
Abstract
Postconditioning has regenerated interest as a mechanical intervention against cerebral ischemia/reperfusion injury, but its molecular mechanisms remain unknown. We previously reported that hypoxic postconditioning (HPC) ameliorated neuronal death induced by transient global cerebral ischemia (tGCI) in hippocampal CA1 subregion of adult rats. This study tested the hypothesis that p38-mitogen-activated protein kinase (p38 MAPK)/mitogen- and stress-response kinase 1 (MSK1) signaling pathway plays a role in the HPC-induced neuroprotection. Male Wistar rats were subjected to 10 min ischemia induced by applying the four-vessel occlusion method. HPC with 120 min was applied at 24 h after reperfusion. Immunohistochemistry and Western blot were used to detect the expression of phosphorylation of p38 MAPK and MSK1, as well as cleaved caspase-3. We found that HPC induced a significant increase of phosphorylated p38 MAPK and MSK1 in neurons of hippocampal CA1 region and a significant decrease in glial cells after tGCI as well. Furthermore, HPC attenuated caspase-3 cleavation triggered by tGCI in CA1 region. Moreover, p38 MAPK inhibition by SB203580 significantly decreased the phosphorylation of MSK1, increased cleaved caspase-3 expression, and abolished the neuroprotection of HPC. These findings suggested that p38 MAPK/MSK1 signaling axis contributed to HPC-mediated neuroprotection against tGCI, at least in part, by regulating the activation of caspase-3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral ischemic postconditioning is defined as a series of brief interruptions of blood flow in the early phase of ischemia/reperfusion that protect neurons from ischemia/reperfusion injury to brain. Presently, several studies have showed that delayed hypoxic postconditioning (HPC) in transient focal cerebral ischemia and in vitro studies [1], and in transient global cerebral ischemia (tGCI) [2] can provide neuroprotection to cerebral ischemia, knowing that moderate hypoxia does not cause neuronal death. Therefore, HPC seems to be safe and amenable in clinical practice. We recently confirmed that postconditioning with 8 % hypoxia for 60–120 min significantly reduced cell death in hippocampal CA1 subregion after 10 min of tGCI and that HPC was effective only when applied 1–2 days after tGCI. Also, the strongest neuroprotection was observed within 120 min of hypoxia and 1 day interval between ischemia and hypoxia [2]. However, the precise intracellular mechanisms underlying the neuroprotection of HPC to tGCI are not completely understood. Recently, evidences have showed that the signaling pathways of mitogen-activated protein kinases (MAPKs) may play important roles in regulating apoptosis, cell death, and cell survival after brain ischemia [3, 4].
Mitogen-activated protein kinase signaling pathways include extracellular signal-regulated kinases (ERKs), c-jun-N-terminal kinase (JNK), and p38 MAPK. In the brain, all these three MAPK pathways may be activated after cerebral ischemia [5]. Lately, we have demonstrated that both phosphorylation of mitogen-activated protein kinase/extra-cellular signal-regulated kinase kinase (MEK) and ERK levels increased after tGCI, whereas HPC can attenuate brain injury through blocking the increase of MEK and ERK phosphorylation [2]. As a member of MAPK family, p38 MAPK was activated under various environmental stress and inflammatory cytokines. The p38 MAPK pathway plays a crucial role in modulating the activity of many transcription factors, leading to biological responses, such as cell proliferation, differentiation, and apoptosis [6, 7]. The p38 pathway may have different roles in various phases of cerebral ischemia. However, whether p38 MAPK and its signaling cascade downstream involved in the induction of neuroprotection by HPC to tGCI remains to be well clarified.
Mitogen- and stress-activated protein kinase-1(MSK1), an identified nuclear serine/threonine kinase, is an important downstream kinase in MAPK signal transduction pathways [8]. MSK1 plays a crucial role in the regulation of gene transcription as a major neurotrophin-activated cyclic AMP response element (CRE)-binding protein (CREB) kinase [9], neuronal synaptic plasticity [10, 11], and cytokine production in the innate immune system [12]. In the heart of rats subjected to ischemic preconditioning, Nagy et al. found that SB203580, a selective inhibitor of p38 MAPK, almost completely blocked the activation of MSK1 [13], suggesting MSK1 is a key regulator of ischemic preconditioning in the heart. Nonetheless, little is known about the role of MSK1 in the brain.
