LTD. NVP-AAM077 can prevent LTD in hippocampus [32, 36], lateral amygdala [34], and nucleus accumbens [37]. However, several conflicting results have been reported. Liu et al. showed that the preferential inhibition of GluN2A did not affect the appearance of LTD [31]. Vasuta et al. found that exercise can significantly alter the contribution of GluN2A to LTD and that NVP-AAM077 prevented LTD in running but not control animals [33]. It was also observed that LTD can be reduced by NVP-AAM007 in P21-P28 mice but not in P12-P18 or P45-90 mice [35].
In view of the switch of GluN2B to GluN2A during development and the dominant expression of GluN2A in synapses, GluN2A could play an important role in the production of both LTP and LTD.
Learn and Memory
Behavioral assays have demonstrated an important role for the GluN2A subunit in mediating physiological functions, such as learning and memory.
Studies have shown that GluN2A is involved in the learning process, and spatial or discrimination learning impairments have been observed in mice lacking the GluN2A subunit [27, 38]. GluN2A is also required for vestibular-cerebellar motor learning through potentiation at the mossy fiber to granule cell synapse [39]. The blockade of GluN2A in the dorsal striatum impairs the learning of complex motor skills [40].
There is increasing evidence to indicate that GluN2A is closely related to hippocampal-dependent spatial memory. Mice lacking GluN2A exhibit impairments in rapidly acquired spatial working memory [41] and also perform poorly in spatial pattern separation tasks [42]. The suppressed expression of GluN2A by peroxisome proliferator-activated receptor α (PPARα) knockdown rendered animals markedly impaired in their consolidation of spatial memory, whereas the introduction of PPARα to the hippocampus of PPARα-null mice increased hippocampal GluN2A and improved spatial learning and memory [43]. Mice treated with dexamethasone for 21 consecutive days showed significantly impaired spatial memory during the Morris water maze task, which could be due to the reduction in the expression of GluN2A/B [44].
GluN2A also plays an important role in fear memory. Contextual fear conditioning is impaired in GluN2A C-terminal-truncated mice [29]. The expression of GluN2A in the amygdala [45], hippocampus [46], retrosplenial cortex [47], or prelimbic medial prefrontal cortex [48] contributes to contextual or trace fear memories. The coactivation of GluN2A and GluN2B induces resistance to fear extinction, and patients with posttraumatic stress disorder could benefit from the antagonism of specific GluN2 subunits [49].
Several results have indicated an important role for GluN2A in auditory activity. Sensory and neuronal injury affects the expression level of GluN2A protein in rats [50]. GluN2A transcripts increase significantly in both the IMAN and Area X of zebra finches, and this increase is critical for vocal learning during the song-learning period between posthatching day 20 and 40 [51].
Pathophysiological Roles
NR2A has been implicated in many common human diseases, such as cerebral ischemia, seizure disorder, Alzheimer’s disease, systemic lupus erythematosus, depression, schizophrenia, Parkinson’s disease, Huntington’s disease, anxiety, and bipolar disorder.
Cerebral Ischemia
Excitotoxicity induced by the overactivation of NMDAR is a critical mechanism that contributes to neuronal damage during cerebral ischemia. It is generally accepted that GluN2B has neurotoxic effects; however, the contribution of GluN2A to excitotoxicity remains controversial.
The regular pattern of GluN2A expression following cerebral ischemia changes could be increase–decrease–increase over time. At early time points, such as 0.5 h for bilateral common carotid artery occlusion in gerbils [52, 53] and 3 h for middle cerebral artery occlusion in rats [54], the expression of GluN2A was increased. From 6 to 24 h after challenge, especially in the rat four-vessel occlusion model, the level of GluN2A was decreased [55–57]. After 48 h, there was a reversal in GluN2A expression [57, 58], and at the 1-week time point, the expression of GluN2A decreased again [57]. The age of rats also influences the expression of GluN2A [59]. In addition, the expression of GluN2A in the penumbra was significantly increased 3 h after focal cerebral ischemia and was then reversed after 24 h [60].
Following cerebral ischemia, there exists a persistent phosphorylation of GluN2A in the hippocampus [61]. The tyrosine phosphorylation of GluN2A occurs as early as 15 min after reperfusion and is sustained for at least 24 h [62–64]. Tyrosine kinases, such as Fyn, Src [65], protein kinase C [66], and Pyk2 [67], are involved in the tyrosine phosphorylation of GluN2A. This phosphorylation of GluN2A could induce the redistribution of NMDA receptors between synaptic lipid rafts and postsynaptic densities [68] and leads to rapid clustering from extrasynaptic to synaptic membrane fractions [69].
Although it is generally agreed that GluN2A plays an important role in ischemic damage, it is still a matter of controversy whether GluN2A mediates prosurvival signals (Table 2). Zhou et al. reported that NMDA-mediated toxicity is caused by the activation of GluN2B- but not GluN2A-containing NMDARs and that the switch from GluN2B to GluN2A in adult rats led to the invulnerability of adult hippocampal slices to NMDA treatment [70]. Subsequently, Liu et al. found that the activation of GluN2B results in excitotoxicity, while the activation of GluN2A promotes neuronal survival both in mature cortical cultures and in an in vivo rat model of focal ischemic stroke [71]. Similarly, Chen et al. found that the GluN2A subtype-specific antagonist NVP-AAM077 enhanced neuronal death following transient global ischemia and abolished the induction of ischemic tolerance; GluN2B was found to have an opposite role [72]. However, other groups have disputed these findings. Morikawa et al. found that, after a 2-h middle cerebral artery occlusion, brain injury volumes revealed a significantly smaller injury size in GluN2A subunit knockout mice [73]. Wang et al. observed that endogenous cdk5 activated by forebrain ischemia can phosphorylate GluN2A at Ser1232 and induce CA1 pyramidal neurons damage [74]. Choo et al. found that activation of NR2A-containing NMDARs was associated with JNK phosphorylation that was neuroprotective in neuronal cultures subjected to excitotoxicity [75]. Zhou et al. reported that overexpression and molecular knockdown of GluN2A exacerbate and attenuate NMDAR-mediated neuronal death, respectively [76]. They proposed that the magnitude and duration of GluN2A activation could be a key factor which determines neuronal fate [77]. It was also found that the downregulation of GluN2A by a GluN2A antisense construct [78], Co 101244 and Zn2+ [79], conantokin G [80], PSD-95 antisense construct [81], or PP2 [82], a potent inhibitor of Src family kinases, significantly reduced excitotoxic cell death. Moreover, enhanced glutamate excitotoxic vulnerability with age is associated with a substantial increase in GluN2A in vitro [83, 84]. Interestingly, von Engelhardt et al. found that 50 nM NVP-AAM077 increased the toxicity induced by submaximal 5 μM NMDA in DIV21 cortical cultures [83].
In a word, GluN2A might mediate both prosurvival and prodeath signalings. The GluN2A-PTEN-TDP-43 (TAR DNA-binding protein-43) [85] and GluN2A-CaMKIV-TORC1 (transducers of regulated CREB activity 1) [86] signaling pathways could mediate the prosurvival effect of GluN2A. In contrast, cdk5 could be a downstream prodeath molecule for GluN2A [74].
