
Abstract
Various types of lipids and their metabolic products associated with the biological membrane play a crucial role in signal transduction, modulation, and activation of receptors and as precursors of bioactive lipid mediators. Dysfunction in the lipid homeostasis in the brain could be a risk factor for the many types of neurodegenerative disorders, including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. These neurodegenerative disorders are marked by extensive neuronal apoptosis, gliosis, and alteration in the differentiation, proliferation, and development of neurons. Sphingomyelin, a constituent of plasma membrane, as well as its primary metabolite ceramide acts as a potential lipid second messenger molecule linked with the modulation of various cellular signaling pathways. Excessive production of reactive oxygen species associated with enhanced oxidative stress has been implicated with these molecules and involved in the regulation of a variety of different neurodegenerative and neuroinflammatory disorders. Studies have shown that alterations in the levels of plasma lipid/cholesterol concentration may result to neurodegenerative diseases. Alteration in the levels of inflammatory cytokines and mediators in the brain has also been found to be implicated in the pathophysiology of neurodegenerative diseases. Although several mechanisms involved in neuronal apoptosis have been described, the molecular mechanisms underlying the correlation between lipid metabolism and the neurological deficits are not clearly understood. In the present review, an attempt has been made to provide detailed information about the association of lipids in neurodegeneration especially in Alzheimer’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids are considered as a group of naturally occurring hydrophobic or amphiphilic small molecules including fats, waxes, sterols, monoglycerides, diglycerides, triglycerides, phospholipids, and others [1]. Biosynthesis of lipid started with the conversion of carbohydrate into triglycerides through the process of lipogenesis in the endoplasmic reticulum. These synthesis processes further give rise to steroids including cholesterol and ergosterol [2, 3]. Fatty acids are considered as important components of biological lipids which are linked with the function of making building blocks of more structurally complex lipids. They also participate in the maintenance of the structure and function of cell membranes. Lipids are involved in a number of biological functions including energy storage, signaling, and acting as structural components of cell membranes [4, 5]. They can be divided into several categories depending on their origin from biological molecules like fatty acids, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, and prenol lipids [4]. Glycerolipids comprise the bulk of storage fat in animal tissues and function as energy storage. These lipids are mainly composed of mono-, di-, and tri-substituted glycerols, the most well known being the fatty acid triesters of glycerol, called triglycerides [6]. The initial steps involved in the metabolization of fats are the hydrolysis of the ester bonds of triglycerides and the release of glycerol and fatty acids from adipose tissue. A phospholipid is composed of two fatty acids, a glycerol unit, a phosphate group, and a polar molecule. It constitutes a major component of cell membranes and forms a semipermeable lipid bilayer that allows only certain molecules to diffuse across the membrane to enter or exit the cell [7].
The glycerophospholipids are usually referred as phospholipids and constitute the main structural component of biological membranes, like the plasma membrane, intracellular membranes of organelles, and neural cell membranes [8]. As glycerophospholipids are involved in the generation of second messengers including arachidonic acid, eicosanoids, platelet-activating factor, and diacylglycerol, they are considered as a reservoir for second messengers. They are also involved in metabolism, cell signaling, signal transduction, apoptosis, and modulation of activities of transporters and membrane-bound enzymes [8, 9]. Past studies reported that a slight alteration in the composition of glycerophospholipids in the brain may lead to various functional neurological disorders [8]. Likewise, glycerophospholipids, sphingomyelins, and sterol lipids including cholesterol and its derivatives are also an important component of membrane lipids that imparts various biological roles including hormone secretion and signal transduction [10].
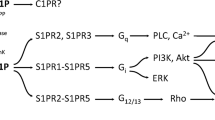
Sphingolipids are the complicated family of compounds that derived from the alipathic amino alcohol sphingosine. The sphingosine backbone is O-linked to amino acid serine and a long-chain fatty acyl CoA and then converted into ceramides, phosphosphingolipids, glycosphingolipids, and other compounds [11]. In the central nervous system, the composition of sphingolipids is quite high and involved in the structural maintenance. Additionally, their metabolites function as second messengers to modulate a wide variety of signaling cascade [11, 12]. Ceramides, a precursor to sphingomyelin, are the simplest sphingolipids that engaged in the formation of lipid rafts essential in the regulation of sphingomyelin pathway of signal transduction, proliferation, differentiation, programmed cell death, and senescence. They also act as the regulator of synaptic function and have been implicated in synapse formation, transmitter release, and plasticity [11, 13, 14]. The occurrence of signaling cascade at cellular level has been found to be linked with lipid signaling [15, 16]. It could occur with the activation of G protein-coupled or nuclear receptors, and also some specific lipids act as signaling molecules and cellular messengers [17]. These signaling molecules include sphingosine-1-phosphate, which is involved in the regulation of signal transmission, cellular growth, and apoptosis [18, 19]. Besides, other signaling molecules including diacylglycerol and the phosphatidylinositol phosphates are involved in the calcium-mediated activation of protein kinase C, and prostaglandins are involved in inflammation and immunity [20, 21].