Cell apoptosis has emerged as one of the future targets for neuroprotective strategies in recent years [14, 15]. Our previous study showed that apoptosis in the hippocampal CA1 subregion after tGCI could be prevented by hypoxic preconditioning [16]. Caspases are well recognized as crucial apoptosis regulators and are generally classified into two groups: the initiator caspases (including caspase-2, -8, -9, and -10) and executioner caspases (consisting of caspases-3, -6, and -7). Caspase-3 is well established as the dominant executioner caspase, and its activation ultimately leads to cell death [17]. It has been documented that activation of p38 MAPK could protect brain endothelial cells from apoptosis in stroke [18]. However, what remains unclear is whether p38 MAPK signaling pathway participates in the HPC-induced neuroprotection by downregulating activation of caspsase-3 to reduce neuronal apoptosis.
Based on the statements above, we hypothesized that p38 MAPK signaling pathway participates in the HPC-induced neuroprotection by upregulating phosphorylated MSK and downregulating activation of caspase-3. The goal of the present study is, therefore, to assess the roles of p38 MPAK and MSK1 after tGCI. Simultaneously, we tried to ascertain if the p38 MAPK/MSK1 signaling pathway contributes to the neuroprotection of HPC in tGCI for the first time.
Materials and Methods
Experiments were performed on adult male Wistar rats weighing 250–300 g (Southern Medical University, Guangdong, China). Animals were treated according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animal (Publication No. 80–23) revised 1996, and experiment protocols were approved and monitored by the Animals Care and Use committee of Guangzhou Medical University.
Transient Global Cerebral Ischemia
Transient global cerebral ischemia was induced using the four-vessel occlusion method [19]. Briefly, the animals were anesthetized with chloral hydrate (350 mg/kg, i.p.). Vertebral arteries were electrocauterized, and common carotid arteries were isolated. A teflon/silastic occluding device was placed loosely around each carotid artery without interrupting carotid blood flow. Forebrain ischemia was induced in the rats which were awake, 24 h after surgery by occluding both common carotid arteries for 10 min. After occlusion, rats that lost their righting reflex within 1 min and whose pupils were dilated were selected for experiments. Rectal temperature was maintained at 37–38 °C throughout the procedure. Sham-operated rats received the same surgical procedures except that the arteries were not occluded.
Hypoxic Postconditioning
Rats were postconditioned by expositing to a 120 min period of systemic hypoxia 24 h after tGCI. Hypoxia was performed as described previously [20]. Briefly, rats were placed in a sealed plastic chamber of 9,000 cm3 through which air containing 8 % O2 and 92 % N2 flowed continuously at the temperature of 23–25 °C. The mixed gas flow rate was 200 mL/min, and no more than three rats were placed in the same chamber at any given time. Sham-operated, hypoxia-treated rats were exposed to 120 min hypoxia 24 h after Sham-operated procedures without ischemia.
Administration of Drug
In order to investigate the role of p38 MAPK/MSK1 signaling pathway on the development of neuroprotection induced by HPC, 10 μL of SB203580 solution (Sigma, St. Louis, MO, USA. 1 mg of SB203580 in 1 mL 3 % dimethylsulfoxide sulfoxide, DMSO) or 3 % DMSO was injected intracerebroventricularly 30 min before HPC (10 μL, i.c.v., bregma: 1.5 mm lateral, 0.8 mm posterior, 4.0 mm deep). To evaluate the toxicity of SB203580 to the cells of hippocampus, six animals without hypoxia or ischemia were treated with 10 μL SB203580 intracerebroventricularly.
Histology
Animals were perfused intracardially with 0.9 % saline, followed by 4 % paraformaldehyde in phosphate buffered saline (PBS) under anesthesia. Brains were removed quickly and further fixed. Postfixed brains were immersed in 15, 30 % sucrose in the same fixative for cytoprotection. Coronal free-floating sections were cut at 30 μm using a cryotome (Thermo, Runcorn, Cheshire, UK). Sections selected from the dorsal hippocampus (between AP 4.8 and 5.8 mm, interaural or AP -3.3 to 3.4 mm, bregma) were used for Nissl staining or Fluoro-Jade B (FJ-B) staining. Nissl staining and FJ-B staining were conducted as described previously (Zhan et al., 2012). Briefly, Nissl staining was performed with 0.1 % Cresyl violet (Sigma), and then sections were dehydrated with 90 and 100 % alcohol and immersed into dimethylbenzene. For FJ-B staining, sections were immersed in 70 % ethanol, washed with distilled water, and treated with 0.06 % potassium permanganate solution for 10 min. Then, the sections were incubated with 0.004 % FJ-B (Millipore, Bedford, MA, USA) in 0.1 % acetic acid for 20 min, washed, and mounted with distrene plasticizer xylene (Sigma).