Seizure Disorder
There may be a connection between increased glutamatergic neurotransmission and seizure activity. The selective increase in the coexpression of GluN2A/2B and GluN1 in dysplastic neurons of human epileptic cortex could contribute to focal seizure onset [87, 88]. In seizures induced by pentylenetetrazole in rats, GluN2A was markedly increased in the cortex during the early seizure development process [89]. In the subiculum of seizure-sensitive gerbils, GluN2A/B immunoreactivity increased significantly at 12 h postictal [90]. It has also been found that GluN2A knockout mice were more resistant to audiogenic-like seizures induced by stimulating the inferior colliculus [91]. Moreover, tyrosine phosphorylation of GluN2A in the rat hippocampus was enhanced following KA-induced status epilepticus [92] or Li/pilocarpine-induced status epilepticus [93].
Seizure activity can also influence the expression of GluN2A and contribute to cognitive changes in seizure patients. In the hippocampus of flurothyl- or kainite-treated neonatal rats, the expression of GluN2A protein was decreased significantly [94, 95]. Recurrent seizures in animal models of early-onset epilepsy resulted in a decrease in the expression of the GluN2A subunit, which could be delayed by at least 5 days but persists for at least 3 to 4 weeks [96]. Alterations in the gene encoding the GluN2A subunit are a major genetic risk factor for idiopathic focal epilepsy [97]. However, other studies have shown opposing results. Except for those with hippocampal sclerosis, all temporal lobe epilepsy patients showed increased GluN2A and GluN2B hybridization densities in dentate granule cells [98]. Following multiple perinatal seizures induced by kainic acid, rat pups showed a robust increase in GluN2A/2B labeling specific to cortical layer V throughout the retrosplenial, parietal, and temporal cortices [99]. Prolonged febrile seizures induce an increase in the hippocampal levels of GluN2A [100]. No differences were found in the expression of GluN2A and GluN2B in the amygdalas of patients with mesial temporal lobe epilepsy [101].
GluN2A might also be involved in the pathological processes of seizures. Ganor et al. found that subpopulations of epilepsy patients show significantly elevated levels of autoantibodies to a peptide of the GluN2A subunit [102]. This type of GluN2A autoantibodies could damage the brain [103].
It is found minocycline could exert an anticonvulsant effect by preventing the increase in GluN2A [104]. Neuropeptide Y could inhibit seizures via the downregulation of the functional expression of GluN2A and GluN2B [105]. Therefore, a pharmacological strategy directed to the GluN2 subunit might help to limit the onset or diffusion of seizures [106].
Alzheimer’s Disease
Many have observed NMDARs dysfunction in Alzheimer’s disease (AD) patients, which is responsible for the cognitive deficits of AD. Sultana et al. observed a significantly decreased level of GluN2A in the hippocampus of subjects with amnestic mild cognitive impairment, a prodromal stage of Alzheimer’s disease [107]. Sze et al. reported that nonphosphorylated and phosphorylated GluN2A were selectively reduced in the entorhinal cortex of AD patients [108]. These researchers also found reductions in GluN2A mRNA levels in the hippocampus [109]. Hynd et al. reported that the transcript and protein expression of both GluN2A and GluN2B was markedly attenuated in susceptible regions in subjects with AD pathology, such as the cingulate gyrus, hippocampus, and superior temporal cortex [110]. However, Mishizen-Eberz et al. found that the expression of GluN2A subunit mRNA and protein were unchanged during AD progression and that neuronal mRNA expression revealed a significant increase in the GluN2A subunit in subjects with moderate neurofibrillary tangle neuropathology [111]. Marcello et al. found that SAP97, which is responsible for the trafficking of GluN1 and GluN2A and is connected to GluN2A and GluN2A localization in hippocampus, is not altered in AD patients [112]. These contradictory results could be due to a small sample size.
A majority of studies show that amyloid-β (Aβ) can alter NMDARs activity, contributing to its neurotoxicity. Aβ25-35 treatment resulted in elevated tyrosine phosphorylation of GluN2A in the CA1 subfield of the rat hippocampus and facilitated the interactions of GluN2A and Src kinases [113]. Aβ oligomers directly activate NMDARs, particularly those with the GluN2A subunit [114]. Amyloid precursor protein mutations associated with familial Alzheimer’s disease enhanced the trafficking of GluN2A to the cell surface [115]. Aβ induces dendritic spine loss via a pathway involving GluN2A-containing NMDARs [116]. However, Liu et al. found that Aβ leads to a loss of synaptic proteins via the suppression of GluN2A function and the activation of GluN2B function [117]. The combined oligomer Aβ(1–40) and stress treatment decreased GluN2A/2B expression in the hippocampus [118].
There is also a relationship between GluN2A and Tau. The blockade of GluN2A induces Tau phosphorylation in rat hippocampal slices [119]. This effect could be related to the GluN2A-PKC-glycogen synthase kinase-3β (GSK3β) signaling pathway [120].
Systemic Lupus Erythematosus
The anti-NMDAR autoantibodies in patients with systemic lupus erythematosus (SLE) is a typical example that suggests that antibodies can alter emotion. DeGiorgio et al. found that lupus antibodies cross-react with GluN2A and GluN2B, gain access to cerebrospinal fluid, and might mediate abnormalities of the central nervous system [121]. Subsequently, autoantibodies directed against GluN2A were found in the sera of patients with SLE [122]. Omdal et al. considered that neuropsychiatric disturbances in SLE are associated with antibodies against GluN2A and GluN2B [123]. A breach in the integrity of the blood–brain barrier could expose neurons to these potentially pathogenic antibodies [124]. Indeed, the levels of anti-NR2 antibodies in the cerebrospinal fluid were significantly elevated in patients with diffuse psychiatric/neuropsychological syndromes, whereas there were no significant differences in serum anti-NR2 levels [125]. The administration of the nonnaturally occurring D form of the DWEYS pentapeptide, a sequence present in the GluN2A and GluN2B subunits, prevents these antibodies from depositing in glomeruli and from mediating neuronal excitotoxicity [126]. An anti-GluN2A antibody could be a predictor for neuropsychiatric systemic lupus erythematosus [127]. However, Harrison et al. reported that no significant association was found between the anti-GluN2A antibody and cognitive dysfunction, depressive symptoms, or anxiety in SLE patients [128].
Anti-GluN2A autoantibodies might induce the apoptosis of GluN2A-expressing neurons [129]. The underlying mechanism could be the promotion of NMDAR-mediated excitotoxicity [130] and enhancement of Ca2+ influx through the inhibition of the binding capacity of zinc [131].
Depression
Growing evidence implicates a role for GluN2A signaling in depression. GluN2A knockout mice [132] or mice expressing mutant GluN2A with a Tyr-1325-Phe mutation, which prevents the phosphorylation of this site [133], showed antidepressant-like profiles in the forced swim test and tail suspension test. GluN2A and its downstream molecules might contribute to chronic mild stress susceptibility [134].
Depression also has an effect on GluN2A. The decreased expression of GluN2A in the perirhinal [135] and prefrontal [136] cortex in major depression patients has been observed. The levels of GluN2A were reduced in different brain regions in prenatally stressed juvenile offspring showing depression-like behavior [137], while the levels of GluN2A in the lateral amygdala were elevated in depressed patients [138]. GRIN2A, which encodes GluN2A, was also found to be hypermethylated in both the prefrontal cortex and hippocampus of recurrent depression patients [139].
