
Abstract
Current perspectives on the molecular underpinnings of major depressive disorder (MDD) posit a mechanistic role of epigenetic DNA modifications in mediating the interaction between environmental risk factors and a genetic predisposition. However, conclusive evidence for differential methylation signatures in the brain’s epigenome of MDD patients as compared to controls is still lacking. To address this issue, we conducted a pilot study including an epigenome-wide methylation analysis in six individuals diagnosed with recurrent MDD and six control subjects matched for age and gender, with a priori focus on the hippocampus and prefrontal cortex as pathophysiologically relevant candidate regions. Our analysis revealed differential methylation profiles of 11 genes in hippocampus and 20 genes in prefrontal cortex, five of which were selected for replication of the methylation status using pyrosequencing. Among these replicated targets, GRIN2A was found to be hypermethylated in both prefrontal cortex and hippocampus. This finding may be of particular functional relevance as GRIN2A encodes the glutamatergic N-methyl-d-aspartate receptor subunit epsilon-1 (NR2A) and is known to be involved in a plethora of synaptic plasticity-related regulatory processes probably disturbed in MDD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Depressive syndromes and their etiologies are highly diverse. As a consequence, knowledge about their molecular underpinnings is still limited, resulting in a relative lack of mechanistically driven treatments [1]. While genome-wide association studies (GWAS) have failed to identify robust and consistent risk modifiers for depression, genetic vulnerability is believed to strongly interact with environmental exposures including stressful life events, and there is accumulating evidence that epigenetic regulation may critically influence the susceptibility for depression by mediating this interplay [1, 2]. Epigenetic regulation refers to the heritable, but reversible modification of gene transcription in the absence of changes to the DNA coding sequence per se [3]. Multiple mechanisms underlying epigenetic regulation have been reported including DNA methylation of cytosine bases [4]. Consistent with evidence for volumetric decreases of the hippocampus (HIP) and other forebrain regions as well as decrements in neurotrophic factors are observations in rodent models of depression that the methylation signatures of genes encoding brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor are altered by stress—especially in the HIP [1, 5]. Even prenatal maternal stress may increase the susceptibility for depression in adolescent offspring via epigenetic regulation [6]. For instance, gestational factors have been proposed to influence adult depression-like behavior in utero by increasing the DNA methylation rate of the alpha calcitonin gene-related peptide in the rodent HIP [7].
Studies of peripheral DNA methylation patterns may also be informative by revealing potential non-invasive biomarkers for depression. Again, these studies have largely focused on BDNF, with a hypermethylation profile of the BDNF gene being of potential relevance as a diagnostic marker in the absence of predictive value for clinical outcome [8, 9].
Interestingly, glutamatergic mechanisms of depression have received less attention from epigenetic research, although there is substantial evidence from rodent models that depressogenic stress induces glutamatergic overactivity as well as overexpression of N-methyl-d-aspartate (NMDA) receptors [10–12]. Further significant support for a proximal role of NMDA receptors in the pathophysiology of depression comes from human studies documenting rapid (within hours) antidepressant responses of the NMDA receptor antagonist ketamine in patients with treatment-resistant depression, possibly via up-regulation of synaptogenesis and synaptic plasticity in HIP and prefrontal cortex (PFC) [13–15].
Given the urgent need for insights into the epigenetics of depression, the rationale of the present study was to conduct an epigenome-wide DNA methylation analysis in postmortem brain specimens obtained from six patients and six healthy controls matched for age and gender, with a priori focus on the HIP and PFC as high-priority candidate regions strongly implicated in the stress-induced neuroplastic changes associated with the disorder and their rapid reversal with ketamine [1, 15]. We hypothesized that our analysis would reveal specific target sites that exhibit differential methylation signatures in patients relative to controls and can be validated as physiologically relevant epigenetic modifications.