The sections from Nissl staining were examined under a light microscope (×660). Survived cells showed well-stained Nissl bodies, whereas damaged cells were either swollen with loss of stainable Nissl material or necrotic with deeply staining dendrites fragmented. Meanwhile, FJ-B stained images were observed with a fluorescence microscope (Leica Microsystems, Wetzlar, Hessen, Germany). Cell counts were conducted by densities as described previously [21]. The cells in the CA1 pyramidal layer were quantitatively analyzed within three non-repeated rectangular areas of 0.037 mm2. Data were quantified bilaterally in sections from each brain and assessed double-blindedly. Also, four sections for each animal were evaluated.
Immunohistochemistry
The animals were sacrificed at 0, 4, 26, 50, and 168 h after reperfusion with or without HPC (n = 6 in each group). Single-label immunohistochemistry was followed by the avidin-biotin-peroxidase complex (ABC) method [20]. Briefly, these sections were first treated with 3 % hydrogen peroxide for 30 min, followed by 5 % normal serum for 1 h, and then they were incubated overnight at 4 °C with primary antibodies including phospho-MSK1 (1:200; Cell Signaling Technology, Beverly, MA, USA), phospho-p38 (1:50; Cell Signaling) and cleaved caspase-3 (1:100; Cell Signaling). After three washes with 0.01 M PBS, the slides were incubated with biotinylated secondary immunoglobulin G antibody for 2 h at room temperature. After being washed with PBS, the sections were incubated with ABC for 30 min at room temperature. The peroxidase reaction was visualized with 0.05 % diaminobenzidine (DAB and 0.01 % hydrogen peroxide). Slices incubated with cleaved caspase-3 were restained with hematoxylin, and they were incubated in ammonia, dehydrated with gradient ethanol, transparentized with xylene, and finally, sealed with neutral gum. Immunopositive cells in which the reaction product was present within a clear and regular-shaped cytoplasmic or nuclear border were quantified under a light microscope (×660). The total number of immunoreactive cells was counted by the total number of four non-repeated random fields (0.037 mm2 per field × 4 = 0.148 mm2 total) in CA1 regions. Finally, the average intensity of cleaved caspase-3 fiber staining in CA1 and CA3 regions was determined using Image-Pro Plus software for Windows, version 6.0 (Media Cybernetics, Inc. Warrendale, PA, USA). Four non-repeated 200× magnification microscopic random fields (141.15 μm2 per field) in the pyramidal layer, stratum radiatum, and stratum lacunosum-moleculare of each subject were assessed in four coronal tissue sections. Measures of staining intensity of fibers were averaged across tissue sections to provide a single mean value for each structure for each rat. These mean values were used for statistical analysis.
Double-fluorescent immunohistochemistry was conducted to demonstrate cell types and the exact position that expressed phospho-p38 and phospho-MSK1. Neuronal nuclei (NeuN), microtubule-associated protein-2 (MAP-2), glial fibrillary acidic protein (GFAP) and Ox-42 were used to identify NeuN, neuronal cell bodies and dendrites, astrocytes, and microglia, respectively. Double-fluorescent immunohistochemistry was performed as described previously [20]. Antibodies used in these studies include NeuN (1:1000; Millipore, Bedford, MA, USA), MAP-2 (1:100; Millipore), GFAP (1:2,000; Millipore), OX-42 (1:50; Millipore), Cy3-conjugated goat antimouse IgG antibody (1:100; Invitrogen, Carlsbad, CA, USA), and FITC-conjugated goat anti-rabbit antibody (1:50; Invitrogen). Slides were analyzed with a confocal laser microscope (Leica Microsystems, Wetzlar, Hessen, Germany).
Western Blotting
The animals were sacrificed at 0, 4, 26, and 50 h after reperfusion with or without HPC (n = 3 in each group). The CA1 subregion protein extraction was performed as described previously [22]. Western blotting analyses were performed as described previously [23]. Primary antibodies included p38 (1:1,000), phospho-p38 (1:500; Cell Signaling), MSK1 (1:1,000; Cell Signaling), phospho-MSK1 (Thr 581) (1:500; Cell Signaling), cleaved caspase-3 (1:800; Cell Signaling), β-actin (1:2,000; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:4,000; Proteintech Group, Inc. Chicago, IL, USA). Densitometric analysis for the quantification of the bands was performed using image analysis software (Quantity One, Bio-Rad Laboratories, Inc. Hercules, CA, USA). Relative optical densities of protein bands were calibrated with β-actin or GAPDH and normalized to those in sham-operated rats.