Schizophrenia
There is growing evidence in support of the hypothesis that hypofunction of NMDARs is involved in the pathophysiology of schizophrenia. Mice lacking the GluN2A exhibit several behavioral abnormalities related to schizophrenia, including hyperlocomotion and cognitive impairments [140]. A microsatellite repeat in the promoter of the GluN2A subunit gene suppresses transcriptional activity and is correlated with chronic outcome in schizophrenia [141]. Reduced GluN2A expression is correlated with negative symptoms in the postmortem cerebellum during chronic schizophrenia [142]. GluN2A is also decreased in the prefrontal cortex during schizophrenia [143, 144]. Dysbindin, a schizophrenia-susceptibility gene that is widely expressed in the forebrain, prevents the expression of GluN2A in the hippocampus [145]. However, several studies did not report changes in GluN2A expression in the dorsolateral prefrontal and anterior cingulate cortex [146], the medial temporal lobe [135], the thalamus [147], and cerebellum [148] of schizophrenia patients.
Parkinson’s Disease
GluN2A might be linked with impaired LTP in patients with Parkinson’s disease and dyskinesia induced by long-term L-DOPA therapy. Hippocampal LTP is altered in both neurotoxic and transgenic models of Parkinson’s disease and these alterations are associated with impaired dopaminergic transmission and a decrease in the GluN2A/GluN2B subunit ratio in synaptic NMDARs [149]. The modulation of the composition of synaptic NMDAR using TAT2A, a cell-permeable peptide targeting GluN2A, during the development of dyskinesias led to a reduction in the percentage of Parkinsonian rats that developed dyskinetic movements [150].
Huntington’s Disease
There is growing evidence indicating the involvement of GluN2A in Huntington’s disease. Variation in the GluN2A receptor gene can affect the age of onset for Huntington disease [151]. The GluN2A expression in the hippocampus of R6/2 transgenic Huntington’s disease mice was found to be decreased [152]. A significant decrease in the percentage of cells expressing GluN2A at all ages is observed in the R6/2 mouse model of Huntington’s disease [153]. However, Jarabek et al. observed no change in GluN2A in the striata of N171-82Q mice, a new transgenic model of Huntington’s disease [154].
Anxiety
A number of different classes of NMDAR antagonists have been shown to exhibit anxiolytic effects in different laboratory tests of anxiety [155]. The underlying mechanism of these antagonists could act through their blocking effect on GluN2A [156]. GluN2A knockout mice exhibit decreased anxiety-like behavior relative to wild-type littermates across multiple tests [132]. Prenatal stress reduced GluN2A expression in the hippocampus, the prefrontal cortex, and striatum in the offspring, and the altered expression of GluN1 and GluN2A could have a potential impact on anxiety-like behavior [157].
Bipolar Disorder
Disturbances in glutamate-mediated synaptic transmission could be involved in the pathophysiology of bipolar disorder (BD). There is a decrease in the expression of GluN2A in the anterior cingulate cortex [158], the perirhinal cortex [135], and hippocampus during bipolar disorder [159].
Perspectives
The absence of a highly selective rodent GluN2A antagonist might be the largest obstacle for studying the pathophysiological role of GluN2A. The selectivity of NVP-AAM077, the most widely used selective GluN2A antagonist, against rodent GluN2A is still a matter of controversy. In contrast, TCN-201 and TCN-213 displayed submicromolar and micromolar potency at GluN1/GluN2A receptor, respectively, although they did not show activity at GluN2B-containing receptor up to 50 μM concentration [160]. These novel antagonists might be useful in deeply understanding the physiological and pathophysiological roles of GluN2A in brain functions.
PDZ-containing proteins are typical scaffolding proteins associated with GluN2A. The human genome contains hundreds of different PDZ ligand-containing proteins, and all of these proteins are likely to be downstream molecules of GluN2A. Therefore, the signaling of GluN2A is likely to be highly complicated. The spatial distribution, cooperation, and relative importance of these signaling proteins remain unclear.
The tyrosine phosphorylation of GluN2A can be regulated by Src, Fyn, or cdk5; however, the phosphatases related to the dephosphorylation of GluN2A have not been reported.
GluN2A, a primary type of NMDAR subunit in the brain, is involved in the pathogenesis of many types of brain diseases. However, the causality between GluN2A and these brain diseases has not been determined. Moreover, only a small number of reports have suggested treatment strategies based on GluN2A or its signaling pathways. In view of the controversy on whether GluN2A mediates prodeath signaling or not, the specific prodeath signaling pathways of GluN2A should be determined in the future. Blocking the actions of the downstream prodeath molecules of GluN2A could be an effective strategy to treat brain diseases.
References
McBain CJ, Mayer ML (1994) N-methyl-D-aspartic acid receptor structure and function. Physiol Rev 74(3):723–760
Foldes RL, Adams SL, Fantaske RP, Kamboj RK (1994) Human N-methyl-D-aspartate receptor modulatory subunit hNR2A: cloning and sequencing of the cDNA and primary structure of the protein. Biochim Biophys Acta 1223(1):155–159
Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B et al (1992) Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 256(5060):1217–1221
Matta JA, Ashby MC, Sanz-Clemente A, Roche KW, Isaac JT (2011) mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 70(2):339–351. doi:10.1016/j.neuron.2011.02.045
Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H (1997) NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci 17(7):2469–2476
Chen N, Luo T, Raymond LA (1999) Subtype-dependence of NMDA receptor channel open probability. J Neurosci 19(16):6844–6854
Krupp JJ, Vissel B, Heinemann SF, Westbrook GL (1996) Calcium-dependent inactivation of recombinant N-methyl-D-aspartate receptors is NR2 subunit specific. Mol Pharmacol 50(6):1680–1688
Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR (1998) Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J Neurophysiol 79(2):555–566
Loftis JM, Janowsky A (2003) The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther 97(1):55–85
Gielen M, Le Goff A, Stroebel D, Johnson JW, Neyton J, Paoletti P (2008) Structural rearrangements of NR1/NR2A NMDA receptors during allosteric inhibition. Neuron 57(1):80–93. doi:10.1016/j.neuron.2007.11.021