Materials and methods
Subjects and tissue acquisition
Postmortem human brain samples were acquired from the Netherlands brain bank (NBB), Netherlands Institute for Neuroscience, Amsterdam. NBB committee approved the experiments. All materials have been collected from donors whose written informed consent for brain autopsy and the use of the material and clinical information for research purposes has been obtained by the NBB. Donors fulfilled Diagnostic and Statistical Manual of Mental Disorders III-R criteria of major depressive disorder (MDD), which was confirmed by an experienced clinician (R.H.). DNA isolation from frozen PFC and HIP tissue specimens was carried out as previously reported [16]. Donors diagnosed with MDD were six individuals (4 females, 2 males) aged 76.3 ± 19.5 years (PFC) and 76.8 ± 19.6 years (HIP). The mean tissue pH values were 6.2 ± 0.1 (PFC) and 6.0 ± 0.9 (HIP), and the mean postmortem intervals were 5.6 ± 1.0 h (PFC) and 6.2 ± 1.6 h (HIP). Control tissue specimens had a mean tissue pH of 6.8 ± 0.3 and mean postmortem interval of 6.1 ± 0.7 h and were obtained from six healthy donors (4 females, 2 males) aged 78.8 ± 14.2 years. Thus, we used PFC and HIP tissue samples from five donors with MDD (S01/168, S06/028, S07/135, S08/090, S08/242) and five healthy controls (S09/134, S09/244, S10/023, S10/109, S10/181). The remaining tissue samples were obtained from four different donors as follows: PFC, donor S97/170 (MDD) and donor *95/026 (control); HIP, donor S09/323 (MDD) and donor S09/007 (control) (Table 1).
Genome-wide DNA methylation analysis
For bisulfite conversion reaction, we used 1 µg of DNA. This leads to the deamination of unmethylated cytosines, which were converted to 6-sulfonyluracil. Then, they were desulfonated to uracil, which ultimately translated into thymidine, while methylated cytosines were not converted. Comparing this converted DNA to the original unconverted sequence enabled detailed evaluation of the location and abundance of methylated cytosine-phosphate-guanosine dinucleotide (CpG) sites. Specifically, DNA was treated with a ZymoResearch (Irvine, CA) bisulfite kit; 200 ng of bisulfite-treated DNA was analyzed using the Infinium Human Methylation 450 K bead arrays spanning approximately over 480,000 CpG sites/sample (Illumina Inc., San Diego, CA). Processing was done according to the manufacturer’s protocol using an automated pipeline, and the arrays were scanned on an Illumina iScan platform (Illumina Inc., San Diego, CA) established at the Life & Brain Center (Bonn, Germany). We used Illumina GenomeStudio software (version 2011.1; Illumina Inc., San Diego, CA) for the extraction of DNA methylation signals from the arrays. Data were extracted as raw signals without background normalization. The methylation of CpG (cytosine guanosine dinucleotide) ranges from 0 (unmethylated, U) to 1 (fully methylated, M) on a continuous scale. The β-values were calculated from the intensity of the M and U alleles ratio of fluorescent signals: β = Max (M, 0)/Max (M, 0) + Max (U, 0) + 100. Further data analysis was carried out along two parallel protocols: We followed a recently proposed search algorithm which encompassed a sequence of operations including quality control, bead number filtering, probe filtering, signal correction (i.e., color-bias adjustment and background correction), subset-based quantile normalization, Infinium I/Infinium II correction and exclusion of samples that potentially contain SNPs (single nucleotide polymorphism) (http://www.ncbi.nlm.nih.gov/SNP/) [17]. This approach was complemented by an additional analysis based on Illumina’s GenomeStudio software. Methylation values were considered as differentially methylated when (1) the absolute difference between β-values means (delta-beta, Δβ) between patients and controls was higher than 0.2, and (2) when adjusted p values were lower than .05. Those CpGs not annotated to a known protein were excluded. We then prioritized the resultant candidate genes for validation with pyrosequencing by applying the following selection criteria: (a) putative link to major depression or brain structure and function in general (PubMed database search); (b) detection of multiple differentially methylated CpG sites in a given gene; and (c) detection of differentially methylated CpG sites in both PFC and HIP (Fig. 1).

Flow chart of methylation analysis. Step 1 Epigenome-wide, sequencing-based microarray experiment. Step 2 Differential methylation analysis using Illumina’s GenomeStudio and the complete pipeline by J. Tost. Step 3 PubMed-based in silico analysis of candidate genes identified in step 2. Step 4 Selection of candidate genes for further evaluation using pyrosequencing
Notably, the GenomeStudio software-based analysis identified a hypermethylation of the gene (GRIN2A) encoding the NMDAR subunit epsilon-1 (NR2A), which was not documented following the algorithm-based analysis. Subsequent pyrosequencing confirmed the GenomeStudio-based results, thus supporting their validity.