Data Analyses
Statistical analyses were performed with the Statistical Package for Social Sciences for Windows, version 11.5 (SPSS, Inc, Chicago, Illinois, USA). Measurement data were summarized by mean ± SD, and the statistical significance was determined by one-way ANOVA or the two-tailed Student’s t-test. p < 0.05 was considered statistically significant.
Result
In the present study, 202 rats were used for experiment. Five rats in the tGCI groups and three rats in the HPC groups died during tGCI. Two rats in the tGCI groups and one rat in the HPC groups died during reperfusion. Two rats died after intracerebroventricular injection.
Phosphorylated p38 MAPK Expression in the tGCI Brain with or Without HPC
Immunohistochemistry study showed phospho-p38 MAPK staining was found mainly in the pyramidal neurons of hippocampus. Phospho-p38 MAPK-positive cells had rounded nuclei with a granular appearance in sham-operated rat brains (Fig. 1A, a and b) and double labeling revealed that these cells were NeuN-positive (Fig. 1C, a–c), indicating a predominantly neuronal localization of phospho-p38 in sham-operated brains. The number of phospho-p38 MAPK-positive neurons significantly increased immediately after tGCI in hippocampal CA1 region. After reperfusion, the peak was reached at 4 h after tGCI and the increased expression lasted for 26 h and then decreased persistently at 50 h and 168 h. Alternatively, phospho-p38 MAPK-positive cells at 50–168 h after reperfusion appeared mostly in cells with elongated, irregular nuclei (Fig. 1A, f and j) and double labeling revealed that these cells were either Ox-42 or GFAP-positive (Fig. 1C, g–l), indicating that phospho-p38 MAPK localized predominantly in microglia and astrocytes at 50–168 h after reperfusion. Compared with tGCI groups, the number of phospho-p38-positive neurons in HPC groups at 50 and 168 h of reperfusion significantly increased. Nevertheless, phospho-p38 MAPK-positive glial cells in HPC groups at 50 h increased lightly in comparison to tGCI groups, but decreased dramatically at 168 h (Fig. 1B).

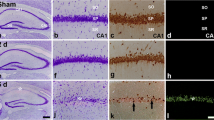
Effect of hypoxic postconditioning on p38 MAPK phosphorylation in the CA1 subregion. A Representative images of immunohistochemistry of phospho-p38 MAPK in the hippocampus after tGCI with or without HPC. The pictures on the low right are magnified from the square areas on the left. Phospho-p38 MAPK-positive neuron-like cells had rounded nuclei, and spindle cell body with elongated axon (bold arrow) and phospho-p38 MAPK-positive glia-like cells appeared mostly in cells with elaborate array of processes and irregular nuclei (arrowhead). Scale bar: a, c, e, g, i, k: 250 μm; b, d, f, h, j, l: 25 μm. Re reperfusion, Re4h 4 h after reperfusion, and HPC6d 6 days after hypoxic postconditioning. B Quantitative analysis of immunoreactive cell counting of phospho-p38 MAPK-positive neuron-like cells and phospho-p38 MAPK-positive glia-like cells in the CA1 subregion. Data are shown as mean ± S.D. *p < 0.05 vs. sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 6 in each group). C Representative photomicrographs show fluorescent double staining of phospho-p38 MAPK (a, d) and NeuN (b, e), phospho-p38 MAPK (g) and Ox-42 (h), and phospho-p38 MAPK (j) and GFAP (k) in the hippocampus after tGCI with or without HPC. The overlapped images show phospho-p38 MAPK and NeuN were almost completely overlapped in the hippocampus of sham (c) and hypoxic preconditioned rats (f). Also, phospho-p38 MAPK fluorescent overlapped either with Ox-42 (i) or GFAP (l) staining at 168 h after reperfusion in CA1 subregion. Scale bar: 150 μm. D Representative images of Western blot using either anti-phospho-p38 MAPK or anti-p38 MAPK antibody in the CA1 subregion of ischemic and hypoxic postconditioned rats. The histogram presents the quantitative analyses of p38 MAPK and phospho-p38 MAPK levels in the CA1 subregion in ischemic and hypoxic postconditioned rats. Data are expressed as percentage of value of sham-operated animals. Each bar represents the mean ± S.D. *p < 0.05 vs. sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 3 in each group)
Figure 1D illustrates the changes of p38 MAPK protein levels detected by Western blot in the rat brains after reperfusion with or without HPC. Phospho-p38 MAPK expression in hippocampal CA1 region increased transiently at 4 h then, decreased to basal expression level at 26 h and persistently at 50 h after reperfusion. Compared with tGCI groups, phospho-p38 MAPK in HPC groups dramatically increased at 24 h after hypoxia. However, total P38 was unchanged after tGCI with or without HPC.