There are growing evidences that cholesterol is of particular importance in the development and progression of the disease. Studies have reported that elevated cholesterol levels increase Aβ in cellular and animal models which further associated with the increased risk of Alzheimer’s disease [12, 22]. Neurodegenerative disorders and other CNS diseases have immense clinical importance, and in the present scenario, there is no effective cure for these diseases and disorders, resulting in an increase economic burden on the society and negative impact on the quality of life. The crucial role of lipids in tissue physiology and cell signaling is demonstrated in many neurological disorders that are associated with deregulated lipid metabolism [13, 23] (Fig. 1). The role of lipids in neurodegenerative diseases and disorders including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and other CNS injuries has been suggested by a number of investigators, but the exact molecular mechanism involved behind this is still unclear. In the present review, we focused on how lipid metabolism is involved in amyloid processing, β-amyloid segregation, and impairment of synaptic transmission, leading to the pathophysiology of Alzheimer’s disease.

Structure of some common lipids (fatty acids, cholesterol, triglyceride, phospholipids) and their components (sphingomyelin, ceramide, sphingosine, sphingosine-1-phosphate) commonly involved in the normal cell physiology and pathophysiology of neurodegeneration
Lipid Rafts and Neurodegeneration
Lipid rafts are plasma membrane microdomains loaded with cholesterol, sphingolipids, sphingomyelin, gangliosides, and glycosylphosphatidylinositol-anchored proteins, which provide a particularly ordered lipid environment and make them resistant to non-ionic detergent extraction using Triton X-100 [12, 24, 25]. It contains around 50 % elevated sphingomyelin and decreased phosphatidylcholine levels as compared to the plasma membrane [26]. In these rafts, cholesterol acts as a dynamic glue that cleaves the raft together and serves as a molecular spacer [27]. Lipids rafts include receptors, channels, recognition molecules, coupling factors, and enzymes which are involved in signal transduction and intracellular trafficking of proteins, lipids, secretory and endocytic pathways, and inflammation and in cell surface proteolysis [28, 29]. In neurons, lipid rafts act as platforms for the signal transduction initiated by several classes of neurotrophic factors and hence are implicated in various physiological and pathological processes [30–32]. Studies also reported that lipid rafts are essential for neuronal cell adhesion, axonal guidance, and synapse communication [33]. Thus, in brain lipid, rafts play a very crucial role as these are structurally unique components of plasma membranes which are involved in the neural development and function and certain neurodegenerative diseases [34–36].
In case of neurological injury such as ischemia and early reperfusion, ceramide has been found to be overexpressed as a result of sphingomyelin metabolism. The overexpression of ceramide leads to the activation of mitochondria-dependent pathways including inhibition of anti-apoptotic proteins and electron transport chain, increase generation of reactive oxygen species (ROS), release of cytochrome c, and activation of caspase-3 and caspase-9, bax, SAPK, and cJNK that are associated with neuronal apoptosis (Fig. 2). In clinical and experimental studies, increased levels of ceramide have also been reported in Alzheimer’s disease [11, 12, 37]. Tau overphosphorylation and β-amyloid peptide accumulation have been found to be associated with the increased levels of ceramide in the brain. Further, its high concentration in the blood may also be associated with the biochemical changes in the brain in the preclinical stage [38].