Statistical analysis of normalized methylation data
Statistical analysis was performed with SPSS Statistical software program for Windows, version 20.0 (SPSS Inc., Chicago, IL). Values are indicated as mean ± SD. Analysis of comparisons between groups was performed by Mann–Whitney U test. Levels of significance were set at p < .05 and p < .01, respectively. To assess the correlation between quantitative values, we determined the linear bivariate correlation coefficient (Pearson’s R) with the corresponding two-tailed significance level (p < .01).
Validation analysis using pyrosequencing
In epigenetic DNA methylation studies, specific targets from genome-wide methylation patterns need to be validated. Pyrosequencing is an ideal validation platform because it rapidly quantifies single and multiple methylation sites. For target validation, we performed pyrosequencing as follows: The DNA from brain samples was amplified using 5 pmol primer each (GRIN2A-PF1: TTTTTGTGTTTTGTGGTGTAT AGATT, GRIN2A-PR1/Bio: ACACTAAAAAATAAATAAATCACACCAAAT), 1 µl of bisulfite-treated DNA and 12,5 µl of Hotstar Plus master mix (Qiagen, Hilden, Germany) in a 25 µl reaction using the following cycler program: 5 min at 95 °C, 45 × (40 s at 95 °C, 40 s at 58 °C, 40 s at 72 °C), and 5 min at 72 °C. For pyrosequencing, we used 20–25 µl of the PCR reaction, the primer GRIN2A-PS1 (GTATGATTTATTTTTTTGTGGTAG), and the Pyromark Q 24 Kit (Qiagen), and performed sequencing according to the manual. For the other targets, the following primers and annealing temperatures were used: OTX2-PF1 (GAAAATAGTTTGT TTTTGGATTTGTGT), OTX2-PR1 Bio (CACATTCAACCCCAACAATAAATAT), OTX2-PS1 (AACAAATCAAACTAAAACTCAA), 56 °C; LYNX1-PF1 (TGGTTGTATGTAGTTT GGAGTGT), LYNX1-PR1/Bio (CCCAAAACCATACCCCTACTACTAATA), LYNX1-PS1 (GTTAGTTTAGTTAGGTTGGAA), 60 °C; MUC4-PF1 (GTTTTTATGGTTAGGTTGAAA TGTTATAGT), MUC4-PR1/Bio (CTCTCCCAACTACTTTCCTAAAC), MUC4-PS1 (TGA AATGTTATAGTTTGGTTATTTA), 60 °C; GPR111-PFI (TTTTAGGTTTAGGTTGATTTG TAAGAA), GPR111-PR1/Bio (ACACTAAAAAATAAATAAATCACACCAAAT), GPR111-PS1 (GTTTTTGTTTTTTGTGAGAG), 56 °C. The degree of methylation at each CpG site was estimated using PyroMarkQ24 software (Qiagen, Hilden, Germany). The rationale for selection of genes to be validated with pyrosequencing from the set of genes identified as being differentially methylated in the microarray experiment was an in silico PubMed-based analysis. Only targets with a known reasonable association with CNS or neuronal functions were selected. Based on these criteria, five genes from the initial microarray experiment qualified for subsequent validation analysis.
Results
Epigenome-wide methylation in brain specimens
In total, we compared 12 specimens of PFC and HIP postmortem tissue obtained from six donors with and without MDD and identified 40 significantly differentially methylated CpG sites in MDD patients that match the above defined selection criteria. More specifically, our results revealed 28 differentially methylated sites in PFC (hypermethylated, 13; hypomethylated, 15) and 12 in HIP (hypermethylated, 5; hypomethylated, 7) (Table 2a, b). All CpGs showed large methylation differences with Δβ-values ranging from 20.6 to 74.2. The distribution of CpGs indicated association with specific gene loci. In PFC, five CpGs were linked to orthodenticle homeobox 2 (OTX2), four CpGs to Zinc finger family member four (ZIC4) and another four in G protein-coupled receptor 133 (GPR133); in HIP, two CpGs were linked to cell surface-associated Mucin 4 (MUC4) and two to GPR111 (Table 2a, b). Interestingly, all CpGs lying within a specific gene locus were either hypermethylated or hypomethylated. Moreover, three loci (MUC4, CPR111 and CCR5) were consistently hypomethylated in both PFC and HIP.