Cull-Candy SG, Leszkiewicz DN (2004) Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004 (255):re16. doi:10.1126/stke.2552004re16
Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459(7247):703–707. doi:10.1038/nature07993
Madry C, Mesic I, Betz H, Laube B (2007) The N-terminal domains of both NR1 and NR2 subunits determine allosteric Zn2+ inhibition and glycine affinity of N-methyl-D-aspartate receptors. Mol Pharmacol 72(6):1535–1544. doi:10.1124/mol.107.040071
Qiu S, Zhang XM, Cao JY, Yang W, Yan YG, Shan L, Zheng J, Luo JH (2009) An endoplasmic reticulum retention signal located in the extracellular amino-terminal domain of the NR2A subunit of N-methyl-D-aspartate receptors. J Biol Chem 284(30):20285–20298. doi:10.1074/jbc.M109.004960
Gardoni F, Schrama LH, van Dalen JJ, Gispen WH, Cattabeni F, Di Luca M (1999) AlphaCaMKII binding to the C-terminal tail of NMDA receptor subunit NR2A and its modulation by autophosphorylation. FEBS Lett 456(3):394–398
Morabito MA, Sheng M, Tsai LH (2004) Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci 24(4):865–876. doi:10.1523/JNEUROSCI.4582-03.2004
Jurado S, Benoist M, Lario A, Knafo S, Petrok CN, Esteban JA (2010) PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J 29(16):2827–2840. doi:10.1038/emboj.2010.160
Ventruti A, Kazdoba TM, Niu S, D’Arcangelo G (2011) Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 189:32–42. doi:10.1016/j.neuroscience.2011.05.050
Tezuka T, Umemori H, Akiyama T, Nakanishi S, Yamamoto T (1999) PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc Natl Acad Sci U S A 96(2):435–440
Kalia LV, Pitcher GM, Pelkey KA, Salter MW (2006) PSD-95 is a negative regulator of the tyrosine kinase Src in the NMDA receptor complex. EMBO J 25(20):4971–4982. doi:10.1038/sj.emboj.7601342
Seabold GK, Burette A, Lim IA, Weinberg RJ, Hell JW (2003) Interaction of the tyrosine kinase Pyk2 with the N-methyl-D-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J Biol Chem 278(17):15040–15048. doi:10.1074/jbc.M212825200
Lim IA, Hall DD, Hell JW (2002) Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem 277(24):21697–21711. doi:10.1074/jbc.M112339200
O’Neill AK, Gallegos LL, Justilien V, Garcia EL, Leitges M, Fields AP, Hall RA, Newton AC (2011) Protein kinase Calpha promotes cell migration through a PDZ-dependent interaction with its novel substrate discs large homolog 1 (DLG1). J Biol Chem 286(50):43559–43568. doi:10.1074/jbc.M111.294603
Sato Y, Tao YX, Su Q, Johns RA (2008) Post-synaptic density-93 mediates tyrosine-phosphorylation of the N-methyl-D-aspartate receptors. Neuroscience 153(3):700–708. doi:10.1016/j.neuroscience.2008.03.006
Xu Y, Zhang B, Hua Z, Johns RA, Bredt DS, Tao YX (2004) Targeted disruption of PSD-93 gene reduces platelet-activating factor-induced neurotoxicity in cultured cortical neurons. Exp Neurol 189(1):16–24. doi:10.1016/j.expneurol.2004.05.013
Gardoni F, Mauceri D, Fiorentini C, Bellone C, Missale C, Cattabeni F, Di Luca M (2003) CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem 278(45):44745–44752. doi:10.1074/jbc.M303576200
Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S et al (1995) Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373(6510):151–155. doi:10.1038/373151a0
Zhao JP, Constantine-Paton M (2007) NR2A−/− mice lack long-term potentiation but retain NMDA receptor and L-type Ca2+ channel-dependent long-term depression in the juvenile superior colliculus. J Neurosci 27(50):13649–13654. doi:10.1523/JNEUROSCI.3153-07.2007
Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V et al (1998) Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell 92(2):279–289
Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M, Sheng M (2010) Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci 30(7):2676–2685. doi:10.1523/JNEUROSCI.4022-09.2010
Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304(5673):1021–1024. doi:10.1126/science.1096615
Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI et al (2007) Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 52(1):60–70. doi:10.1016/j.neuropharm.2006.07.013
Vasuta C, Caunt C, James R, Samadi S, Schibuk E, Kannangara T, Titterness AK, Christie BR (2007) Effects of exercise on NMDA receptor subunit contributions to bidirectional synaptic plasticity in the mouse dentate gyrus. Hippocampus 17(12):1201–1208. doi:10.1002/hipo.20349
Muller T, Albrecht D, Gebhardt C (2009) Both NR2A and NR2B subunits of the NMDA receptor are critical for long-term potentiation and long-term depression in the lateral amygdala of horizontal slices of adult mice. Learn Mem 16(6):395–405. doi:10.1101/lm.1398709
de Marchena J, Roberts AC, Middlebrooks PG, Valakh V, Yashiro K, Wilfley LR, Philpot BD (2008) NMDA receptor antagonists reveal age-dependent differences in the properties of visual cortical plasticity. J Neurophysiol 100(4):1936–1948. doi:10.1152/jn.90290.2008
Kollen M, Dutar P, Jouvenceau A (2008) The magnitude of hippocampal long term depression depends on the synaptic location of activated NR2-containing N-methyl-D-aspartate receptors. Neuroscience 154(4):1308–1317. doi:10.1016/j.neuroscience.2008.04.045
Chergui K (2011) Dopamine induces a GluN2A-dependent form of long-term depression of NMDA synaptic responses in the nucleus accumbens. Neuropharmacology 60(6):975–981. doi:10.1016/j.neuropharm.2011.01.047
Brigman JL, Feyder M, Saksida LM, Bussey TJ, Mishina M, Holmes A (2008) Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn Mem 15(2):50–54. doi:10.1101/lm.777308
Andreescu CE, Prestori F, Brandalise F, D’Errico A, De Jeu MT, Rossi P, Botta L, Kohr G et al (2011) NR2A subunit of the N-methyl D-aspartate receptors are required for potentiation at the mossy fiber to granule cell synapse and vestibulo-cerebellar motor learning. Neuroscience 176:274–283. doi:10.1016/j.neuroscience.2010.12.024
Lemay-Clermont J, Robitaille C, Auberson YP, Bureau G, Cyr M (2011) Blockade of NMDA receptors 2A subunit in the dorsal striatum impairs the learning of a complex motor skill. Behav Neurosci 125(5):714–723. doi:10.1037/a0025213
Bannerman DM, Niewoehner B, Lyon L, Romberg C, Schmitt WB, Taylor A, Sanderson DJ, Cottam J et al (2008) NMDA receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J Neurosci 28(14):3623–3630. doi:10.1523/JNEUROSCI.3639-07.2008
Kannangara TS, Eadie BD, Bostrom CA, Morch K, Brocardo PS, Christie BR (2014) GluN2A−/− Mice Lack Bidirectional Synaptic Plasticity in the Dentate Gyrus and Perform Poorly on Spatial Pattern Separation Tasks. Cereb Cortex. doi:10.1093/cercor/bhu017