Lipid raft-mediated signaling of sphingomyelin and ceramide allied with normal functions and apoptosis. Sphingomyelin and its secondary messengers are implicated in the regulation of cell survival and neural apoptosis. Amyloid fibril aggregation alters the ceramide concentration, resulting in the mitochondrial dysfunctions, enhanced generation of reactive oxygen species (ROS), and activation of cytochrome c. Increased level of ceramide as a result of sphingomyelin hydrolysis is also associated with the activation of proapoptotic factors and generation of ROS that further activate SAPK kinase, Jun N-terminal kinases (JNKs 1/2), and caspase cascade, including the activation of caspase-3 and caspase-9. The outflow signaling through the JNKs and caspases ultimately triggers the onset of neuronal apoptosis, while the ceramide-induced activation of CAPK also activates MAPK kinase and extracellular signal-regulated kinases (ERKs 1/2). MAPK cascade strongly stimulates ERK but weakly inhibits JNK and therefore is involved in cell growth, differentiation, and the protective mechanism [49, 51]
The deregulation of lipid metabolism affects the tissue physiology and cell signaling which leads to the various neurological disorders, including bipolar disorders, schizophrenia, and neurodegenerative diseases like Alzheimer’s, Parkinson’s, and Huntington’s diseases [39].
Lipid Integration in Alzheimer’s Disease
Alzheimer’s disease is a progressive neurodegenerative disorder associated with loss of neurons. It is characterized by the presence of excessive amounts of neuritic plaques containing amyloid β protein and abnormal tau protein filaments in the form of neurofibrillary tangles [40–42]. Amyloid β protein has been associated to induce oxidative stress as a result of increased levels of lipid peroxidation products including hydroxynonenal (HNE), which can react further with various amino acids of associated proteins and alter their structure and function in the brain. The disease rapidly progresses to complete loss of mental powers, particularly loss of memory and normal emotional behavior. Dementia is a characteristic feature of Alzheimer’s disease associated with loss of intellectual ability, leading to disruption of multiple higher cortical functions including memory, reasoning, orientation, learning capacity, and emotional stability [43, 44]. Degeneration of cholinergic neurons, particularly in the basal forebrain, has been found to be associated with loss of the neurotransmitter acetylcholine [45, 46]. Depletion of acetylcholine level in Alzheimer’s disease patients appears to be a critical element in producing dementia [47]. In addition, direct association of lipid with Alzheimer’s disease and the role of inflammatory mediators in the production and accumulation of β-amyloid protein, ROS, and lipid peroxide have also been reported [48, 49]. A lot of evidence on roles of lipids in Alzheimer’s disease suggest that the alterations in the composition of brain lipid profile including phospholipid, sphingomyelin, ceramide, and ganglioside could be a causative factor in neuropathological or neurological disorders [11, 12, 50]. Lipids are involved in a variety of signaling events and functions; alterations in the levels of their composition in brain cells could modulate the signaling cascade and ultimately affect the neural function which further leads to Alzheimer’s disease [23, 50, 51]. Lukiw and Bazan [52] reported the role of docosahexaenoic acid (DHA), a dietary essential polyunsaturated fatty acid (PUFA) enriched in fish oil, in maintaining the cell structure and function in the nervous system. They further suggested that the decline in the level of DHA leads to the oxidative stress, apoptosis, and cognitive and neuronal dysfunctions in Alzheimer’s disease. Studies also reported that neuroprotection D1 (NPD1), the first identified DHA-derived mediator, significantly allied with cell survival and repair through the activation of anti-apoptotic and anti-inflammatory signaling [53].
The first reports of alterations in glycolipids including levels of gangliosides, cerebrosides, and cerebroside sulfatide in Alzheimer’s disease came out in the late 1960s [50, 54]. Further, in the 1990s, brain phospholipid content was also reported to be decreased in Alzheimer’s disease [55]. Decreased phospholipid species in specific brain regions including the hippocampus and parahippocampal gyrus of Alzheimer’s disease brains were suggested due to increases in phospholipid turnover as evidenced by increase in the phospholipid catabolite glycerophosphocholine [55]. Bassett and Montine [56] reported that apolipoprotein E present in the central nervous system is principally involved in the regulation of lipid transport and its metabolism through known receptor-mediated processes; hence, it is more vulnerable to oxidation than other lipoproteins which may contribute to neurodegeneration in the Alzheimer’s disease brain. A number of other studies consistently reported the decreased levels of sphingomyelins and increased levels of ceramides in Alzheimer’s disease brains as a result of sphingomyelin hydrolysis [37, 57, 58].
