
Abstract
Rationale
Sigma-1 receptor (Sig-1R) agonists showed anti-amnesic properties in Alzheimer’s disease models and anti-inflammatory properties in cerebrum ischaemia models. The agonist of Sig-1R was reported to up-regulate brain-derived neurotrophic factor (BDNF) levels in the hippocampus of mice. Here, we investigate whether the activation of Sig-1R attenuates the learning and memory impairment induced by ischaemia/reperfusion and how it affects the expression of BDNF.
Objectives
Bilateral common carotid artery occlusion (BCCAO) was induced for 20 min in C57BL/6 mice.
Materials and methods
Sig-1R agonist, PRE084, sigma 1/2 non-selective agonist, DTG, Sig-1R antagonist and BD1047 were injected once daily throughout the experiment. Behavioural tests were performed from day 8. On day 22 after BCCAO, mice were sacrificed for biochemical analysis.
Results
PRE084 and DTG ameliorated learning and memory impairments in the Y maze, novel object recognition, and water maze tasks and prevented the decline of synaptic proteins and BDNF expression in the hippocampus of BCCAO mice. Furthermore, PRE084 and DTG up-regulated the level of NMDA receptor 2A (NR2A), calcium/calmodulin-dependent protein kinase type IV (CaMKIV) and CREB-specific co-activator transducer of regulated CREB activity 1 (TORC1). Additionally, the effects of PRE084 and DTG were antagonised by the co-administration of BD1047.
Conclusions
Sig-1R activation showed an attenuation in the ischaemia/reperfusion model and the activation of Sig-1R increased the expression of BDNF, possibly through the NR2A-CaMKIV-TORC1 pathway, and Sig-1R agonists might function as neuroprotectant agents in vascular dementia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sigma-1 receptor (Sig-1R) is recognised as a chaperone protein of the endoplasmic reticulum (ER) and is putatively located on membranes forming focal contacts between the ER and mitochondria (Hayashi and Su 2007). Sig-1R is highly expressed in the brain (Alonso et al. 2000; Langa et al. 2003). In the nervous system, Sig-1R regulates neurogenesis; K + channels; inositol 1,4,5-triphosphate receptor (IP3R)-mediated Ca2+ signalling; and drug addiction (Maurice and Lockhart 1997; Hayashi and Su 2001, 2004; Aydar et al. 2002; Matsumoto et al. 2003). Sig-1Rs are well known to exert anti-depressant-like effects (Yagasaki et al. 2006; Takebayashi et al. 2004). Agonists of Sig-1R have been shown to have anti-amnesic properties in amnesia models, including amyloid-induced AD models (Villard et al. 2009), cholinergic deficit models (Zou et al. 2000) and aging-related memory loss models (Maurice 2001; Phan et al. 2003). Recently, the effects of Sig-1R on brain ischaemia have gained attention from researchers. The endogenous ligand of Sig-1R, DHEA, exerted neuroprotective effects by reducing the ischaemia-induced neuronal death and improving spatial learning deficits 3 to 48 h after ischaemia (Li et al. 2009). However, the detailed role of Sig-1R in ischaemia-induced learning and memory impairments is still unclear.
Brain-derived neurotrophic factor (BDNF) plays key roles in the maintenance and repair of the nervous system (Lewin and Barde 1996) and has emerged as an important regulator of synaptic plasticity and learning and memory (Cunha et al. 2010; Korte et al. 1995). A recent report suggested that chronic anti-depressant treatment modulated the BDNF signalling pathway through the Sig-1R chaperone (Yagasaki et al. 2006). Kikuchi-Utsumi and Nakaki (2008) found that chronic administration of Sig-1R agonist SA4503 regulated the region-specific BDNF functions in the rat brains. Recently, Fujimoto et al. (2012) reported that SA4503 increased Sig-1R protein and concomitantly increased the expression of endogenous BDNF. Overexpression of Sig-1R potentiated the conversion of the precursor pro-BDNF to mature BDNF and enhanced the secretion of mature BDNF into extracellular spaces (Fujimoto et al. 2012). These studies suggest that Sig-1R is a potential modulator for the BDNF signal pathway. Therefore, in this study, we hypothesise that Sig-1R stimulation will modulate BDNF in the hippocampus, thereby enhancing synaptic plasticity and improving deficits of learning and memory in brain ischaemia/reperfusion.
Methods
Animals
Equal numbers of male and female C57BL/6 mice (11 weeks old, n = 96 in total) were used in the experiments. C57BL/6 mice were chosen because this strain is one of the most susceptible mouse strains to neuronal damage after cerebral ischaemia due to their poorly developed anastomosis between the carotid artery (posterior communicating artery) (Fujii et al. 1997; Yang et al. 1997). The animals were purchased from Beijing HFK Bioscience Co., Ltd. The animals were kept in a polycarbonate cage (15.0 cm × 30.0 cm × 10.0 cm) with six mice per cage and maintained under standard housing conditions (room temperature 25 ± 2 °C and humidity 50 ± 5 %) with a 12-h light and 12-h dark cycle. Food and water were available ad libitum. An acclimation period of at least 1 week was provided before initiating the experiment. All animal studies were performed in strict accordance with the P.R. China legislation on the use and the care of laboratory animals and with the guidelines established by the Institute for Experimental Animals at Shenyang Pharmaceutical University.
Surgical procedure
Bilateral common carotid artery occlusion (BCCAO) animal models were widely used to study the effects of brain ischaemia. BCCAO results in a systemic decrease of blood flow in the whole brain, which clinically resembles transient global cerebral ischaemia followed by a number of pathological changes, such as excitotoxicity, loss of ATP in the neurons and neurological deficits (Huang and McNamara 2004; Kim et al. 2007). In this study, we used the BCCAO model to investigate the effects of Sig-1R on extensive brain injury by ischaemia/reperfusion. The mice were anaesthetised with chloral hydrate (300 mg/kg i.p.). After a lateral neck incision, brain ischaemia was induced by BCCAO for 20 min as described (Wu et al. 2001; Zhao et al. 2005; Yamamoto et al. 2009). Body temperature was kept at 37 ± 0.5 °C using a heating pad during and for 1 h after the surgery. Sham-operated animals underwent all of the procedures, except that their arteries were not occluded. After surgery, the mice were housed individually and were allowed to recover for 7 days before the start of the behavioural assays.