Phosphorylated MSK1 Expression in the tGCI Brain with or Without HPC
Phospho-MSK1 staining detected by immunohistochemistry was found mainly in the pyramidal neurons of hippocampus. This protein was expressed in the cell nuclei and proximal axons of pyramidal cells and gilal cells (Fig. 2A). The general nuclear staining was positive for neuronal marker NeuN (Fig. 2C, a–c), indicating a predominant neuronal localization of phospho-MSK1 in Sham brains. After reperfusion, the number of phospho-MSK1-positive neurons in hippocampal CA1 region of tGCI brains increased transiently at 4 h and then, decreased to basal expression level at 26–50 h and persistently at 168 h. Compared with tGCI groups, the number of phospho-MSK1-positive neurons in HPC groups significantly increased immediately and continued rising at least 24 h after hypoxia. The phospho-MSK1-positive cells with an elaborate array of processes in shape were consistent with glial morphology verified as such by Ox-42 and GFAP double staining (Fig. 2C, g–l), indicating that microglias and astrocytes were phospho-MSK1 immunopositive. P-MSK1-positive glial cells in tGCI increased persistently and peaked at 168 h. Nevertheless, compared with the rats of corresponding reperfusion time points in tGCI groups, p-MSK1-positive glial cells in HPC groups decreased after hypoxia except for a transient increase at 24 h (Fig. 2B).

Effect of hypoxic postconditioning on MSK1 phosphorylation in the CA1 subregion. A Representative images of immunohistochemistry of phospho-MSK1 in the hippocampus after tGCI with or without HPC. The pictures on the low right are magnified from the square areas on the left. Phospho-MSK1-positive neuron-like cells had rounded nuclei, and spindle cell body with elongated axon (bold arrow) and phospho-p38 MAPK-positive glia-like cells appeared mostly in cells with elaborate array of processes and irregular nuclei (arrowhead). Scale bar: a, c, e, g, i, k: 250 μm; b, d, f, h, j, l: 25 μm. B Quantitative analysis of immunoreactive cell counting of phospho-MSK1-positive neuron-like cells and phospho-MSK1-positive glia-like cells in the CA1 subregion. Data are shown as mean ± S.D. *p < 0.05 vs. sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 6 in each group). C Representative photomicrographs show fluorescent double staining of phospho-MSK1 (a, d) and NeuN (b, e), phospho-MSK1 (g) and Ox-42 (h), and phospho-MSK1 (j) and GFAP (k) in the hippocampus after tGCI with or without HPC. The overlapped images show phospho-MSK1 and NeuN were almost completely overlapped in the hippocampus of sham (c) and hypoxic preconditioned rats (f). Also, phospho-MSK1 fluorescent overlapped either with Ox-42 (i) or GFAP (l) staining 168 h after reperfusion in CA1 subregion. Scale bar: 150 μm. D Representative images of Western blot using either anti-phospho-MSK1 or anti-MSK1 antibody in the CA1 subregion of ischemic and hypoxic postconditioned rats. The histogram presents the quantitative analyses of MSK1 and phospho-MSK1 levels in the CA1 subregion in ischemic and hypoxic postconditioned rats. Data are expressed as percentage of value of sham-operated animals. Each bar represents the mean ± S.D. *p < 0.05 vs. sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 3 in each group)
Western bolt analysis showed that phospho-MSK1 expression in CA1 was significantly upregulated at 26 h after tGCI compared with Sham-operated group and then, returned to baseline level at 50 h. The results indicated that activation of MSK1 was later than that of p38 MAPK in CA1 of rats after tGCI. Compared with tGCI groups, phospho-MSK1 in HPC groups increased significantly 24 h after hypoxia. However, no significant change in the levels of total MSK1 was observed in ischemic brains with or without HPC (Fig. 2D).