Oxidative Stress Markers and Alzheimer’s Disease
Oxidative stress refers to the impairment in the antioxidant defense mechanisms as a result of excessive production of free radicals including superoxide radical, hydrogen peroxide, hydroxyl radical, nitric oxide, and peroxynitrite [59]. These are associated with the increased levels of lipid peroxidation, DNA, and protein oxidation products in brains and lead to neurodegenerative diseases including Alzheimer’s disease [60–62]. The presence of a high content of PUFA in the brain makes it a highly oxidative organ consuming 20 % oxygen of the body instead of only 2 % of the total body weight; therefore, lipid peroxidation is thought to be a major cause of oxidative damage in Alzheimer’s disease [23, 55, 63]. Brain cells are more vulnerable toward oxidative damage than any other cells due to their low activity of antioxidant enzymes and high number of mitochondria. Mitochondrial dysfunction and oxidative metabolism play an important role in the pathogenesis of Alzheimer’s disease and other neurodegenerative diseases. Transition metals have been found to be accumulated in the brain and hence increase the susceptibility to free radical generations through the process of Fenton’s reaction. PUFA is involved in the modulation of iron transporters that facilitate iron uptake and thus induce neuronal apoptosis [64, 65]. The increased levels of ROS cause oxidative damage to nucleic acids, proteins, carbohydrates, and lipids and are linked with the accumulation of oxidized products. These oxidized products such as lipid peroxides give rise to reactive α- and β-unsaturated aldehydes such as malondialdehyde, 4-HNE, and acrolein [60, 66]. High levels of HNE, an oxidative product formed as a result of enhanced lipid peroxidation, have been found in the Alzheimer’s brain tissue [67–69]. DHA, another primary lipid peroxidation target in oxidative damage, has been found to oxidize non-enzymatically into various neuroprostanes and may be involved in the generation of ROS and enhanced oxidative stress linked with cognitive decline and neuronal dysfunction [52]. The presence of acrolein, an α- and β-unsaturated aldehydic product of lipid peroxidation in neurofibrillary tangles in the Alzheimer’s disease brain, has also been reported [60]. Increased levels of reactive oxygen and nitrogen species as a result of oxidative events in Alzheimer’s brain may initiate the apoptotic cascade that significantly leads to cellular dysfunction and ultimately death [70, 71]. Autopsied brain from Alzheimer’s disease patients indicated an increase in the level of protein oxidation, lipid peroxidation, and its toxic metabolite HNE and a decrease in the levels of PUFA. The levels of antioxidant enzymes including glutamine synthetase, creatinine kinase, and glutathione peroxidase were also found to be decreased in the brain in Alzheimer’s disease [61]. The role of Aβ in the production of free radicals and in the pathophysiology of Alzheimer’s disease has also been demonstrated by various investigators [41, 45].
Immunohistochemical and histopathological studies showed that the presence of oxidative stress products in neurofibrillary tangles and senile plaques as a result of oxidative stress is the important hallmark of Alzheimer’s disease [71–73]. In addition to this, neuroinflammation was also found to be implicated in the pathogenesis of Alzheimer’s disease. These inflammatory cytokines are activated as a result of enhanced oxidative stress and linked with neurodegeneration in Alzheimer’s diseases [39, 74]. Recently, Bazan et al. [75] showed the protective role of DHA and its mediator NPD1 in the neuroinflammation and stroke-mediated brain damage through the discerning regulation of apoptotic and anti-apoptotic proteins. They further reported the protective role of NPD1 in Alzheimer’s disease via the downregulation of amyloidogenic processing of the amyloid-β precursor protein and expression of pro-inflammatory genes.