Heat map
All CpG sites differing between MDD patients and controls were graphically illustrated using a heat map (Fig. 2a, b). Hierarchical clustering analysis revealed clear separation of patients and controls in both regions of interest. One patient who had received treatment with a 1,200-mg dose of carbamazepine over the past 3 years (S97/170) exhibited a slightly different methylation pattern relative to the mean of the patient sample (Fig. 2b).

Heat map displays highly methylated loci in red and sparsely methylated loci in blue. Hierarchical clustering of the samples after normalization revealed a clear separation of patients versus controls in PFC (a) and HIP (b). CTL controls, HIP hippocampus, MDD major depressive disorder, PFC prefrontal cortex
Pyrosequencing in brain samples
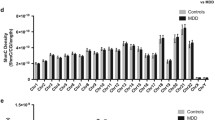
Pyrosequencing-based validation of the five resultant targets from genome-wide methylation patterns identified three CpGs linked to GRIN2A as significantly hypermethylated in both PFC (sum score: MDD, 26.1 ± 4.93; controls, 18.8 ± 2.31; p = .037) and HIP (sum score: MDD, 31.0 ± 5.73; controls, 22.5 ± 2.91; p = .025) (Fig. 3). On the single CpG level, differences between patients and controls remained significant, with p values ranging from .037 to .016. CpG site No. 1 was identically annotated on the applied methylation chip, no. 2–3 were not annotated on the chip, but were newly designed for pyrosequencing and were located adjacent to no. 1. The results of the pyrosequencing confirmed the values of the corresponding CpG (No. 1) on the array. Pyrosequencing of MUC4, LYNX1, OTX2 and GPR111 also corroborated the different methylation levels of those CpGs identically annotated on the microarray (Fig. 3). Moreover, several CpGs not annotated on the microarray were detected as differentially methylated when comparing patients and controls (Fig. 3). Only in one case, pyrosequencing failed to reproduce the microarray data: In HIP, CpG 1 of MUC4 was hypomethylated on the array, but not in the pyrosequencing analysis.

DNA methylation analysis using pyrosequencing with bisulfite-treated DNA obtained from brain. Pyrosequencing confirmed CpG methylation differences in genes nominated from microarray analysis. Underscored numbers denote CpGs corresponding to annotation of the microarray; those CpG sites not underscored were not annotated on the microarray and evaluated by pyrosequencing additionally. CTL controls, HIP hippocampus, MDD major depressive disorder, PFC prefrontal cortex
Correlation of microarray with pyrosequencing data
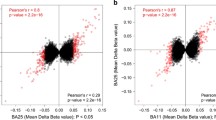
As mentioned above, DNA methylation values of the target genes were additionally evaluated by an independent validation method using pyrosequencing. Here, the microarray data of MUC4, LYNX1 and OTX2 showed high correlation with the pyrosequencing data ranging from 0.92 to 0.94 (Pearson’s correlation). Weaker correlations were found for the methylation values of GRIN2A in HIP (0.8; Pearson’s correlation).
Discussion
Our epigenome-wide DNA methylation analysis in postmortem HIP and PFC specimens confirmed our hypothesis of differential DNA methylation profiles in MDD. Based on our analysis algorithm (Fig. 1), we identified five genes as potentially informative targets for replication of our microarray data with pyrosequencing. Due to the fact that we applied stringent criteria for the statistical analysis of our array data, the number of significant CpGs was rather small. Since we investigated whole-brain samples including different cell types, further potential targets might have escaped our analysis. An alternative approach comparing neuronal versus non-neuronal profiles might have detected some additional CpGs.