Roy A, Jana M, Corbett GT, Ramaswamy S, Kordower JH, Gonzalez FJ, Pahan K (2013) Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor alpha. Cell Rep 4(4):724–737. doi:10.1016/j.celrep.2013.07.028
Tongjaroenbuangam W, Ruksee N, Mahanam T, Govitrapong P (2013) Melatonin attenuates dexamethasone-induced spatial memory impairment and dexamethasone-induced reduction of synaptic protein expressions in the mouse brain. Neurochem Int 63(5):482–491. doi:10.1016/j.neuint.2013.08.011
Walker DL, Davis M (2008) Amygdala infusions of an NR2B-selective or an NR2A-preferring NMDA receptor antagonist differentially influence fear conditioning and expression in the fear-potentiated startle test. Learn Mem 15(2):67–74. doi:10.1101/lm.798908
Gao C, Gill MB, Tronson NC, Guedea AL, Guzman YF, Huh KH, Corcoran KA, Swanson GT et al (2010) Hippocampal NMDA receptor subunits differentially regulate fear memory formation and neuronal signal propagation. Hippocampus 20(9):1072–1082. doi:10.1002/hipo.20705
Corcoran KA, Donnan MD, Tronson NC, Guzman YF, Gao C, Jovasevic V, Guedea AL, Radulovic J (2011) NMDA receptors in retrosplenial cortex are necessary for retrieval of recent and remote context fear memory. J Neurosci 31(32):11655–11659. doi:10.1523/JNEUROSCI.2107-11.2011
Gilmartin MR, Kwapis JL, Helmstetter FJ (2013) NR2A- and NR2B-containing NMDA receptors in the prelimbic medial prefrontal cortex differentially mediate trace, delay, and contextual fear conditioning. Learn Mem 20(6):290–294. doi:10.1101/lm.030510.113
Leaderbrand K, Corcoran KA, Radulovic J (2014) Co-activation of NR2A and NR2B subunits induces resistance to fear extinction. Neurobiol Learn Mem 113:35–40. doi:10.1016/j.nlm.2013.09.005
Wang Z, Ruan Q, Wang D (2005) Different effects of intracochlear sensory and neuronal injury stimulation on expression of synaptic N-methyl-D-aspartate receptors in the auditory cortex of rats in vivo. Acta Otolaryngol 125(11):1145–1151
Heinrich JE, Singh TD, Sohrabji F, Nordeen KW, Nordeen EJ (2002) Developmental and hormonal regulation of NR2A mRNA in forebrain regions controlling avian vocal learning. J Neurobiol 51(2):149–159
Kang TC, Hwang IK, Park SK, An SJ, Yoon DK, Moon SM, Lee YB, Sohn HS et al (2001) Chronological changes of N-methyl-D-aspartate receptors and excitatory amino acid carrier 1 immunoreactivities in CA1 area and subiculum after transient forebrain ischemia. J Neurocytol 30(12):945–955
Won MH, Kang T, Park S, Jeon G, Kim Y, Seo JH, Choi E, Chung M et al (2001) The alterations of N-Methyl-D-aspartate receptor expressions and oxidative DNA damage in the CA1 area at the early time after ischemia-reperfusion insult. Neurosci Lett 301(2):139–142
Gappoeva MU, Izykenova GA, Granstrem OK, Dambinova SA (2003) Expression of NMDA neuroreceptors in experimental ischemia. Biochemistry (Mosc) 68(6):696–702
Zhang L, Hsu JC, Takagi N, Gurd JW, Wallace MC, Eubanks JH (1997) Transient global ischemia alters NMDA receptor expression in rat hippocampus: correlation with decreased immunoreactive protein levels of the NR2A/2B subunits, and an altered NMDA receptor functionality. J Neurochem 69(5):1983–1994
Hsu JC, Zhang Y, Takagi N, Gurd JW, Wallace MC, Zhang L, Eubanks JH (1998) Decreased expression and functionality of NMDA receptor complexes persist in the CA1, but not in the dentate gyrus after transient cerebral ischemia. J Cereb Blood Flow Metab 18(7):768–775. doi:10.1097/00004647-199807000-00008
Liu Z, Zhao W, Xu T, Pei D, Peng Y (2010) Alterations of NMDA receptor subunits NR1, NR2A and NR2B mRNA expression and their relationship to apoptosis following transient forebrain ischemia. Brain Res 1361:133–139. doi:10.1016/j.brainres.2010.09.035
Dos-Anjos S, Martinez-Villayandre B, Montori S, Regueiro-Purrinos MM, Gonzalo-Orden JM, Fernandez-Lopez A (2009) Transient global ischemia in rat brain promotes different NMDA receptor regulation depending on the brain structure studied. Neurochem Int 54(3–4):180–185. doi:10.1016/j.neuint.2008.09.016
Gurd JW, Bissoon N, Beesley PW, Nakazawa T, Yamamoto T, Vannucci SJ (2002) Differential effects of hypoxia-ischemia on subunit expression and tyrosine phosphorylation of the NMDA receptor in 7- and 21-day-old rats. J Neurochem 82(4):848–856
Park S, Jung Y (2010) Combined actions of Na/K-ATPase, NCX1 and glutamate dependent NMDA receptors in ischemic rat brain penumbra. Anat Cell Biol 43(3):201–210. doi:10.5115/acb.2010.43.3.201
Matsumoto S, Shamloo M, Isshiki A, Wieloch T (2002) Persistent phosphorylation of synaptic proteins following middle cerebral artery occlusion. J Cereb Blood Flow Metab 22(9):1107–1113. doi:10.1097/00004647-200209000-00008
Takagi N, Shinno K, Teves L, Bissoon N, Wallace MC, Gurd JW (1997) Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptor subunits 2A and 2B. J Neurochem 69(3):1060–1065
Liu Y, Zhang G, Gao C, Hou X (2001) NMDA receptor activation results in tyrosine phosphorylation of NMDA receptor subunit 2A(NR2A) and interaction of Pyk2 and Src with NR2A after transient cerebral ischemia and reperfusion. Brain Res 909(1–2):51–58
Takagi N, Sasakawa K, Besshoh S, Miyake-Takagi K, Takeo S (2003) Transient ischemia enhances tyrosine phosphorylation and binding of the NMDA receptor to the Src homology 2 domain of phosphatidylinositol 3-kinase in the rat hippocampus. J Neurochem 84(1):67–76
Chen M, Hou X, Zhang G (2003) Tyrosine kinase and tyrosine phosphatase participate in regulation of interactions of NMDA receptor subunit 2A with Src and Fyn mediated by PSD-95 after transient brain ischemia. Neurosci Lett 339(1):29–32
Cheung HH, Teves L, Wallace MC, Gurd JW (2003) Inhibition of protein kinase C reduces ischemia-induced tyrosine phosphorylation of the N-methyl-d-aspartate receptor. J Neurochem 86(6):1441–1449
Liu Y, Zhang GY, Yan JZ, Xu TL (2005) Suppression of Pyk2 attenuated the increased tyrosine phosphorylation of NMDA receptor subunit 2A after brain ischemia in rat hippocampus. Neurosci Lett 379(1):55–58. doi:10.1016/j.neulet.2004.12.054
Besshoh S, Bawa D, Teves L, Wallace MC, Gurd JW (2005) Increased phosphorylation and redistribution of NMDA receptors between synaptic lipid rafts and post-synaptic densities following transient global ischemia in the rat brain. J Neurochem 93(1):186–194. doi:10.1111/j.1471-4159.2004.03009.x
Zhang F, Guo A, Liu C, Comb M, Hu B (2013) Phosphorylation and assembly of glutamate receptors after brain ischemia. Stroke 44(1):170–176. doi:10.1161/STROKEAHA.112.667253
Zhou M, Baudry M (2006) Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci 26(11):2956–2963. doi:10.1523/JNEUROSCI.4299-05.2006
Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J et al (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 27(11):2846–2857. doi:10.1523/JNEUROSCI.0116-07.2007
Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH et al (2008) Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39(11):3042–3048. doi:10.1161/STROKEAHA.108.521898
Morikawa E, Mori H, Kiyama Y, Mishina M, Asano T, Kirino T (1998) Attenuation of focal ischemic brain injury in mice deficient in the epsilon1 (NR2A) subunit of NMDA receptor. J Neurosci 18(23):9727–9732
Wang J, Liu S, Fu Y, Wang JH, Lu Y (2003) Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci 6(10):1039–1047. doi:10.1038/nn1119
Choo AM, Geddes-Klein DM, Hockenberry A, Scarsella D, Mesfin MN, Singh P, Patel TP, Meaney DF (2012) NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochem Int 60(5):506–516. doi:10.1016/j.neuint.2012.02.007
Zhou X, Ding Q, Chen Z, Yun H, Wang H (2013) Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem 288(33):24151–24159. doi:10.1074/jbc.M113.482000
Zhou X, Chen Z, Yun W, Wang H (2015) NMDA receptor activity determines neuronal fate: location or number? Rev Neurosci 26(1):39–47. doi:10.1515/revneuro-2014-0053
Mattar PA, Holmes KD, Dekaban GA (2003) An antisense construct reduces N-methyl-D-aspartate receptor 2A expression and receptor-mediated excitotoxicity as determined by a novel flow cytometric approach. J Neurosci Res 74(5):782–793. doi:10.1002/jnr.10793
Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB (2009) Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A 106(24):9854–9859. doi:10.1073/pnas.0903546106
Alex AB, Saunders GW, Dalpe-Charron A, Reilly CA, Wilcox KS (2011) CGX-1007 prevents excitotoxic cell death via actions at multiple types of NMDA receptors. Neurotoxicology 32(4):392–399. doi:10.1016/j.neuro.2011.03.002
Hou XY, Zhang GY, Wang DG, Guan QH, Yan JZ (2005) Suppression of postsynaptic density protein 95 by antisense oligonucleotides diminishes postischemic pyramidal cell death in rat hippocampal CA1 subfield. Neurosci Lett 385(3):230–233. doi:10.1016/j.neulet.2005.05.054
Hou XY, Liu Y, Zhang GY (2007) PP2, a potent inhibitor of Src family kinases, protects against hippocampal CA1 pyramidal cell death after transient global brain ischemia. Neurosci Lett 420(3):235–239. doi:10.1016/j.neulet.2007.03.048
von Engelhardt J, Coserea I, Pawlak V, Fuchs EC, Kohr G, Seeburg PH, Monyer H (2007) Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology 53(1):10–17. doi:10.1016/j.neuropharm.2007.04.015
Brewer LD, Thibault O, Staton J, Thibault V, Rogers JT, Garcia-Ramos G, Kraner S, Landfield PW et al (2007) Increased vulnerability of hippocampal neurons with age in culture: temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res 1151:20–31. doi:10.1016/j.brainres.2007.03.020
Zheng M, Liao M, Cui T, Tian H, Fan DS, Wan Q (2012) Regulation of nuclear TDP-43 by NR2A-containing NMDA receptors and PTEN. J Cell Sci 125(Pt 6):1556–1567. doi:10.1242/jcs.095729
Xu Q, Ji XF, Chi TY, Liu P, Jin G, Gu SL, Zou LB (2015) Sigma 1 receptor activation regulates brain-derived neurotrophic factor through NR2A-CaMKIV-TORC1 pathway to rescue the impairment of learning and memory induced by brain ischaemia/reperfusion. Psychopharmacology (Berl) 232(10):1779–1791. doi:10.1007/s00213-014-3809-6
Ying Z, Babb TL, Mikuni N, Najm I, Drazba J, Bingaman W (1999) Selective coexpression of NMDAR2A/B and NMDAR1 subunit proteins in dysplastic neurons of human epileptic cortex. Exp Neurol 159(2):409–418. doi:10.1006/exnr.1999.7188
Babb TL, Ying Z, Mikuni N, Nishiyama K, Drazba J, Bingaman W, Wyllie E, Wylie CJ et al (2000) Brain plasticity and cellular mechanisms of epileptogenesis in human and experimental cortical dysplasia. Epilepsia 41(Suppl 6):S76–81
Zhu LJ, Chen Z, Zhang LS, Xu SJ, Xu AJ, Luo JH (2004) Spatiotemporal changes of the N-methyl-D-aspartate receptor subunit levels in rats with pentylenetetrazole-induced seizures. Neurosci Lett 356(1):53–56
Suh JG, Ryoo ZW, Won MH, Oh YS, Kang TC (2001) Differential alteration of NMDA receptor subunits in the gerbil dentate gyrus and subiculum following seizure. Brain Res 904(1):104–111
Sakamoto T, Mishina M, Niki H (2002) Mutation of NMDA receptor subunit epsilon 1: effects on audiogenic-like seizures induced by electrical stimulation of the inferior colliculus in mice. Brain Res Mol Brain Res 102(1–2):113–117
Moussa RC, Ikeda-Douglas CJ, Thakur V, Milgram NW, Gurd JW (2001) Seizure activity results in increased tyrosine phosphorylation of the N-methyl-D-aspartate receptor in the hippocampus. Brain Res Mol Brain Res 95(1–2):36–47
Niimura M, Moussa R, Bissoon N, Ikeda-Douglas C, Milgram NW, Gurd JW (2005) Changes in phosphorylation of the NMDA receptor in the rat hippocampus induced by status epilepticus. J Neurochem 92(6):1377–1385. doi:10.1111/j.1471-4159.2005.02977.x
Bo T, Jiang Y, Cao H, Wang J, Wu X (2004) Long-term effects of seizures in neonatal rats on spatial learning ability and N-methyl-D-aspartate receptor expression in the brain. Brain Res Dev Brain Res 152(2):137–142. doi:10.1016/j.devbrainres.2004.06.011
Cornejo BJ, Mesches MH, Coultrap S, Browning MD, Benke TA (2007) A single episode of neonatal seizures permanently alters glutamatergic synapses. Ann Neurol 61(5):411–426. doi:10.1002/ana.21071
Swann JW, Le JT, Lee CL (2007) Recurrent seizures and the molecular maturation of hippocampal and neocortical glutamatergic synapses. Dev Neurosci 29(1–2):168–178. doi:10.1159/000096221
Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, Geider K, Laube B et al (2013) Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 45(9):1067–1072. doi:10.1038/ng.2728
Mathern GW, Pretorius JK, Mendoza D, Leite JP, Chimelli L, Born DE, Fried I, Assirati JA et al (1999) Hippocampal N-methyl-D-aspartate receptor subunit mRNA levels in temporal lobe epilepsy patients. Ann Neurol 46(3):343–358
Gashi E, Avallone J, Webster T, Friedman LK (2007) Altered excitability and distribution of NMDA receptor subunit proteins in cortical layers of rat pups following multiple perinatal seizures. Brain Res 1145:56–65. doi:10.1016/j.brainres.2007.01.110
Gibbs S, Chattopadhyaya B, Desgent S, Awad PN, Clerk-Lamalice O, Levesque M, Vianna RM, Rebillard RM et al (2011) Long-term consequences of a prolonged febrile seizure in a dual pathology model. Neurobiol Dis 43(2):312–321. doi:10.1016/j.nbd.2011.02.013
de Moura JC, Tirapelli DP, Neder L, Saggioro FP, Sakamoto AC, Velasco TR, Panepucci RA, Leite JP et al (2012) Amygdala gene expression of NMDA and GABA(A) receptors in patients with mesial temporal lobe epilepsy. Hippocampus 22(1):92–97. doi:10.1002/hipo.20863