Environmental factors including exposure to heavy metals and pesticides have also been found to be linked with enhanced vulnerability to the incidences of Alzheimer’s disease in humans [76–79]. Studies demonstrated that exposure to arsenic activates the JNK3 and p38 MAPK associated with the formation of neurotic plaques and neurofibrillary tangles [80, 81], which may cause apoptosis in cortical neurons and contribute to Alzheimer’s disease [82, 83]. We have also reported that arsenic accumulates in the brain regions and enhanced the levels of oxidative stress products and inhibition of antioxidant defense system in rats. Further, this enhanced oxidative stress leads to the death of neuronal cells in the hippocampal area and caused cholinergic dysfunctions [84]. In an another study, enhanced oxidative stress as a measure of lipid peroxidation following exposure to lambda-cyhalothrin, a type II synthetic pyrethroid, and monocrotophos, an organophosphate pesticide in rats, and its association with cholinergic dysfunctions have also been reported [85, 86]. Besides toxicants, exposure to high levels of man-made electromagnetic radiations and their adverse health effects has become a matter of concern for the health scientists. Some effects linked to electromagnetic pollution are decreased testosterone levels in men, miscarriages in pregnant women, birth defects in babies, Alzheimer’s disease, cataracts, depression and suicides, chronic fatigue, and others [87]. In spite of that, a few studies have suggested that radiation emitted by a cell phone may interact with brain activity and behavior [88–90], gene expression and DNA, cell growth, proliferation and tumors [91, 92], hormones, proteins, and enzymes [93–95]. Recently, Kesari et al. [96] reported that exposure to electromagnetic radiation could be a risk factor childhood leukemia, brain tumors, genotoxic effects, immune system deregulation, allergic and inflammatory responses, infertility, cardiovascular and neurological effects, and neurodegenerative diseases. They further suggested that these deleterious health effects are associated with increased ROS, which may enhance the effect of microwave radiations and cause neurodegeneration.
Conclusion
The lack of neuronal repair in the central nervous system is the main basis of the severity of injury, functional deficits, and associated neurodegenerative disorders. The alterations in lipid metabolism and lipid rafts due to the enhanced oxidative stress and impaired enzymatic cascade are also associated with these diseases. These alterations result in the transformation of membrane flexibility and permeability which allow the accumulation of lipid peroxides and compromised energy metabolism that linked with the neurodegeneration. Deposition of amyloid plaques as a result of altered lipid rafts and generation of ROS to cause lipid peroxidation and damage to the biological membrane is found in Alzheimer’s disease. The modulation in lipid raft synthesis and inhibitors against myelin inhibitory protein could be the constructive tool for neuronal loss therapy and may open new avenues for the treatment of neurological disorders and diseases. The present review has mainly focused on the involvement of lipids, their metabolites, and oxidative stress in the pathogenesis of neurodegeneration especially on Alzheimer’s disease. As the production of amyloid precursor protein mainly regulated by the lipid rafts and its interaction with other molecules affects the signaling cascade, the review examined the mechanism underlying them. Further studies are required on lipid rafts to open new vistas in the regulation of signaling pathways and myelin inhibitory protein for the treatment of neurodegenerative diseases.
References
Campbell NA, Mitchell LG, Reece JB (1999) Biology, 5th edn. Benjamin/Cummings Publ. Co., Inc, Menlo Park
Schroepfer G (1981) Sterol biosynthesis. Annu Rev Biochem 50:585–621
Lees N, Skaggs B, Kirsch D, Bard M (1995) Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae—a review. Lipids 30(3):221–226
Fahy E, Subramaniam S, Murphy R et al (2009) Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res 50(Supplement):S9–S14
Subramaniam S, Fahy E, Gupta S et al (2011) Bioinformatics and systems biology of the lipidome. Chem Rev 111(10):6452–6490