From a functional perspective, the observed intragenic methylation changes in GRIN2A may be most relevant due to their key role in determining NMDA receptor function. In general, promoter sequence methylation is thought to downregulate expression of the gene product, whereas gene body methylation is positively correlated with expression activity [18]. This suggests that the observed hypermethylation of the GRIN2A gene body may lead to overexpression of NR2A [19]. Consistent with this, elevated expression of NR2A has indeed been documented in the amygdala and locus coeruleus (LC) of MDD patients, but not in the hippocampus or PFC, which may have methodological reasons [20–23]. Notwithstanding these discrepancies, different combinations of specific NR2 subunits are known to result in NMDA receptors with different functional characteristics [22]. For instance, NMDA receptors containing NR2A subunits mediate faster glutamate neurotransmission than NR2B-containing NMDA receptors [24]. Potential overexpression of GRIN2A may thus promote vulnerability for MDD via up-regulating NMDA receptor-dependent glutamatergic signaling. This hypothesis is in accord with findings that functional inactivation of NR2A in knockout mice reduced anxiety- and depression-related behaviors [25]. Related to this, functional inhibitors of NMDA receptor activity including the non-competitive antagonist ketamine [13, 14, 26–28] or the glycine transporter-I antagonist sarcosine (N-methylglycine) [29] have been identified as rapid-acting antidepressants in controlled trials, further supporting a putative proximal contribution of GRIN2A overexpression to the pathophysiology of MDD.
From a mechanistic perspective, GRIN2A overexpression due to stress-induced glutamatergic overactivity may interfere with a plethora of neuroplastic processes including the formation and maintenance of dendritic spines [30]. Furthermore, GRIN2A receptor was found up-regulated in human PFC of MDD patients [31]. This is consistent with recent proposals based on preclinical studies that blockade of NMDA receptors with ketamine is synaptogenic and induces synaptic plasticity within 30 min, thus rapidly reversing the deleterious changes caused by depressogenic stress [15, 32, 33].
Notably, candidate gene approach-based population and family association studies have also implicated GRIN2A in mood disorders [34], and experiments in rodents have identified NR2A-containing NMDA receptors as additional molecular target of the serotonin reuptake inhibitor (SSRI) fluoxetine [35]. Depressogenic stress has been shown to increase GRIN2A expression in rodent HIP, an effect that was normalized after treatment with the dual serotonin-noradrenaline reuptake inhibitor (SNRI) duloxetine [19]. Thus, activity at NMDA receptors may contribute to the mechanism of action of many commonly used antidepressant treatments [25]. One intriguing hypothesis is that the latency of antidepressant drug effects (ranging from hours for ketamine and deep brain stimulation, to weeks for monoamine reuptake inhibitors) is determined by how proximal these agents influence, and interact with, synaptogenesis and synaptic plasticity in HIP and PFC. Another important implication of our findings is that directly targeting the NR2A subunit with selective antagonists could have instantaneous efficacy as first line or adjunct therapy of MDD.
Among the five candidate genes replicated via pyrosequencing was also OTX2, which encodes a transcription factor that is involved in forebrain development and represents a key regulator of brain plasticity even in the mature forebrain [36]. Polymorphisms located in the OTX2 gene may confer vulnerability for mood disorders [37]. Little is known about the specific contribution of OTX2 to the pathophysiology of MDD, but our findings of six hypomethylated CpG sites within this gene strongly support a potential role in the neuroplastic changes associated with the disorder.
Another replicated candidate was LYNX1, which encodes a protein that enhances nicotinic acetylcholine receptor (nAChR) function in the presence of acetylcholine and regulates cortical plasticity. In rodents, expression of LYNX1 maintains stability of mature cortical networks in the presence of cholinergic innervation [38] and is enriched in interneuron populations in visual cortex. These interneurons are thought to regulate the convergence of GABAergic and nicotinic systems, which is known to be affected in psychiatric disorders [39]. The observed hypermethylation signature of this gene in the HIP may interfere with episodic memory formation and thus contribute to the cognitive impairments associated with MDD [40].
Regarding GPR111 and GPR113 (G Protein-Coupled Receptor 111 and 113, respectively), their possible contribution to the pathophysiology of MDD remains elusive. However, at least in mice, the loss of Gpr111 or Gpr115 function did not result in detectable abnormalities, suggesting that genes of this GPR group could perhaps function redundantly [41]. Thus, the differential methylation of GPR111 observed in our study may be functionally irrelevant due to compensation by GPR115. In this context, we note that gastric and duodenal neuroendocrine tumors show significant overexpression of GPR113 compared with normal tissue [42], but studies of GPR113 expression in MDD are still lacking.