Y maze task
The spontaneous alteration behaviour in a Y maze is assessed as a spatial working memory task (Yamamoto et al. 2009). The apparatus consists of three identical arms made of wood: Each arm was 40 cm long, 12 cm high, 5 cm wide at the bottom and 10 cm wide at the top. This behavioural test was performed 8 days after the BCCAO, according to a previous study (Lu et al. 2009), with minor modifications. Each mouse was placed at the end of one fixed arm and was allowed to move freely through the maze during a 5-min session. The total number of arm entries (N) and the sequence of entries were recorded. A successful alternation was defined as entries into all three arms on consecutive choices. The floor was cleaned with ethanol (10 %) after each individual trial to eliminate olfactory cues. The number of successive alternations was recorded. Alternation behaviour was calculated as number of alternations/(N–2) × 100 %.
Novel object recognition test
The novel object recognition test was performed on the 10th day after the BCCAO, as a non-aversive learning paradigm that relies on the spontaneous exploratory behaviour of animals. The experimental arena consisted of a square box (L 45 × W 45 × H 15 cm) with wood walls. Two days prior to the experiments, the mice received a habituation of 5 min in the empty open-field arena, twice per day (Damgaard et al. 2010; McClean et al. 2011). On the day of the experiment, each mouse received a 3-min habituation. Two identical objects, A1 and A2, were placed at the centre of the box in a symmetrical position approximately 15 cm away from the black wall. The animals were singly placed in the box and allowed to explore for 5 min (training session), and the total time spent exploring each of the two objects (when the animal’s snout was directly pointed toward the object at a distance ≤2 cm) was recorded. One hour after the training session, the mice were placed back into the arena and exposed to the familiar object A1 and a novel object B for an additional 5 min. After a 24-h retention interval, the mice were placed again in the same box with the novel object B replaced with another novel object C and were allowed to explore freely for 5 min. The placement of the novel and familiar objects alternated between the left and right to reduce potential bias for a particular location. Throughout the experiments, the objects were used in a counterbalanced manner in terms of their physical complexity and emotional neutrality. Any faeces and urine were removed with paper towels, and the floor was cleaned with 10 % ethanol after each individual trial to eliminate olfactory cues. The exploration time(s) for each object in the training session and the retention sessions were recorded. The preferential index (PI) was calculated as time spent exploring novel object/total exploration time. The discrimination index (DI) was calculated as [(time spent exploring novel object − time spent exploring familiar object)/total exploration time].
Morris water maze task
The maze was a circular pool (100 cm in diameter and 40 in height) filled with water at 25 ± 1 °C to avoid hypothermia. A small escape platform (10 cm in diameter) was placed at a fixed position in the centre of one quadrant and hidden 1 cm beneath the water surface. The room contained a number of fixed visual cues on the walls.
Acquisition phase
In the acquisition trial phase, mice were trained twice a day for five consecutive days (days 1–5) with an intertrial interval of 3 h. Four points equally spaced along the circumference of the pool (north, south, east and west) served as the starting position, which was randomised during the two trials each day. If an animal did not reach the platform within 60 s, it was guided to the platform where it had to remain for 10 s before being returned to its home cage. The mice were kept dry between trials in a plastic holding cage filled with paper towels. Escape latency (time to find the hidden platform), distance swum and swimming speed were recorded (n = 10–12 per group). All of the trials were completed between 09:00 and 18:00 (McClean et al. 2011).
Probe trial
One day after finishing the acquisition task (day 6), a probe trial was performed to assess the spatial memory (after a 24-h delay). The platform was removed from the maze, and each mouse was allowed to explore freely for 60 s. Spatial acuity was expressed as the amount of time spent in the exact area where the escape platform was located (McClean et al. 2011).
Immunohistochemical staining
Immunohistochemical staining was performed 22 days after the BCCAO as described previously (Andsberg et al. 2002). Briefly, sections were incubated with rabbit anti-BDNF antibody (1:100, Santa Cruz) and mouse anti-NeuN antibody (1:100, Millipore) at 4 °C overnight. After washings (three times) with PBS, the sections were incubated with biotin-labelled goat anti-rabbit/mouse antibody (1:200, Santa Cruz) at 37 °C for 30 min. The sections were treated with avidin-biotin enzyme reagent (Santa Cruz), and DAB was used to visualise the positive signal. The intensity of each section was quantified using Image Pro-Plus 6.0 software by Olympus IX 71.
Western blotting
The mice were sacrificed 22 days after the BCCAO, and the hippocampi were dissected for Western blotting analysis. The protein (40 μg) was run on a 12 % gradient SDS–PAGE gel and electrophoretically transferred to polyvinyl difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5 % skim milk for 2 h at room temperature, the membranes were incubated with primary antibodies at 4 °C overnight. The membranes were incubated with horseradish peroxidase-labelled anti-mouse IgG or anti-rabbit IgG antibodies (Santa Cruz) for 2 h at room temperature. Protein bands were visualised with an ECL Western blotting kit (Kangwei Biotechnology, China). The intensity was quantified by densitometry using Quantity One 4.6.2 software (Bio-Rad, Hercules, CA, USA) and corrected with the corresponding β-actin level. The results were expressed as a percentage of the control. The following antibodies were used: anti-synaptophysin (SYP, 1:1000; sc-17750, Santa Cruz), anti-GAP-43 (1:400; sc-17790, Santa Cruz), anti-PSD95 (1:1000; ab18258, Abcam), anti-Nogo-A (1:400; sc-25660, Santa Cruz), anti-CaMKIV (1:400; sc-55501, Santa Cruz), anti-TORC1 (1:300; sc-67146, Santa Cruz), anti-NeuN (1:300; MAB377, Milipore), anti-TrkB (1:800; BS6683, Bioworld Technology), anti-TrkB (phospho-Y706, 1:200; BS4201, Bioworld Technology), anti-ERK1/2 (1:500; sc-292838, Santa Cruz), anti-p-ERK1/2 (Thr 202, 1:400; sc-101760, Santa Cruz), anti-CREB-1 (1:400; sc-25785, Santa Cruz), anti-p-CREB-1 (Ser 133, 1:400; sc-101663, Santa Cruz) and β-actin (1:400; sc-47778, Santa Cruz), anti-BDNF (1:500; sc-546, Santa Cruz), anti-NR2A (1:300; Cell Signaling) and anti-VEGF (1:400; sc-7269, Santa Cruz), to detect protein levels in the hippocampus of mice.