Ganor Y, Goldberg-Stern H, Lerman-Sagie T, Teichberg VI, Levite M (2005) Autoimmune epilepsy: distinct subpopulations of epilepsy patients harbor serum autoantibodies to either glutamate/AMPA receptor GluR3, glutamate/NMDA receptor subunit NR2A or double-stranded DNA. Epilepsy Res 65(1–2):11–22. doi:10.1016/j.eplepsyres.2005.03.011
Levite M, Ganor Y (2008) Autoantibodies to glutamate receptors can damage the brain in epilepsy, systemic lupus erythematosus and encephalitis. Expert Rev Neurother 8(7):1141–1160. doi:10.1586/14737175.8.7.1141
Ahmadirad N, Shojaei A, Javan M, Pourgholami MH, Mirnajafi-Zadeh J (2014) Effect of minocycline on pentylenetetrazol-induced chemical kindled seizures in mice. Neurol Sci 35(4):571–576. doi:10.1007/s10072-013-1552-0
Dong C, Zhao W, Li W, Lv P, Dong X (2013) Anti-epileptic effects of neuropeptide Y gene transfection into the rat brain. Neural Regen Res 8(14):1307–1315. doi:10.3969/j.issn.1673-5374.2013.14.007
Berretta N, Ledonne A, Mango D, Bernardi G, Mercuri NB (2012) Hippocampus versus entorhinal cortex decoupling by an NR2 subunit-specific block of NMDA receptors in a rat in vitro model of temporal lobe epilepsy. Epilepsia 53(5):e80–84. doi:10.1111/j.1528-1167.2012.03420.x
Sultana R, Banks WA, Butterfield DA (2010) Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res 88(3):469–477. doi:10.1002/jnr.22227
Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ (2001) N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J Neurol Sci 182(2):151–159
Bi H, Sze CI (2002) N-methyl-D-aspartate receptor subunit NR2A and NR2B messenger RNA levels are altered in the hippocampus and entorhinal cortex in Alzheimer’s disease. J Neurol Sci 200(1–2):11–18
Hynd MR, Scott HL, Dodd PR (2004) Differential expression of N-methyl-D-aspartate receptor NR2 isoforms in Alzheimer’s disease. J Neurochem 90(4):913–919. doi:10.1111/j.1471-4159.2004.02548.x
Mishizen-Eberz AJ, Rissman RA, Carter TL, Ikonomovic MD, Wolfe BB, Armstrong DM (2004) Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout progression of Alzheimer’s disease pathology. Neurobiol Dis 15(1):80–92
Marcello E, Epis R, Saraceno C, Gardoni F, Borroni B, Cattabeni F, Padovani A, Di Luca M (2012) SAP97-mediated local trafficking is altered in Alzheimer disease patients’ hippocampus. Neurobiol Aging 33(2):422 e421–410. doi:10.1016/j.neurobiolaging.2010.09.015
Wu GM, Hou XY (2010) Oligomerized Abeta25-35 induces increased tyrosine phosphorylation of NMDA receptor subunit 2A in rat hippocampal CA1 subfield. Brain Res 1343:186–193. doi:10.1016/j.brainres.2010.04.055
Texido L, Martin-Satue M, Alberdi E, Solsona C, Matute C (2011) Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium 49(3):184–190. doi:10.1016/j.ceca.2011.02.001
Innocent N, Cousins SL, Stephenson FA (2012) NMDA receptor/amyloid precursor protein interactions: a comparison between wild-type and amyloid precursor protein mutations associated with familial Alzheimer’s disease. Neurosci Lett 515(2):131–136. doi:10.1016/j.neulet.2012.03.029
Tackenberg C, Grinschgl S, Trutzel A, Santuccione AC, Frey MC, Konietzko U, Grimm J, Brandt R et al (2013) NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis 4:e608. doi:10.1038/cddis.2013.129
Liu J, Chang L, Roselli F, Almeida OF, Gao X, Wang X, Yew DT, Wu Y (2010) Amyloid-beta induces caspase-dependent loss of PSD-95 and synaptophysin through NMDA receptors. J Alzheimers Dis 22(2):541–556. doi:10.3233/JAD-2010-100948
Huang HJ, Liang KC, Chang YY, Ke HC, Lin JY, Hsieh-Li HM (2010) The interaction between acute oligomer Abeta(1–40) and stress severely impaired spatial learning and memory. Neurobiol Learn Mem 93(1):8–18. doi:10.1016/j.nlm.2009.07.010
Allyson J, Dontigny E, Auberson Y, Cyr M, Massicotte G (2010) Blockade of NR2A-containing NMDA receptors induces Tau phosphorylation in rat hippocampal slices. Neural Plast 2010:340168. doi:10.1155/2010/340168
De Montigny A, Elhiri I, Allyson J, Cyr M, Massicotte G (2013) NMDA reduces Tau phosphorylation in rat hippocampal slices by targeting NR2A receptors, GSK3beta, and PKC activities. Neural Plast 2013:261593. doi:10.1155/2013/261593
DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B (2001) A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med 7(11):1189–1193. doi:10.1038/nm1101-1189
Husebye ES, Sthoeger ZM, Dayan M, Zinger H, Elbirt D, Levite M, Mozes E (2005) Autoantibodies to a NR2A peptide of the glutamate/NMDA receptor in sera of patients with systemic lupus erythematosus. Ann Rheum Dis 64(8):1210–1213. doi:10.1136/ard.2004.029280
Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren SI (2005) Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol 12(5):392–398. doi:10.1111/j.1468-1331.2004.00976.x
Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B (2006) Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci U S A 103(3):678–683. doi:10.1073/pnas.0510055103
Arinuma Y, Yanagida T, Hirohata S (2008) Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum 58(4):1130–1135. doi:10.1002/art.23399
Bloom O, Cheng KF, He M, Papatheodorou A, Volpe BT, Diamond B, Al-Abed Y (2011) Generation of a unique small molecule peptidomimetic that neutralizes lupus autoantibody activity. Proc Natl Acad Sci U S A 108(25):10255–10259. doi:10.1073/pnas.1103555108
Gono T, Kawaguchi Y, Kaneko H, Nishimura K, Hanaoka M, Kataoka S, Okamoto Y, Katsumata Y et al (2011) Anti-NR2A antibody as a predictor for neuropsychiatric systemic lupus erythematosus. Rheumatology (Oxford) 50(9):1578–1585. doi:10.1093/rheumatology/keq408
Harrison MJ, Ravdin LD, Lockshin MD (2006) Relationship between serum NR2a antibodies and cognitive dysfunction in systemic lupus erythematosus. Arthritis Rheum 54(8):2515–2522. doi:10.1002/art.22030
Wang L, Zhou D, Lee J, Niu H, Faust TW, Frattini S, Kowal C, Huerta PT et al (2012) Female mouse fetal loss mediated by maternal autoantibody. J Exp Med 209(6):1083–1089. doi:10.1084/jem.20111986
Faust TW, Chang EH, Kowal C, Berlin R, Gazaryan IG, Bertini E, Zhang J, Sanchez-Guerrero J et al (2010) Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc Natl Acad Sci U S A 107(43):18569–18574. doi:10.1073/pnas.1006980107
Gono T, Takarada T, Fukumori R, Kawaguchi Y, Kaneko H, Hanaoka M, Katsumata Y, Yoneda Y et al (2011) NR2-reactive antibody decreases cell viability through augmentation of Ca(2+) influx in systemic lupus erythematosus. Arthritis Rheum 63(12):3952–3959. doi:10.1002/art.30616
Boyce-Rustay JM, Holmes A (2006) Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology 31(11):2405–2414. doi:10.1038/sj.npp.1301039
Taniguchi S, Nakazawa T, Tanimura A, Kiyama Y, Tezuka T, Watabe AM, Katayama N, Yokoyama K et al (2009) Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J 28(23):3717–3729. doi:10.1038/emboj.2009.300