We did not perform an additional analysis of our main findings on the protein level to show that the observed methylation changes lead to altered expression levels. This represents a limitation of our study. Another limitation of our study is the small sample size of postmortem brains and the fact that in two cases PFC and HIP were not obtained from the same donor.
A comparison of our five targets to those identified by candidate gene approach-based meta-analyses [2] yielded no overlap in any of the suggested sites. This is perhaps not surprising, given that the GWAS studies published to date also failed to replicate any of candidate variants proposed by these meta-analyses [2]. On the other hand, the data incorporated in these genetic studies were all derived from peripheral DNA as opposed to the present study, which carried out an epigenome-wide analysis of cerebral DNA isolated from HIP and PFC, thus having the chance of unravelling putative pathophysiological pathways much more directly.
Previous epigenetic studies, which have largely focused on neurotrophic pathways in bipolar disorder, have yielded rather conflicting results, with one study reporting a hypermethylation of the BDNF gene promoter region in PFC [43] and another study yielding no evidence for altered methylation profiles in this particular region [44]. Consistent with the latter, our analysis also detected no methylation changes in this specific locus. While such discrepancies between studies may likely reflect the heterogeneity of clinical phenotypes and underlying etiologies [1], there is also evidence for potential pharmacotherapy-related effects. For instance, Asai et al. [45] identified a diverse pattern of carbamazepine-induced CpG hypermethylations and hypomethylations. Among these, we found no overlap with our candidate CpGs. The only carbamazepine-treated patient included in our study exhibited three hypomethylated CpGs and one hypermethylated CpG compared to the sample mean, which might be related to a possible influence of medication.
In conclusion, our epigenome-wide profiling of postmortem HIP and PFC specimens identified widespread methylation changes, five of which were selected for replication using pyrosequencing. Among these candidates, GRIN2A is of particular functional relevance as it encodes the NMDAR subunit epsilon-1 (NR2A) and is involved in a plethora of synaptogenesis and synaptic plasticity-related regulatory processes probably disturbed in MDD. An important implication of our findings is that targeting the NR2A subunit with selective antagonists might have rapid efficacy as first line or adjunct therapy of MDD.
References
Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455:894–902
Flint J, Kendler KS (2014) The genetics of major depression. Neuron 81:484–503
Henikoff S, Matzke MA (1997) Exploring and explaining epigenetic effects. Trends Genet 13:293–295
Gibney ER, Nolan CM (2010) Epigenetics and gene expression. Heredity 105:4–13
Vialou V, Feng J, Robison AJ, Nestler EJ (2013) Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol 53:59–87
Dulawa SC (2014) Epigenetic programming of depression during gestation. BioEssays 36:353–358
Jiao J, Opal MD, Dulawa SC (2013) Gestational environment programs adult depression-like behavior through methylation of the calcitonin gene-related peptide gene. Mol Psychiatry 18:1273–1280
Fuchikami M et al (2011) DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One 6:e23881
Tadić A (2013) Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol Psychiatry. doi:10.1038/mp.2013.58
Moghaddam B (1993) Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem 60:1650–1657
Bartanusz V (1995) Stress-induced changes in messenger RNA levels of N-methyl-d-aspartate and AMPA receptor subunits in selected regions of the rat hippocampus and hypothalamus. Neuroscience 66:247–252
Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ (1996) Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci 16:274–282
Berman RM et al (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354
Zarate CA Jr et al (2006) A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864
Zarate C (2013) New paradigms for treatment-resistant depression. Ann N Y Acad Sci 1292:21–31
Kaut O, Schmitt I, Wüllner U (2012) Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 13:87–91
Touleimat N, Tost J (2012) Complete pipeline for Infinium Human Methylation 450 K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 3:325–341
Hellman A, Chess A (2007) Gene body-specific methylation on the active X chromosome. Science 315:1141–1143