Statistical analysis
The data were expressed as the mean ± SEM. Statistical analysis was conducted using SPSS 16.0. Differences between more than two groups were determined by one-way ANOVA followed by post hoc tests. The analysis of acquisition phase in the water maze test was determined by two-way ANOVA. p < 0.05 was considered significant.
Results
The effects of Sig-1R activation on the impairment of spontaneous alteration behaviour induced by brain ischaemia/reperfusion in the Y maze test
The effect of Sig-1R on the impairment of spontaneous alternation behaviour was evaluated on the 8th day after the BCCAO using the Y maze test. No significant differences in the total number of arm entries were found among the groups (F7, 88 = 1.426, p = 0.205; Fig. 1a). As shown in Fig. 1b, the BCCAO group exhibited a significantly reduced spontaneous alternation behaviour compared with the sham group (F7, 88 = 9.820, p < 0.001; post hoc, p < 0.01). PRE084 (3 and 1 mg/kg) dose-dependently attenuated the BCCAO-induced impairment of spontaneous alternation behaviour (F7, 88 = 9.820, p < 0.001; PRE084 at 3 mg/kg, p < 0.05; PRE084 at 1 mg/kg, p < 0.01; Fig. 1b). The post hoc analysis showed that after co-administration with the antagonist BD1047 (1 mg/kg), the effect of PRE084 (1 mg/kg) was completely blocked (p < 0.05; Fig. 1b). DTG also improved the impairment of spontaneous alternation behaviour (p < 0.01), whereas BD1047 (1 mg/kg) completely antagonised this improvement (p < 0.01; Fig. 1b).

Effects of Sig-1R activation on the impairment of spontaneous alteration behaviour induced by brain ischaemia/reperfusion in Y maze test. a Total number of arm entries; b spontaneous alternation behaviour (%). All results are presented as mean ± SEM. n = 12; ## p < 0.01 vs sham; *p < 0.05, **p < 0.01 vs BCCAO; &p < 0.05 vs PRE084 (1 mg/kg), $$ p < 0.01 vs DTG (1 mg/kg)
The effects of Sig-1R activation on visual recognition impairment induced by brain ischaemia/reperfusion in the novel object recognition test
We evaluated the effect of Sig-1R activation on the impairment of visual recognition through a novel object recognition test. The level of exploratory preferences for the novel object B (both the PI and DI were calculated, DI not shown) in the BCCAO mice group was significantly decreased compared with that in the sham mice group (F7, 87 = 9.398, p < 0.001; post hoc, p < 0.01; Fig. 2a). A strong increase in the PI was observed in PRE084 (3 and 1 mg/kg)-treated mice in the 1-h test (PRE084 at 3 mg/kg, p < 0.01; PRE084 at 1 mg/kg, p < 0.05; Fig. 2a). However, restored recognition memory was not observed after co-administration with the antagonist BD1047 (p < 0.01; Fig. 2a). BD1047 also reversed the improvement of the recognition memory of the DTG (1 mg/kg) group after BCCAO (p < 0.01; Fig. 2a). Similar results were observed in the BCCAO mice when the test was performed 24 h after training (F7, 87 = 11.932, p < 0.001; post hoc, p < 0.001; Fig. 2c). The data showed that PRE084 (3 and 1 mg/kg) significantly increased the PI (p < 0.001 for both dose; Fig. 2c) in a dose-dependent manner. Compared with PRE084 (1 mg/kg), co-administration with BD1047 decreased the PI (p < 0.001; Fig 2c). The DTG group increased the PI after the BCCAO (p < 0.01; Fig. 2c), but co-administration with BD1047 showed a significant inversion after DTG (1 mg/kg) treatment (p < 0.05; Fig. 2c). No significant differences in the total exploring time after 1 or 24 h were found among the groups (1 h: F7, 87 = 0.555, p = 0.791; 24 h: F7, 87 = 1.152, p = 0.339; Fig. 2b, d). These results indicate that Sig-1R activation could restore the recognition memory deficit induced by brain ischaemia/reperfusion.

Effects of Sig-1R activation on visual recognition impairment induced by brain ischaemia/reperfusion in the the novel object recognition test. a Preferential index of 1 h; b total exploring time of 1 h; c preferential index of 24 h; d total exploring time of 24 h; all results are presented as mean ± SEM. n = 11–12; ## p < 0.01, ### p < 0.001 vs sham; *p < 0.05, **p < 0.01, ***p < 0.001 vs BCCAO; &&p < 0.01, &&&p < 0.001 vs PRE084 (1 mg/kg), $ p < 0.05, $$ p < 0.01 vs DTG (1 mg/kg)
The effects of Sig-1R activation on learning and memory deficits induced by the BCCAO in the Morris water maze test
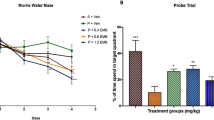
To determine whether Sig-1R activation improves spatial memory deficits induced by brain ischaemia/reperfusion in mice, we assessed their performance using the Morris water maze test. During the acquisition trial phase, there was a significant increase in escape latency in the BCCAO mice (two-way ANONA, group, F7,429 = 10.136, p < 0.001; Fig. 3a). Mice in the PRE084 (3 and 1 mg/kg) groups required significantly less time to find the platform than the BCCAO group (p < 0.01 for 3 mg/kg, p < 0.05 for 1 mg/kg; Fig. 3a). In the probe test, as no significant differences in swimming speeds were shown among the groups (F7,85 = 0.760, p = 0.622; Fig. 3b), the exploration time and the exploration distance spent in the target quadrant were significantly reduced in the BCCAO mice compared with the sham group (exploration time F7,85 = 2.228, p < 0.05, post hoc, p < 0.05; exploration distance F7,85 = 2.230, p < 0.05, post hoc, p < 0.05; Fig. 3c, d ). Mice in the PRE084 (3 and 1 mg/kg) groups spent more time (p < 0.01 for 3 mg/kg, p < 0.05 for 1 mg/kg; Fig. 3c) and swam longer distances (p < 0.01 for 3 mg/kg, p < 0.05 for 1 mg/kg; Fig. 3d) in the target quadrant than the BCCAO group. DTG (1 mg/kg) also increased the exploration time (p < 0.05; Fig. 3c) as well as the exploration distance (p < 0.05; Fig. 3c) in the target quadrant. Taken together, these data suggest that Sig-1R activation could ameliorate learning and memory deficits in brain ischaemia/reperfusion in mice.