Han X, Shao W, Liu Z, Fan S, Yu J, Chen J, Qiao R, Zhou J et al (2015) iTRAQ-based quantitative analysis of hippocampal postsynaptic density-associated proteins in a rat chronic mild stress model of depression. Neuroscience 298:220–292. doi:10.1016/j.neuroscience.2015.04.006
Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH (2007) Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 32(9):1888–1902. doi:10.1038/sj.npp.1301312
Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B (2009) Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry 33(1):70–75. doi:10.1016/j.pnpbp.2008.10.005
Sun H, Guan L, Zhu Z, Li H (2013) Reduced levels of NR1 and NR2A with depression-like behavior in different brain regions in prenatally stressed juvenile offspring. PLoS One 8(11):e81775. doi:10.1371/journal.pone.0081775
Karolewicz B, Szebeni K, Gilmore T, Maciag D, Stockmeier CA, Ordway GA (2009) Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int J Neuropsychopharmacol 12(2):143–153. doi:10.1017/S1461145708008985
Kaut O, Schmitt I, Hofmann A, Hoffmann P, Schlaepfer TE, Wullner U, Hurlemann R (2015) Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur Arch Psychiatry Clin Neurosci 265(4):331–341. doi:10.1007/s00406-014-0572-y
Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T (2001) Hyperfunction of dopaminergic and serotonergic neuronal systems in mice lacking the NMDA receptor epsilon1 subunit. J Neurosci 21(2):750–757
Itokawa M, Yamada K, Yoshitsugu K, Toyota T, Suga T, Ohba H, Watanabe A, Hattori E et al (2003) A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 13(5):271–278. doi:10.1097/01.fpc.0000054082.64000.63
Pinacho R, Villalmanzo N, Roca M, Iniesta R, Monje A, Haro JM, Meana JJ, Ferrer I et al (2013) Analysis of Sp transcription factors in the postmortem brain of chronic schizophrenia: a pilot study of relationship to negative symptoms. J Psychiatr Res 47(7):926–934. doi:10.1016/j.jpsychires.2013.03.004
Beneyto M, Meador-Woodruff JH (2008) Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 33(9):2175–2186. doi:10.1038/sj.npp.1301604
Bitanihirwe BK, Lim MP, Kelley JF, Kaneko T, Woo TU (2009) Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 9:71. doi:10.1186/1471-244X-9-71
Tang TT, Yang F, Chen BS, Lu Y, Ji Y, Roche KW, Lu B (2009) Dysbindin regulates hippocampal LTP by controlling NMDA receptor surface expression. Proc Natl Acad Sci U S A 106(50):21395–21400. doi:10.1073/pnas.0910499106
Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH (2006) Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry 11(8):737–747. doi:10.1038/sj.mp.4001844, 705
Dracheva S, Byne W, Chin B, Haroutunian V (2008) Ionotropic glutamate receptor mRNA expression in the human thalamus: absence of change in schizophrenia. Brain Res 1214:23–34. doi:10.1016/j.brainres.2008.03.039
Schmitt A, Koschel J, Zink M, Bauer M, Sommer C, Frank J, Treutlein J, Schulze T et al (2010) Gene expression of NMDA receptor subunits in the cerebellum of elderly patients with schizophrenia. Eur Arch Psychiatry Clin Neurosci 260(2):101–111. doi:10.1007/s00406-009-0017-1
Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, Di Filippo M, Pendolino V et al (2012) Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain 135(Pt 6):1884–1899. doi:10.1093/brain/aws101
Gardoni F, Sgobio C, Pendolino V, Calabresi P, Di Luca M, Picconi B (2012) Targeting NR2A-containing NMDA receptors reduces L-DOPA-induced dyskinesias. Neurobiol Aging 33(9):2138–2144. doi:10.1016/j.neurobiolaging.2011.06.019
Arning L, Kraus PH, Valentin S, Saft C, Andrich J, Epplen JT (2005) NR2A and NR2B receptor gene variations modify age at onset in Huntington disease. Neurogenetics 6(1):25–28. doi:10.1007/s10048-004-0198-8
Luthi-Carter R, Apostol BL, Dunah AW, DeJohn MM, Farrell LA, Bates GP, Young AB, Standaert DG et al (2003) Complex alteration of NMDA receptors in transgenic Huntington’s disease mouse brain: analysis of mRNA and protein expression, plasma membrane association, interacting proteins, and phosphorylation. Neurobiol Dis 14(3):624–636
Ali NJ, Levine MS (2006) Changes in expression of N-methyl-D-aspartate receptor subunits occur early in the R6/2 mouse model of Huntington’s disease. Dev Neurosci 28(3):230–238. doi:10.1159/000091921
Jarabek BR, Yasuda RP, Wolfe BB (2004) Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington’s mouse model. Brain 127(Pt 3):505–516. doi:10.1093/brain/awh058
Barkus C, McHugh SB, Sprengel R, Seeburg PH, Rawlins JN, Bannerman DM (2010) Hippocampal NMDA receptors and anxiety: at the interface between cognition and emotion. Eur J Pharmacol 626(1):49–56. doi:10.1016/j.ejphar.2009.10.014
Dere E, Topic B, De Souza Silva MA, Fink H, Buddenberg T, Huston JP (2003) NMDA-receptor antagonism via dextromethorphan and ifenprodil modulates graded anxiety test performance of C57BL/6 mice. Behav Pharmacol 14(3):245–249. doi:10.1097/01.fbp.0000069580.37661.05
Sun H, Jia N, Guan L, Su Q, Wang D, Li H, Zhu Z (2013) Involvement of NR1, NR2A different expression in brain regions in anxiety-like behavior of prenatally stressed offspring. Behav Brain Res 257:1–7. doi:10.1016/j.bbr.2013.08.044
Woo TU, Walsh JP, Benes FM (2004) Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry 61(7):649–657. doi:10.1001/archpsyc.61.7.649
McCullumsmith RE, Kristiansen LV, Beneyto M, Scarr E, Dean B, Meador-Woodruff JH (2007) Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res 1127(1):108–118. doi:10.1016/j.brainres.2006.09.011
Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F et al (2010) Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther 335(3):636–644. doi:10.1124/jpet.110.172544
Acknowledgments
The authors acknowledge support from the Natural Science Foundation of China (NSFC 81200886, NSFC 81402886), the Natural Science Foundation of Hebei Province (H2014208004), the Science and Technology Project of Hebei Province (13397703D), the Key Basic Research Program of the Application Foundation Research Project of Hebei Province (14967719D, 15962704D), the State Key Laboratory Breeding Base—Hebei Key Laboratory of Molecular Chemistry for Drug, and Hebei Research Center of Pharmaceutical and Chemical Engineering.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sun, Y., Cheng, X., Zhang, L. et al. The Functional and Molecular Properties, Physiological Functions, and Pathophysiological Roles of GluN2A in the Central Nervous System. Mol Neurobiol 54, 1008–1021 (2017). https://doi.org/10.1007/s12035-016-9715-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9715-7