Calabrese F, Guidotti G, Molteni R, Racagni G, Mancini M, Riva MA (2012) Stress-induced changes of hippocampal NMDA receptors: modulation by duloxetine treatment. PLoS One 7(5):e37916
Karolewicz B (2009) Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int J Neuropsychopharmacol 12:143–153
Karolewicz B, Stockmeier CA, Ordway GA (2005) Elevated levels of the NR2C subunit of the NMDA receptor in the locus coeruleus in depression. Neuropsychopharmacology 30:1557–1567
Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B (2009) Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry 33:70–75
Beneyto M, Meador-Woodruff JH (2008) Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 33:2175–2186
Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11:327–335
Boyce-Rustay JM, Holmes A (2006) Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic-and antidepressant-like effects in mice. Neuropsychopharmacology 31:2405–2414
Diazgranados N et al (2010) A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry 67:793–802
Murrough JW et al (2013) Neurocognitive effects of ketamine in treatment-resistant major depression: association with antidepressant response. Psychopharmacology (Berl) Epub ahead of print 2013 Sep 11. PubMed PMID:24022236
Murrough JW et al (2013) Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry 170:1134–1142
Huang CC et al (2013) Inhibition of glycine transporter-I as a novel mechanism for the treatment of depression. Biol Psychiatry 74:734–741
Akashi K et al (2009) NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J Neurosci 35:10869–10882
Goswami DB, Jernigan CS, Chandran A, Iyo AH, May WL, Austin MC, Stockmeier CA, Karolewicz B (2013) Gene expression analysis of novel genes in the prefrontal cortex of major depressive disorder subjects. Prog Neuropsychopharmacol Biol Psychiatry 43:126–133
Li N et al (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964
Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72
Cherlyn SY et al (2010) Genetic association studies of glutamate, GABA and related genes in schizophrenia and bipolar disorder: a decade of advance. Neurosci Biobehav Rev 34:958–977
Kiss JP et al (2012) GluN2B-containing NMDA receptors as possible targets for the neuroprotective and antidepressant effects of fluoxetine. Neurochem Int 60:170–176
Spatazza J et al (2013) Choroid-plexus-derived Otx2 homeoprotein constrains adult cortical plasticity. Cell Rep 3:1815–1823
Sabunciyan S et al (2007) Polymorphisms in the homeobox gene OTX2 may be a risk factor for bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 144B:1083–1086
Morishita H, Miwa JM, Heintz N, Hensch TK (2010) Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science 330:1238–1240
Demars MP, Morishita H (2014) Cortical parvalbumin and somatostatin GABA neurons express distinct endogenous modulators of nicotinic acetylcholine receptors. Mol Brain 27:75
Millan MJ et al (2012) Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 11:141–168
Prömel S, Waller-Evans H, Dixon J, Zahn D, Colledge WH, Doran J, Carlton MB, Grosse J, Schöneberg T, Russ AP, Langenhan T (2012) Characterization and functional study of a cluster of four highly conserved orphan adhesion-GPCR in mouse. Dev Dyn 241:1591–1602
Sherman SK, Maxwell JE, Carr JC, Wang D, O’Dorisio MS, O’Dorisio TM, Howe JR (2014) GIPR expression in gastric and duodenal neuroendocrine tumors. J Surg Res 190:587–593
Rao JS, Keleshian VL, Klein S, Rapoport SI (2012) Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry 2:e132
Mill J et al (2008) Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 82:696–711
Asai T et al (2013) Effect of mood stabilizers on DNA methylation in human neuroblastoma cells. Int J Neuropsychopharmacol 16:2285–2294
Acknowledgments
R.H. was supported by a Starting Independent Researcher Grant (“NEMO–Neuromodulation of Emotion”) jointly provided by the Ministry of Innovation, Science, Research & Technology of the German State of North Rhine-Westphalia (MIWFT) and the University of Bonn
Conflict of interest
The authors report no competing biomedical financial interests or personal affiliations in connection with the content of this manuscript.
Ethical standard
This study has been approved by the local ethics committee and has been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All persons gave their informed consent prior to their inclusion in the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kaut, O., Schmitt, I., Hofmann, A. et al. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur Arch Psychiatry Clin Neurosci 265, 331–341 (2015). https://doi.org/10.1007/s00406-014-0572-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-014-0572-y