Effects of Sig-1R activation on learning and memory induced by brain ischaemia/reperfusion in the Morris water maze test. a The escape latency in training period; b swimming speed in probe test; c exploration time in probe test; d exploration distance in probe test; all results are presented as mean ± SEM. n = 11–12; # p < 0.05 vs sham; *p < 0.05, **p < 0.01 vs BCCAO
The effects of Sig-1R activation on neuronal cell damage in the hippocampus induced by BCCAO
In BCCAO mice, NeuN-positive cells decreased compared with the sham group (F7,32 = 2.827, p < 0.05, post hoc, p < 0.01; Fig. 4a, b, i). Treatment with PRE084 (3 and 1 mg/kg) significantly reduced neuronal cell loss (p < 0.05 both; Fig. 4d, e, i) as did the DTG (p < 0.05; Fig. 4g, i). Co-treatment with BD1047 (1 mg/kg) reversed the effect of PRE084 (p < 0.05; Fig. 4h, i). Furthermore, the degenerating neurons were further quantified by Western blotting. Compared with the sham group, the NeuN protein levels in BCCAO mice showed a dramatic decrease (F7,16 = 5.895, p < 0.01, post hoc, p < 0.01; Fig. 4j). In the PRE084 (3 and 1 mg/kg)-treated group, the NeuN level was significantly increased (p < 0.01 both; Fig. 4j). DTG also increased the NeuN level (p < 0.05; Fig. 4j). However, this increase was totally blocked by BD1047 (1 mg/kg) (p < 0.05 both; Fig. 4j).

Effects of Sig-1R activation on neuronal cell damage in the hippocampus induced by BCCAO. NeuN-positive cell in CA1 region of hippocampus: a sham, b BCCAO, c PRE084 (0.3 mg/kg), d PRE084 (1 mg/kg), e PRE084 (3 mg/kg), f PRE084 (1 mg/kg) + BD1047 (1 mg/kg), g DTG (1 mg/kg), h DTG (1 mg/kg) + BD1047 (1 mg/kg); i graphic representation of NeuN-positive cell number, n = 5; j Western blot analysis of NeuN in hippocampus, n = 3. All results are presented as mean ± SEM. ## p < 0.01 vs sham; **p < 0.01 vs BCCAO; &p < 0.05 vs PRE084 (1 mg/kg), $ p < 0.05 vs DTG (1 mg/kg)
The effects of Sig-1R activation on the expression of SYP, GAP-43, PSD95 and NogoA in the hippocampus
To investigate the effect of Sig-1R on synaptic plasticity in BCCAO mice, the expressions of synapse-associated proteins, synaptophysin (SYP), growth-associated protein-43 (GAP-43), postsynaptic density 95 (PSD95) and NogoA were detected. Western blot analysis showed that PRE084 (3 and 1 mg/kg) strongly increased the levels of SYP, GAP-43 and PSD95 in the hippocampus compared with BCCAO mice (SYP: F7,16 = 3.536, p < 0.05, post hoc, p < 0.05 for both doses; GAP-43: F7,16 = 7.542, p < 0.001, post hoc, p < 0.05 for both doses; PSD95: F7,16 = 4.751, p < 0.01, post hoc, p < 0.01 for both doses; Fig. 5a–c), and similar results were observed in the DTG group (1 mg/kg) (SYP: F7,16 = 3.528, p < 0.05, post hoc, p < 0.01; GAP-43: F7,16 = 7.542, p < 0.001, post hoc, p < 0.05; PSD95: F7,16 = 4.751, p < 0.01, post hoc, p < 0.01; Fig. 5a–c). However, the restoration of the SYP, GAP-43 and PSD95 expressions was abolished by co-administration with BD1047 (1 mg/kg) (SYP: F7,16 = 3.536, p < 0.05, post hoc, p < 0.05 both; GAP-43 F7,16 = 7.542, p < 0.001, post hoc, p < 0.01 both; PSD95 F7,16 = 4.751, p < 0.01, post hoc, p < 0.01 and p < 0.05, respectively; Fig. 5a–c). The level of Nogo-A as an inhibitor of axonal growth showed no differences among these groups (F7,24 = 1.362, p = 0.266; Fig. 5d).