Coleman RA, Lee DP (2004) Enzymes of triglyceride synthesis and their regulation. Prog Lipid Res 43(2):134–176
Zachowski A (1993) Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J 294(Pt 1):1–14
Farooqui AA, Horrocks LA, Farooqui T (2000) Glycerophospholipids in brain: their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem Phys Lipids 106(1):1–29
Berridge MJ, Irvine RF (1989) Inositol phosphates and cell signalling. Nat J 341(1):197–205
Bach D, Wachtel E (2003) Phospholipid/cholesterol model membranes: formation of cholesterol crystallites. Biochim Biophys Acta 1610(2):187–197
Haughey NJ, Bandaru VVR, Bai M, Mattson MM (2010) Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim Biophys Acta 1801(8):878–886
Jana A, Hogan EL, Pahan K (2009) Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci 278(1–2):5–15
Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM Jr (2002) Lipid rafts in neuronal signaling and function. Trends Neurosci 8:412–417
Mencarelli C, Martinez-Martinez P (2013) Ceramide function in the brain: when a slight tilt is enough. Cell Mol Life Sci 70:181–203
Wang X (2004) Lipid signaling. Curr Opinions Plant Biol 7(3):329–336
Dinasarapu AR, Saunders B, Ozerlat I, Azam K, Subramaniam S (2011) Signaling gateway molecule pages a data model perspective. Bioinformatics 27(12):1736–1738
Eyster KM (2007) The membrane and lipids as integral participants in signal transduction. Adv Physiol Educ 31(1):5–16
Hinkovska-Galcheva V, VanWay SM, Shanley TP, Kunkel RG (2008) The role of sphingosine-1-phosphate and ceramide-1-phosphate in calcium homeostasis. Curr Opin Investig Drugs 9(11):1192–1205
Saddoughi SA, Song P, Ogretmen B (2008) Roles of bioactive sphingolipids in cancer biology and therapeutics. Subcell Biochem 49:413–440
Klein C, Malviya AN (2008) Mechanism of nuclear calcium signaling by inositol 1,4,5-trisphosphate produced in the nucleus, nuclear located protein kinase C and cyclic AMP-dependent protein kinase. Front Biosci 13(13):1206–1226
Boyce JA (2008) Eicosanoids in asthma, allergic inflammation, and host defense. Cur Mo Med 8(5):335–349
Cutler RG, Kelly J, Storie K et al (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101:2070–2075
Sultana R, Perluigi M, Butterfield DA (2013) Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med 62:157–169
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Brown DA, London E (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 275:17221–17224
Pike LJ (2008) The challenge of lipid rafts. J Lipid Res 50:S323
Korade Z, Kenworthy AK (2008) Lipid rafts, cholesterol, and the brain. Neuropharmacology 55(8):1265–1273
Brown DA, London E (1998) Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14:111–136
Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39
Kamiguchi H (2006) The region-specific activities of lipid rafts during axon growth and guidance. J Neurochem 98(2):330–335
Grider MH, Park D, Spencer DM, Shine HD (2009) Lipid raft-targeted Akt promotes axonal branching and growth cone expansion via mTOR and Rac1, respectively. J Neurosci Res 87:3033–3042
Munderloh C, Solis GP, Bodrikov V et al (2009) Reggies/flotillins regulate retinal axon regeneration in the zebra fish optic nerve and differentiation of hippocampal and N2a neurons. J Neurosci 29:6607–6615
Petro KA, Schengrund CL (2009) Membrane raft disruption promotes axonogenesis in N2a neuroblastoma cells. Neurochem Res 34:29–37
Cordy JM, Hooper NM, Turner AJ (2006) The involvement of lipid rafts in Alzheimer's disease. Mol Membr Biol 23(1):111–122
Cheng H, Vetrivel KS, Gong et al (2007) Mechanisms of disease: new therapeutic strategies for Alzheimer’s disease—targeting APP processing in lipid rafts. Nat Clin Pract Neurol 3:374–382
Vetrivel KS, Thinakaran G (2010) Membrane rafts in Alzheimer’s disease β-amyloid production. Biochim Biophys Acta 1801:860–867
Han X, Holtzman MD, McKeel DW Jr, Kelley J, Morris JC (2002) Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem 82:809–818
Car H, Zendzian-Piotrowska M, Fiedorowicz A, Prokopiuk S, Sadowska A, Kurek K (2012) The role of ceramides in selected brain pathologies: ischemia/hypoxia, Alzheimer disease. Postepy Hig Med Dosw 66:295–303
Wenk GL (2003) Neuropathologic changes in Alzheimer's disease. J Clin Psychiatry 64(9):7–10
Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118(1):5–36