Cleaved Caspase-3 Expression in the tGCI Brain with or Without HPC
To confirm spatial distribution of cleaved caspase-3 after tGCI with or without HPC, immunochemistry was carried out. Cleaved caspase-3 positive staining in the hippocampal CA1 from sham-operated animals was very weak (Fig. 3A, b). At the earlier time points of reperfusion (0, 4, and 26 h), most pyramidal cells had normal morphology, and the immunoreactivity was predominantly present in the cell processes (Fig. 3A, e). However, at 7 d postischemia, the majority of cells in CA1 region showed pyknotic changes, and the cleaved caspase-3 staining exhibited mostly in nuclei and cell processes and weakly in the cytoplasm (Fig. 3A, h). As shown in Fig. 2B and C, the density of cleaved caspase-3 positive fiber in CA1 region from tGCI groups began to increase at 4 h, and it reached its maximum at 26 h and returned to the sham level at 50 h postischemia. However, HPC eliminated the changes in the density of cleaved caspase-3 fiber in CA1 region observed after tGCI, and the density of cleaved caspase-3 fiber in the HPC groups did not significantly change compared with the sham-operated group. Furthermore, the number of cleaved caspase-3 positive nucleus increased persistently up to 26 h after tGCI with or without HPC, and the expression of cleaved caspase-3 in nucleus from the HPC groups at 50 h and 168 h of reperfusion were significantly lower in comparison with the tGCI groups. We also noticed that immunoreactivity of cleaved caspase-3 was exclusively localized in the stratum lucidum (mossy fibers layer) of the CA3 subregion (Fig. 3A, c,f,i), and no significant change in the density of cleaved caspase-3 fiber was observed in ischemic brains with or without HPC (Fig. 3D).

Effect of hypoxic postconditioning on cleaved caspase-3 in the CA1 subregion. A Immunohistochemistry for cleaved caspase-3 in the rat brains of 26 and 168 h after tGCI and sham operation. Details from the CA1 region (b, e, h) showing the immunoreactivity of cleaved caspase-3 in the cell processes (bold arrow) or in nuclei (arrowhead). Details from the CA3 region (c, f, i) showing intense staining in fibers. Scale bar: a, d, g: 250 μm; b, c, e, f, h, i: 25 μm. B Quantitative analysis of cleaved caspase-3 positive fiber density in the CA1 subregion. C Quantitative analysis of cleaved caspase-3 positive nucleus counting in the CA1 subregion. D Quantitative analysis of cleaved caspase-3 positive fiber density in the CA3 subregion. Data are shown as mean ± S.D. *p < 0.05 vs. sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 6 in each group). E Representative images of Western blot using anti-cleaved caspase-3 antibody in the CA1 subregions of ischemic and hypoxic preconditioned rats. The histogram presents the quantitative analyses of cleaved caspase-3. Data are expressed as percentage of value of sham-operated animals. Each bar represents the mean ± S.D. *p < 0.05 vs. sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 3 in each group)
Western bolt analysis showed that cleaved caspase-3 expression in CA1 region increased persistently after tGCI with or without HPC. Compared with the tGCI groups, the expression of cleaved caspase-3 in CA1 from the HPC groups significantly decreased at 24 h after HPC (Fig. 3E).
SB203580 Downregulated the Expression of Phosphorylated p38 MAPK and MSK1 and Abolished the Neuroprotection Effect of HPC
In order to ascertain whether p38 MAPK/MSK1 signaling pathway contributes to the neuroprotection induced by HPC, experiments with SB203580 were carried on. Western blot analysis was shown in Fig. 4C and D. The results showed that the activation (phosphorylated/non-phosphorylated) of p38 MAPK and MSK1 in CA1 region 24 h after HPC was significantly inhibited by prior intraventricular administration of SB203580 30 min before HPC compared with those of the HPC group or vehicle group. Delayed neuronal death was evaluated on day 7 after ischemia with or without SB203580 treatment. No significant neuronal loss was detected in CA1 region of rats subjected to sham operation (Fig. 4A, a–d), while the obvious neuronal destruction and loss of pyramidal neurons in CA1 region of the tGCI group were noticed (Fig. 4A, e–h), with surviving cells and NeuN-positive cells significantly decreased, and FJ-B-positive cells significantly increased at meanwhile (Fig. 4B). In the rats of HPC group, surviving cells and NeuN-positive cells significantly increased and FJ-B-positive cells significantly decreased compared with the tGCI group (Fig. 4B). Simple intraventricular administration of SB203580 in sham-operated rats had no neurotoxic effects on the pyramidal neurons of the hippocampus (Fig. 4A, m–p). However, intraventricular administration of SB203580 significantly inhibited the protection of HPC against neuronal damage induced by tGCI (Fig. 4A, u–x). The vehicle group, in which 3 % DMSO was also administrated in the same way did not show obvious changes in the histological characteristics compared with HPC group (Fig. 4A, q–t). To explore whether activation of caspase-3 was associated with p38 MAPK signaling pathway inactivation, we further observed the effect of SB203580 on caspase-3 activation in CA1 region after HPC. We found that the protein level of cleaved caspase-3 in SB203580 + HPC group increased remarkably compared with HPC group or vehicle group (Fig. 4E).