Effects of Sig-1R activation on the expression of SYP, GAP-43, PSD95, NogoA in the hippocampus. Changes of the expression of SYP (a), GAP-43 (b), PSD95 (c) and NogoA (d) among the groups, shown as percentage of control. All results are presented as mean ± SEM. n = 3; # p < 0.05, ## p < 0.01 vs sham; *p < 0.05, **p < 0.01 vs BCCAO; &&p < 0.01 vs PRE084 (1 mg/kg), $ p < 0.05, $$ p < 0.01 vs DTG (1 mg/kg)
Sig-1R activation up-regulates the expression of BDNF in the hippocampus
BDNF immunoreactivity in the hippocampus CA1 region was significantly decreased in the BCCAO group compared with that in the sham group 22 days after the BCCAO (F7,32 = 15.572, p < 0.001, post hoc, p < 0.001; Fig. 6a, b, i), whereas BDNF immunoreactivity in the PRE084 (3 and 1 mg/kg)- and DTG (1 mg/kg)-treated groups was higher than that in the BCCAO group (p < 0.001; Fig. 6d, f, g, i). After co-administration with BD1047, the restored immunoreactivity was decreased (p < 0.001; Fig. 6f, h, i). Western blotting analysis showed consistent results with immunohistochemical staining. Compared with the sham group, the protein levels of BDNF in the hippocampus decreased in BCCAO mice (F7,16 = 13.803, p < 0.001, post hoc, p < 0.01; Fig. 6j). Treatments with PRE084 (3 and 1 mg/kg) or DTG (1 mg/kg) increased the hippocampal BDNF levels significantly (PRE084: p < 0.01 for both doses; DTG: p < 0.001; Fig. 6j). The improvement of the agonists (PRE084 and DTG) on BDNF expression was totally blocked by the antagonist BD1047 (p < 0.05 and p < 0.01, respectively; Fig. 6j). The quantification of the BDNF protein levels and other behavioural data were assessed by the Pearson correlation coefficients, as presented in Table 1. The correlations are significant, suggesting a close relationship between BDNF protein levels and learning and memory behaviours.

Sig-1R activation up-regulates the expression of BDNF in the hippocampus. BDNF immunoreactivity in CA1 region of hippocampus: a sham, b BCCAO, c PRE084 (0.3 mg/kg), d PRE084 (1 mg/kg), e PRE084 (3 mg/kg), f PRE084 (1 mg/kg) + BD1047 (1 mg/kg), g DTG (1 mg/kg), h DTG (1 mg/kg) + BD1047 (1 mg/kg); i graphic representation of BDNF immunoreactivity, n = 5; j Western blot analysis of BDNF in hippocampus, n = 3. All results are presented as mean ± SEM. ## p < 0.01,### p < 0.001 vs sham; **p < 0.01, ***p < 0.001 vs BCCAO; &p < 0.05, &&&p < 0.001 vs PRE084 (1 mg/kg), $$ p < 0.01, $$$ p < 0.001vs DTG (1 mg/kg)
Sig-1R activation increases the phosphorylation of TrkB, ERK and CREB in the hippocampus
To further examine the effect of Sig-1R activation on BDNF signalling, we detected the phosphorylation of TrkB and the downstream event, the phosphorylation of ERK, with Western blotting. The ratio of phosphorylated TrkB and TrkB in BCCAO mice showed a dramatic decrease (F7,16 = 12.26, p < 0.001, post hoc, p < 0.001; Fig. 7a) compared with the sham group. In the PRE084 (3 and 1 mg/kg)-treated group, this ratio was significantly increased (p < 0.01 both; Fig. 7a). DTG also increased the phosphorylated TrkB levels (p < 0.001; Fig. 7a). However, the increase was reversed by BD1047 (1 mg/kg) (p < 0.05 both; Fig. 7a). Compared to BCCAO mice, PRE084- (3 and 1 mg/kg) and DTG (1 mg/kg)-treated mice had improved ratios of phosphorylated ERK (p-ERK) and ERK (PRE084: F7,16 = 3.831, p < 0.05, post hoc, p < 0.01 for 3 mg/kg, p < 0.05 for 1 mg/kg; DTG: p < 0.05; Fig. 7b). However, co-administration of BD1047 (1 mg/kg) blocked the improvement of PRE084 (p < 0.05; Fig. 7b). Furthermore, the level of phosphorylated CREB (p-CREB), the main regulator of BDNF, was significantly decreased in the BCCAO mice (F7,16 = 8.563, p < 0.05, post hoc, p < 0.01; Fig. 7c). PRE084 (3 and 1 mg/kg) and DTG increased the depressed ratio of p-CREB/CREB in the BCCAO mice (PRE: p < 0.001 for both doses; DTG: p < 0.001; Fig. 7c). The increase in the ratio of p-CREB/CREB by PRE084 and DTG treatments was blocked by co-administration of BD1047 (p < 0.01; Fig. 7c).

Sig-1R activation increases phosphorylated of TrkB, ERK and CREB in the hippocampus. Changes of the ratio of p-TrkB/TrkB (a), p-ERK/ERK (b) and p-CREB-1/CREB-1 (c), among the groups, shown as percentage of control. All results are presented as mean ± SEM. n = 3; ## p < 0.01, ### p < 0.001 vs sham; *p < 0.05, **p < 0.01, ***p < 0.001 vs BCCAO; &p < 0.05, &&p < 0.01,&&&p < 0.001 vs PRE084 (1 mg/kg), $$ p < 0.01, $$$ p < 0.001 vs DTG (1 mg/kg)
Sig-1R targets in the NR2A-CaMKIV-TORC1 pathway
Sig-1R regulates the ratio of p-CREB and CREB to increase the transcription of BDNF and the dephosphorylation of CREB-specific co-activator transducers of regulated CREB activity 1 (TORC1), resulting in the activation of CREB and its downstream gene targets. Thus, we investigated the level of TORC1 and its upstream regulators, including NR2A and CaMKIV, with Western blotting (Fig. 8a). The level of TORC1 was reduced in BCCAO mice compared with the sham group (F7, 16 = 6.131, p < 0.01, post hoc, p < 0.01; Fig. 8b). PRE084 (3 and 1 mg/kg) increased the reduction in mice (p < 0.01 and p < 0.05, respectively; Fig. 8b). DTG (p < 0.01; Fig. 8b) also increased the reduction in mice, while the encasement was reversed by the co-administration of BD1047 in both groups (p < 0.05 and p < 0.01, respectively; Fig. 8b). The levels of NR2A and CaMKIV were similar to those of TORC1 (Fig. 8a–c). These results suggest that Sig-1R is involved in the NR2A-CaMKIV-TORC1 signalling pathway.