Honjo K, Black SE, Verhoeff NP (2012) Alzheimer’s disease, cerebrovascular disease, and the β-amyloid cascade. Can J Neurol Sci 39(6):712–728
Breunig JJ, Guillot-Sestier MV, Town T (2013) Brain injury, neuroinflammation and Alzheimer's disease. Front Aging Neurosci 5:26
Fadil H, Borazanci A, Ait Ben Haddou E, Yahyaoui M, Korniychuk E, Jaffe SL, Minagar A (2009) Early onset dementia. Int Rev Neurobiol 84:245–262
Jorge RE, Robinson RG (2011) Treatment of late-life depression: a role of non-invasive brain stimulation techniques. Int Rev Psychiatry 23(5):437–444
Selkoe DJ (1994) Alzheimer’s disease: a central role for amyloid. J Neuropathol Exp Neurol 53(5):438–447
Hamlin AS, Windels F, Boskovic Z, Sah P, Coulson EJ (2013) Lesions of the basal forebrain cholinergic system in mice disrupt idiothetic navigation. PLoS One 8(1):e53472
Akiyama H, Barger S, Barnum S et al (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Ariga T, McDonald MP, Yu RK (2008) Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease—a review. J Lipid Res 49:1157–1175
Ariga T, Jarvis WD, Yu RK (1998) Role of sphingolipid-mediated cell death in neurodegenerative diseases. J Lipid Res 39:1–16
Cherayil GD (1968) Fatty acid composition of brain glycolipids in Alzheimer’s disease, senile dementia, and cerebrocortical atrophy. J Lipid Res 9:207–214
Hicks DA, Nalivaeva NN, Turner AJ (2012) Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signalling. Front Physiol 3(189):1–18
Lukiw WJ, Bazan NG (2008) Docosahexaenoic acid and the aging brain. J Nutr 138:2510–2514
Bazan NG (2006) Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photo-receptors. Trends Neurosci 29:263–271
Suzuki K, Katzman R, Korey SR (1965) Chemical studies on Alzheimer’s disease. J Neuropathol Exp Neurol 24:211–224
Nitsch RM, Blusztajn JK, Pittas AG et al (1992) Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci U S A 89:1671–1675
Bassett CN, Montine TJ (2003) Lipoproteins and lipid peroxidation in Alzheimer's disease. J Nutr Health Aging 7(1):24–29
He X, Huang Y, Li B, Gong CX, Schuchman EH (2010) Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging 31(3):398–408
Satoi H, Tomimoto H, Ohtani R et al (2005) Astroglial expression of ceramide in Alzheimer’s disease brains: a role during neuronal apoptosis. Neuroscience 130:657–666
Markesbery WR (1999) The role of oxidative stress in Alzheimer disease. Arch Neurol 56:1449–1451
Lovell MA, Xie C et al (2000) Acrolein, a product of lipid peroxidation, inhibits glucose and glutamate uptake in primary neuronal cultures. Free Radic Biol Med 29(8):714–720
Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer's disease. Brain Pathol 9(1):133–146
Hardas SS, Sultana R, Clark AM et al (2013) Oxidative modification of lipoic acid by HNE in Alzheimer’s disease brain. Redox Biol 1:80–85
Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer's disease amyloid β peptide. Biochim Biophys Acta 1768:1976–1990
Schonfeld E, Yasharel I, Yavin E, Brand A (2007) Docosahexaenoic acid enhances iron uptake by modulating iron transporters and accelerates apoptotic death in pc12 cells. Neurochem Res 32:1673–1684
Brand A, Schonfeld E, Isharel I, Yavin E (2008) Docosahexaenoic acid-dependent iron accumulation in oligodendroglia cells protects from hydrogen peroxide-induced damage. J Neurochem 105:1325–1335
Poli G, Schaur RJ (2000) 4-Hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life 50(4–5):315–321
Sayre LM, Zelasko DA, Harris PL et al (1997) 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem 68:2092–2097
Montine KS, Kim PJ, Olson SJ, Markesbery WR, Montine TJ (1997) 4-Hydroxy-2-nonenal pyrrole adducts in human neurodegenerative disease. J Neuropathol Exp Neurol 56:866–871
Williams TI, Lynn BC, Markesbery WR, Lovell MA (2006) Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging 27:1094–1099
Stadtman ER, Berlett BS (1997) Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol 10:485–494
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45:358–368
Su JH, Anderson AJ, Cummings BJ, Cotman CW (1994) Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 5(18):2529–2533
Pappolla MA, Omar RA, Kim KS, Robakis NK (1992) Immunohistochemical evidence of oxidative stress in Alzheimer’s disease. Am J Pathol 140:621–628