Effect of SB203580 treatment on phosphorylation of protein kinases, cleaved caspase-3, and neuronal cells damage in CA1 subregion after tGCI with or without postconditioning. A Representative microphotographs of cresyl violet staining, immunostaining of NeuN, and FJ-B staining in the hippocampus at 7 days after tGCI with or without SB203580 treatment. Sham group (a–d); tGCI group (e–h); tGCI + hypoxia group (i–l); sham + SB group (m–p), infusion with SB203580 without ischemia or hypoxia; tGCI + vehicle + hypoxia group, tGCI with vehicle infusion before HPC (q–t); tGCI + SB + hypoxia group, tGCI with SB203580 infusion before HPC (u–x). Scale bar: a, e, i, m, q: 250 μm; b–d, f–h, j–l, n–p, r–t: 25 μm. B Quantitative analyses of neurons survival, NeuN-positive cells, and FJ-B-positive cells in CA1 subregion. Each bar represents the mean ± S.D. *p < 0.05 vs. sham-operated animals, # p < 0.05 vs. tGCI group, and &p < 0.05 vs. tGCI + hypoxia group. p38 MAPK inhibitor, SB203580 inhibited the activation (phosphorylated/non-phosphorylated) of p38 MAPK (C) and MSK1 (D), and increased cleaved caspase-3 (e) in CA1 subregion at 24 h after HPC. Each bar represents the mean ± S.D. *p < 0.05 vs. sham animals and # p < 0.05 vs. tGCI group and &p < 0.05 vs. tGCI + hypoxia group (n = 6 in each group)
Taken together, the results indicated that SB203580 can weaken the protection of HPC against neuronal damage induced by tGCI.
Discussion
In the current study, HPC induced significant increases of p-p38 MAPK expression after tGCI in the rat hippocampal CA1. The role of p38 MAPK in brain ischemia remains highly controversial at present. Some studies showed that activation of p38 MAPK might facilitate neuronal death after brain ischemic insult [24, 25]. On the contrary, the others indicated the activation of p38 MAPK protected neurons from ischemic stimulation [26, 27]. These contradictory results might be related to several reasons as follows. One of the reasons is the difference in the time of the phosphorylated p38 MAPK expression. Lethal brain ischemic insult induced upregulation of phosphorylated p38 MAPK early and transiently. However, sublethal brain stimulation induced upregulation of phosphorylated p38 MAPK relatively later and persistently. In our studies, upregulation of phosphorylated p38 MAPK in the CA1 subregion increased transiently at 4 h after reperfusion, while in the HPC groups, it initiated increase at 24 h and lasted for 144 h after hypoxia. Another reason is the distinct cell types that expressed of the p38 MAPK. Microglia and astrocytes in hippocampal CA1 region were activated after tGCI [28]. Our present results demonstrated that phosphorylated p38 MAPK levels increased in glias after tGCI. The increase in phosphorylated p38 MAPK levels in microglia could be involved in microglial activation after global forebrain ischemia [29]. After activation of microglia and astrocytes, neurotoxic substances were released. They may contribute to the delayed cell death after ischemia [29, 30]. Third, the distinct isoforms of p38 MAPK may have different functions in brain after ischemia. There are four different p38 MAPK isoforms, even as p38α, p38β, p38γ, and p38δ. Both p38α and p38β have highly similar three-dimensional configuration. These two different isoforms serve as the major forms of p38 MAPK in the brain [31]. p38α was detected mainly in neurons, while p38β expression in glia cells was most evident in CA1 region of hippocampus and in red nucleus [32]. A recent study showed that p38α inhibition or deficiency promoted macrophage apoptosis, suggesting that p38α plays a prosurvival role [33]. However, p38β, expressed with a notable expression level in glia cells in many brain regions, may play a role in inflammation in pathological conditions [32]. In the previous study, exposure to moderate hypoxia (5 % O2) progressively stimulated phosphorylation and activation of p38α. However, hypoxia had no effect on enzyme activity of p38β [34]. In our present study, phosphorylated p38 MAPK increased significantly in neurons of hippocampal CA1 region after HPC. Therefore, we speculated that continuous activation of p38 MAPK in neurons, and probably activation of p38α, could provide neuroprotection. HPC induced neuroprotection against tGCI via continuous activation of p38 MAPK in neurons, probably activation of p38α, and downregulation of p38 MAPK in glias, and probably downregulation of p38β.