Sig-1R activation involved in NR2A-CaMKIV-TORC1 pathway though Western blotting. a Western blotting bands of NR2A, CaMKIV and TORC1 in hippocampus and the analysis of NR2A, b statistic analysis of CaMKIV, c statistic analysis of TORC1. All results are presented as mean ± SEM, n = 3; ## p < 0.01 vs sham; *p < 0.05, **p < 0.01, ***p < 0.001 vs BCCAO; &p < 0.05, &&p < 0.01 vs PRE084 (1 mg/kg), $ p < 0.05, $$ p < 0.01 vs DTG (1 mg/kg)
The relationship between Sig-1R activation and VEGF
As VEGF is important for ischaemia restoration, we detected the expression of VEGF in the hippocampus and found no significant differences among the groups (F7,16 = 0.223, p = 0.974; Figs. 9 and 10).

Western blot analysis of the expression of VEGF in hippocampus. All results are presented as mean ± SEM. n = 3

The proposed effects of Sig-1R activation in brain ischaemia injury
Discussion
Abundant evidence has suggested that ischaemia/reperfusion injury in BCCAO causes delayed neuronal death in the vulnerable CA1 region of the hippocampus and extensive neuronal excitotoxicity (Penton-Rol et al. 2011). Our results show that Sig-1R agonists recused the degenerating neurons in BCCAO mice, while the antagonist BD1047 reversed the effect of the agonists, which suggests that Sig-1R was involved in the rescue of neuronal loss in the hippocampus as a means of neuroprotection. Ischaemia/reperfusion injuries also result in the reduction of synaptic proteins and the impairment of spatial memory in mice (Yamamoto et al. 2009; Zhao et al. 2005). In this study, we found that ischaemia/reperfusion injuries resulted in spatial memory impairments in the Y maze and Morris water maze, consistent with other reports (Watanabe and Utsuki 2011; Zhang et al. 2010). In this study, the Sig-1R agonist, PRE084, and the non-selected sigma 1\2 receptor agonist, DTG, dramatically improved the learning and memory deficits in BCCAO mice. We further found that the effects of PRE084 and DTG on the spatial memory impairments were blocked by the Sig-1R-selected antagonist BD1047. Previous studies reported that DHEA, an endogenous ligand of Sig-1R, improved spatial memory impairment from transient cerebral ischaemia in rats (Li et al. 2009). As reported, when administered during a 3- to 48-h time period after ischaemia, the sigma agonist DHEA exerts neuroprotection (Li et al. 2009), which is consistent with our 24-h therapeutic time period. Additionally, neuronal damage in the CA1 region was not seen until 12 h after ischaemia and then developed over 48 h of recirculation. Therefore, treatment with Sig-1R agonists within 24 h after BCCAO still had a neuroprotective effect in this study (Olsson et al. 2003). These results suggest that the activation of Sig-1R can ameliorate spatial memory impairments resulting from brain ischaemia.
Synaptic plasticity has traditionally been associated with higher brain functioning, including learning and memory processing. In particular, various forms of neuronal plasticity within the hippocampal formation are thought to underlie spatial learning and memory development (Bannerman et al. 2014). A loss of synaptic proteins was found in mice with brain ischaemia (Yamamoto et al. 2009; Zhao et al. 2005). In this study, we found that the Sig-1R agonists PRE084 and DTG attenuated the decrease of synaptic protein (SYP, GAP-43 and PSD95) levels in BCCAO mice, whereas the attenuation was abolished by the Sig-1R antagonist BD1047, suggesting that Sig-1R activation might rescue the synapse loss resulting from ischaemia/reperfusion.
BDNF has emerged as an important regulator of synaptic plasticity and learning and memory (Korte et al. 1995; Cunha et al. 2010). The binding of BDNF to TrkB results in the phosphorylation of tyrosine residues in the cytoplasmic domain of the receptor, triggering the activation of three major signalling cascades, including mitogen-activated protein kinase kinases (MEKs)-extracellular signal-regulated kinases (ERKs), phospholipase Cγ1 (PLCγ1)-inositol-1,4,5-triphosphate (IP3)/diacylglycerol (DAG) and phosphoinositide 3-kinase (PI3K)-Akt pathways (Nagahara and Tuszynski 2011). BDNF directly participated in synaptogenesis by increasing the number and size of dendritic spines in the hippocampal neurons (El-Husseini et al. 2000; Tyler and Pozzo-Miller 2003). Similarly, studies have demonstrated that the expression of synaptic proteins SYP, GAP-43 and PSD95 is dependent on the binding of BDNF to TrkB receptors and the subsequent activation of the ERK and phosphatidylinositol-3 kinase (PI3K) pathways (Li and Keifer 2012; Robinet and Pellerin 2011). In this study, immunocytochemistry and Western blot analysis revealed that Sig-1R activation attenuated the decrease in BDNF levels, and TrkB and ERK activation in the hippocampus of BCCAO mice. These results suggest that increased BDNF by Sig-1R agonists may increase the phosphorylation of TrkB and ERK, thus improving synaptic plasticity and learning and memory. BDNF could regulate affective behaviours through diverse mechanisms, including the modulation of neurogenesis, synaptic functioning and structural plasticity in the hippocampus (Pattwell et al. 2012; Bath et al. 2012). We further found significant correlations between the quantitation of BDNF protein levels and performance in the Y maze as well as the index of preference in new object organisation. These results suggest that Sig-1R activation might rescue the loss of neurons and synaptic proteins resulting from brain ischaemia/reperfusion and thus improving the learning and memory deficit through regulating BDNF signalling. Other factors, such as vascular endothelial growth factor (VEGF), might also regulate the level of p-ERK (Kilic et al. 2006). We investigated whether VEGF contributed to the decrease of p-ERK in the model group and the increase of p-ERK in the Sig-1R agonists treated groups. However, there were no changes in the VEGF level, suggesting that the decrease of hippocampal p-ERK in the BCCAO mice might not be related to VEGF.