Farooqui AA, Horrocks LA, Farooqui T (2007) Modulation of inflammation in brain: a matter of fat. J Neurochem 101(3):577–599
Bazan NG, Calandria JM, Gordon WC (2013) Docosahexaenoic acid and its derivative neuroprotectin D1 display neuroprotective properties in the retina, brain and central nervous system. Nestle Nutr Inst Workshop Ser 77:121–131
Giasson BI, Sampathu DM, Wilson CA et al (2002) The environmental toxin arsenite induces tau hyperphosphorylation. Biochemistry 41(51):15376–15387
Borenstein AR, Copenhaver CI, Mortimer JA (2006) Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord 20(1):63–72
Jones N (2010) Alzheimer disease: risk of dementia and Alzheimer disease increases with occupational pesticide exposure. Nat Rev Neurol 6(7):353
Wingo TS, Rosen A, Cutler DJ, Lah JJ, Levey AI (2012) Paraoxonase-1 polymorphisms in Alzheimer's disease, Parkinson's disease, and AD-PD spectrum diseases. Neurobiol Aging 33(1):204, e13–15
Namgung U, Xia Z (2001) Arsenic induces apoptosis in rat cerebellar neurons via activation of JNK3 and p38 MAP kinases. Toxicol Appl Pharmacol 174:130–138
Hull M, Lieb K, Fiebich BL (2002) Pathways of inflammatory activation in Alzheimer’s disease: potential targets for disease modifying drugs. Curr Med Chem 9:83–88
Gong G, O’bryant SE (2010) The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis Assoc Disord 24:311–316
Gharibzadeh S, Hoseini SS (2008) Arsenic exposure may be a risk factor for Alzheimer's disease. J Neuropsychiatry Clin Neurosci 20(4):501
Yadav RS, Chandravanshi LP, Shukla RK et al (2011) Neuroprotective efficacy of curcumin in arsenic induced cholinergic dysfunctions in rats. Neurotoxicology 32:760–768
Ansari RW, Shukla RK, Yadav RS et al (2012) Cholinergic dysfunctions and enhanced oxidative stress in the neurobehavioral toxicity of lambda-cyhalothrin in developing rats. Neurotox Res 22:292–309
Sankhwar ML, Yadav RS, Shukla RK et al (2012) Impaired cholinergic mechanisms following exposure to monocrotophos in young rats. Hum Exp Toxicol 31(6):606–616
Eberhardt JL, Persson BR, Brun AE, Salford LG, Malmgren LO (2008) Blood-brain barrier permeability and nerve cell damage in rat brain 14 and 28 days after exposure to microwaves from GSM mobile phones. Electromagn Biol Med 27:215–229
Eulitz C, Ullsperger P, Freude G, Elbert T (1998) Mobile phones modulate response patterns of human brain activity. Neuroreport 9:3229–3232
Freude G, Ullsperger P, Eggert S, Ruppe I (1998) Effects of microwaves emitted by cellular phones on human slow brain potentials. Bioelectromagnetics 19:384–387
Koivisto M, Krause CM, Revonsuo A, Laine M, Hamalainen H (2000) The effects of electromagnetic field emitted by GSM phones on working memory. Neuroreport 11:1641–1643
Smythe JW, Costall B (2003) Mobile phone use facilitates memory in male, but not female, subjects. Neuroreport 14:243–246
Trosic I, Busljeta I, Modlic B (2004) Investigation of the genotoxic effect of microwave irradiation in rat bone marrow cells: in vivo exposure. Mutagenesis 19:361–364
Lee TM, Ho SM, Tsang LY et al (2001) Effect on human attention of exposure to the electromagnetic field emitted by mobile phones. Neuroreport 12:729–731
Krause CM, Sillanmaki L, Koivisto M et al (2000) Effects of electromagnetic fields emitted by cellular phones on the electroencephalogram during a visual working memory task. Int J Radiat Biol 76:1659–1667
Krause CM, Sillanmaki L, Koivisto M et al (2000) Effects of electromagnetic field emitted by cellular phones on the EEG during a memory task. Neuroreport 11(2000b):761–764
Kesari KK, Siddiqui MH, Meena R, Verma HN, Kumar S (2013) Cell phone radiation exposure on brain and associated biological systems. Indian J Exp Biol 51(3):187–200
Acknowledgements
The authors are thankful to Dr. Harisingh Gour Central University, Sagar (MP), India and SRM University, Barabanki (UP), India for providing the opportunity to work and for their support and interest. Neeraj Kumar Tiwari is also thankful to the Uttar Pradesh Council of Science and Technology (UPCST), Lucknow, India for providing the Young Scientist Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yadav, R.S., Tiwari, N.K. Lipid Integration in Neurodegeneration: An Overview of Alzheimer’s Disease. Mol Neurobiol 50, 168–176 (2014). https://doi.org/10.1007/s12035-014-8661-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8661-5