Sig-1R activation increased the hippocampal BDNF levels, possibly contributing to the restoration of synaptic loss and spatial memory deficits resulting from brain ischaemia. How Sig-1R controlled the expression of BDNF is unclear. CREB is activated by phosphorylation and plays a central role in BDNF transcription (Tao et al. 1998). Stroke-surviving neurons have sustained concentrations of phospho-CREB and elevated concentrations of BDNF (Kokaia et al. 1995; Walton and Dragunow 2000). In this study, we found that ischaemia/reperfusion resulted in a decrease in the levels of phospho-CREB, accompanied by a decrease of BNDF expression. This can be explained by the entry of calcium through extrasynaptic NMDA receptors, triggered by bath glutamate exposure or hypoxic/ischaemic conditions, activated a general and dominant CREB shut-off pathway that blocked the induction of BDNF expression (Hardingham et al. 2002; Hardingham and Bading 2010). In contrast, synaptic NMDA receptors have an opposite effect on extrasynaptic NMDA receptors, CREB functioning and gene regulation (Hardingham et al. 2002; Peng et al. 2006; Liu et al. 2007). Calcium entry through synaptic NMDA receptors induced CREB activity and BDNF gene expression (Hardingham et al. 2002). In hippocampal neurons, extrasynaptic NMDA receptors are composed predominantly of NMDA receptor 1 (NR1) and NMDA receptor 2B (NR2B) subunits, whereas synaptic NMDA receptors also contain NMDA receptor 2A (NR2A) subunits (Hardingham and Bading 2010). Intracellular calciumin neurons via NR2A-containing NMDA receptors enhance the activity of CaMKIV. Activated CaMKIV phosphorylates SIK2, which dephosphorylates TORC1, leading to its translocation into the nucleus. Then, TORC1 induces CREB target genes, such as BDNF (Hardingham et al. 2002; Hardingham and Bading 2010). Sig-1R serves as an inter-organelle signalling modulator locally at the mitochondrion-associated ER membrane (MAM) (Hayashi and Su 2007) and remotely at the plasmalemma/plasma membrane to regulate functional proteins (Su et al. 2010). In our study, Sig-1R activation attenuated a decrease in the level of hippocampal NR2A receptors, suggesting that Sig-1R activation may restore or increase synaptic NMDA receptors in mice with brain ischaemia. We also found that Sig-1R activation increases the levels of CAMKIV, TORC1 and phospho-CREB with an increase of NR2A expression in mice with brain ischaemia. The Pearson correlation is significant between BNDF and NR2A. Taken together, these data suggest that the NR2A-CaMKIV-TORC1-CREB pathway is possibly involved in the regulation of Sig-1R on BDNF expression.
In summary, we identified that Sig-1R activation attenuated the learning and memory impairment caused by brain ischaemia/reperfusion and up-regulated the expression of synaptic proteins. These results suggest that the NR2A-TORC1-CREB pathway may contribute to the increase of BDNF levels through Sig-1R activation. However, the causal relationship between BDNF induction and the increase in NR2A-CaMKIV-TORC1 protein levels needs to be evaluated. In future studies, we will block the NR2A receptor or silence the NR2A gene to validate the causal relationship. These findings might provide a new target for the development of novel therapeutics in dementia induced by ischaemia.
References
Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, Maurice T (2000) Immunocytochemical localization of the sigma (1) receptor in the adult rat central nervous system. Neuroscience 97:155–170
Andsberg G, Kokaia Z, Klein RL, Muzyczka N, Lindvall O, Mandel RJ (2002) Neuropathological and behavioral consequences of adeno-associated viral vector-mediated continuous intrastriatal neurotrophin delivery in a focal ischemia model in rats. Neurobiol Dis 9:187–204
Aydar E, Palmer CP, Klyachko VA, Jackson MB (2002) The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34:399–410
Bannerman DM, Sprengel R, Sanderson DJ, McHugh SB, Rawlins JN, Monyer H, Seeburg PH (2014) Hippocampal synaptic plasticity, spatial memory and anxiety. Nat Rev Neurosci 15:181–192
Bath KG, Jing DQ, Dincheva I, Neeb CC, Pattwell SS, Chao MV, Lee FS, Ninan I (2012) BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology 37:1297–1304
Cunha C, Brambilla R, Thomas KL (2010) Simple role for BDNF in learning and memory? Front Mol Neurosci 3:1–14
Damgaard T, Larsen DB, Hansen SL, Grayson B, Neill JC, Plath N (2010) Positive modulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors reverses sub-chronic PCP-induced deficits in the novel object recognition task in rats. Behav Brain Res 207:144–150
El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290:1364–1368
Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA (1997) Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke 28:1805–1810
Fujimoto M, Hayashi T, Urfer R, Mita S, Su TP (2012) Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synaps 66:630–639
Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signaling: implications for neurodegenerative disorders. Nat Neurosci 11:682–696
Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414
Hayashi T, Su TP (2001) Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc Natl Acad Sci U S A 98:491–496
Hayashi T, Su TP (2004) Sigma-1 receptor ligands: potential in the treatment of neuropsychiatric disorders. CNS Drugs 18:269–284
Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131:596–610
Huang Y, McNamara JO (2004) Ischemic stroke: “acidotoxicity” is a perpetrator. Cell 118:665–666
Kikuchi-Utsumi K, Nakaki T (2008) Chronic treatment with a selective ligand for the sigma-1 receptor chaperone, SA4503, up-regulates BDNF protein levels in the rat hippocampus. Neurosci Lett 440:19–22
Kilic E, Kilic U, Wang Y, Bassetti CL, Marti HH, Hermann DM (2006) The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J 22:307–314
Kim DH, Li H, Yoo KY, Lee BH, Hwang IK, Won MH (2007) Effects of fluoxetine on ischemic cells and expressions in BDNF and some antioxidants in the gerbil hippocampal CA1 region induced by transient ischemia. Exp Neurol 204:748–758
Kokaia Z, Zhao Q, Kokaia M, Elmer E, Metsis M, Smith ML, Siesjö BK, Lindvall O (1995) Regulation of brain-derived neurotrophic factor geneexpression after transient middle cerebral artery occlusion with and without brain damage. Exp Neurol 136:73–88
Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 92:8856–8860
Langa F, Codony X, Tovar V et al (2003) Generation and phenotypic analysis of sigma receptor type I (σ1) knockout mice. Eur J Neurosci 18:2188–2196
Lewin GR, Barde YA (1996) Physiology of the neurotrophins. Annu Rev Neurosci 19:289–317
Li W, Keifer J (2012) Rapid enrichment of presynaptic protein in boutons undergoing classical conditioning is mediated by brain-derived neurotrophic factor. Neuroscience 203:50–58
Li Z, Cui S, Zhang Z, Zhou R, Ge Y, Sokabe M, Chen L (2009) DHEA-neuroprotection and -neurotoxicity after transient cerebral ischemia in rats. J Cerebr Blood F Met 29:287–296
Liu Y, Wang TP, Aarts M et al (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 27:2846–2857
Lu P, Mamiya T, Lu LL et al (2009) Silibinin prevents amyloid b peptide-induced memory impairment and oxidative stress in mice. Br J Pharmacol 157:1270–1277
Matsumoto RR, Liu Y, Lerner M, Howard EW, Brackett DJ (2003) σ receptors: Potential medications development target for anti-cocaine agents. Eur J Pharmacol 469:1–12
Maurice T (2001) Beneficial effect of the sigma1 receptor agonist PRE-084 against the spatial learning deficits in aged rats. Eur J Pharmacol 431:223–227
Maurice T, Lockhart BP (1997) Neuroprotective and anti-amnesic potentials of σ (sigma) receptor ligands. Prog Neuropsychopharmacol Biol Psychiatry 21:69–102
McClean PL, Parthsarathy V, Faivre E, Hölscher C (2011) The Diabetes Drug Liraglutide Prevents Degenerative Processes in a Mouse Model of Alzheimer's Disease. J Neurosci 31:6587–6594
Nagahara AH, Tuszynski MH (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 10:209–219
Olsson T, Wieloch T, Smith M-J (2003) Brain damage in a mouse model of global cerebral ischemia Effect of NMDA receptor blockade. Brain Res 982:260–269
Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I (2012) The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci 32:2410–2421
Peng PL, Zhong X, Tu W et al (2006) ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49:719–733
Penton-Rol G, Marin-Prida J, Pardo-Andreu G et al (2011) C-phycocyanin is neuroprotective against global cerebral ischemia/reperfusion injury in gerbils. Brain Res Bull 86:42–52
Phan VL, Urani A, Sandillon F, Privat A, Maurice T (2003) Preserved sigma1 (sigma1) receptor expression and behavioral efficacy in the aged C57BL/6 mouse. Neurobiol Aging 24:865–881
Robinet C, Pellerin L (2011) Brain-derived neurotrophic factor enhances the hippocampal expression of key postsynaptic proteins < i > in vivo</i > including the monocarboxylate transporter MCT2. Neuroscience 192:155–163
Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE (2010) The sigma-1 receptor chaperone as an interorganelle signaling modulator. Trends Pharmaco Sci 31:557–566
Takebayashi M, Hayashi T, Su TP (2004) A Perspective on the new mechanism of antidespressants: neuritogenesis through Sigma-1 receptor. Pharmacopsychiatry 37:208–213
Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20:709–726
Tyler WJ, Pozzo-Miller L (2003) Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J Physiol 553:497–509
Villard V, Espallergues J, Keller E et al (2009) Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1-41 against amyloid beta (25-35)-induced toxicity in mice. Neuropsychopharmacology 34:1552–1566
Walton MR, Dragunow M (2000) Is CREB a key to neuronal survival? Trends Neurosci 23:48–53
Watanabe S, Utsuki N (2011) Effect of ischemia on spatial learning in mice. In: CARLS series of advanced study of logic and sensibility. Keio Univserty Press, Tokyo, pp 7-14
Wu C, Zhan RZ, Qi S, Fujihara H, Taga K, Shimoji K (2001) A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J Neurosci Methods 107:101–106
Yagasaki Y, Numakawa T, Kumamaru E, Hayashi T, Su TP, Kunugi H (2006) Chronic antidepressants potentiate via sigma-1 receptors the brain-derived neurotrophic factor-induced signaling for glutamate release. J Biol Chem 281:12941–12949
Yamamoto Y, Shioda N, Han F et al (2009) Nobiletin improves brain ischemia-induced learning and memory deficits through stimulation of CaMKII and CREB phosphorylation. Brain Res 1295:218–229
Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M (1997) C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res 752:209–218
Zhang LM, Fu FH, Zhang XM, Zhu M, Wang T, Fan HY (2010) Escin attenuates cognitive deficits and hippocampal injury after transient global cerebral ischemia in mice via regulating certain inflammatory genes. Neurochem Int 57:119–127
Zhao Q, Murakami Y, Tohda M, Watanabe H, Matsumoto K (2005) Preventive Effect of Chotosan, a Kampo Medicine, on Transient Ischemia-Induced Learning Deficit Is Mediated by Stimulation of Muscarinic M1 But Not Nicotinic Receptor. Biol Pharm Bull 28:1873–1878
Zou LB, Yamada K, Sasa M, Nakata Y, Nabeshima T (2000) Effects of σ1 receptor agonist SA4503 and neuroactive steroids on performance in a radial arm maze task in mice. Neuropharmacology 39:1617–1627
Acknowledgments
This work was supported by National Science and Technology Major Special Project on Major New Drug Innovation of China (No. 2009ZX09103-119 and No. 2009ZX09301-012).
Conflict of interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, Q., Ji, XF., Chi, TY. et al. Sigma 1 receptor activation regulates brain-derived neurotrophic factor through NR2A-CaMKIV-TORC1 pathway to rescue the impairment of learning and memory induced by brain ischaemia/reperfusion. Psychopharmacology 232, 1779–1791 (2015). https://doi.org/10.1007/s00213-014-3809-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-014-3809-6