Mitogen- and stress-response kinase has been shown to be activated via both ERKs and p38 MAPK pathways [35]. MSK activation depends largely on the nature of the stimuli. Growth factors appear to activate MSKs predominantly through the ERK pathway, whereas stress such as ischemia appears to signal via the p38 MAPK cascade [8]. As indicated by the Western blot experiments, our results showed that cerebral ischemia activated phospho-p38 MAPK as early as 4 h after reperfusion, and the phosphorylation of MSK1 occurred with delayed kinetics when compared with p38 MAPK. This suggested that p38 MAPK might be responsible for the activation of MSK1 in CA1 region after tGCI. Furthermore, consistent with the expression of phospho-p38 MAPK, tGCI-induced MSK1 activation was located predominantly in neurons at early stages (from 0 to 26 h after reperfusion) and predominantly in glial cells at late stages (from 50 to 168 h after reperfusion). Also, HPC increased the expression of phospho-MSK1 in neurons after tGCI. At last, we investigated the phosphorylation of p38 MAPK and MSK1 in the presence of inhibitor to ascertain the involvement of p38 MAPK in MSK1 activation by HPC. MSK1 phosphorylation was prevented in the presence of the p38 MAPK-selective inhibitor SB203580. These data strongly suggested that under our experiment conditions, the activation of MSK1 relied on p38 MAPK activation.
The detailed mechanism about the effects of p38/MSK1 signaling pathway on HPC still unknown. p38/MSK1 signaling pathway may mediate its neuroprotective effects through phosphorylation of effector proteins, notably transcription factors which in turn regulate cell survival. MSK1 directly targeted transcription factors, such as CREB and nuclear factors-κB, thereby enhancing their transcriptional activity. In vitro, MSK1 was far more efficient in phosphorylating CREB at Ser 133 than other kinases [8]. Also, CREB was the first in vivo substrate of MSK1 to be identified. It has been shown that carotid artery occlusion preconditioning before hypoxia was characterized by a robust and sustained phosphorylation of CREB which provided complete neuroprotection in rats [36]. Consistent with these results, it has been reported that CREB phosphorylation acted as a trigger in the neuroprotection mediated by ischemic preconditioning. This was associated with a significant increase in B-cell lymphoma 2 (Bcl-2) expressions and subsequent protection against ischemia [37]. However, these studies did not involve whether the neuroprotection of CREB was mediated through p38/MSK1 signaling pathway. Previous studies have indicated that the activation of p38/MSK1 signaling pathway resulted in the phosphorylation of CREB which may have mediated cardioprotection after ischemic preconditioning [13]. Therefore, it is possible that the phosphorylation of CREB induced by HPC via activation of p38/MSK1 signaling pathway can increase the expression of Bcl-2, leading to cell survival after tGCI. Further experiments will be required to investigate this interesting mechanism.
We previously reported that apoptotic DNA fragment significantly increased in the hippocampal CA1 subregion at 24–48 h postischemia [16]. In this study, we found that caspase-3 activation in the CA1 region preceded the DNA fragmentation, which suggests that the apoptotic process was involved in the delayed neuronal death. Furthermore, our results indicated that tGCI induced at least two distinct phases of caspase-3 activation in the hippocampus: an early phase in which cleaved caspase-3 was localized mostly in dendrites at 4–26 h postischemia, and a late phase in which cleaved caspase-3 was found predominantly in nuclei and weakly in the cytosol. The finding that localization of the activated process is drastically altered over time indicated that the activation of caspase-3 following ischemia is not simply an event within a single cell but rather part of a pathological signal transduction involving cell-to-cell interactions. Given the cytoarchitecture of neurons in which processes reach out for a long distance, it is possible that the activation of apoptotic program might have differential effects that depend on spatial subcellular localization of the activated caspase-3. Specifically, neurons in which caspase-3 is initially activated in the nucleus will be more likely to exhibit oligonucleosomal DNA degradation, while neurons in which caspase-3 is initially activated in a distant process will be more likely to exhibit dendritic and axonal cytoskeletal proteolysis. Further experiments will be required to investigate this interesting possibility.
A role for p38 MAPK in caspase-3 activation has been reported in human neutrophils [38]. However, the mechanism of this activation is still poorly understood. p38 MAPK might regulate caspase-3 activation through direct molecular interaction. In neutrophils, p38 MAPK can directly phosphorylate and inhibit caspase-3 activity and apoptosis [38]. Another possibility is that p38 MAPK might phosphorylate and activate its substrate, such as MSK1 that interact with and activate caspase-3. In this study, we found that phosphorylated p38 MAPK was significantly increased after ischemia, whereas the activation of caspase-3 displayed lower activity. In addition, HPC increased the expression of phosphorylated p38 MAPK and MSK1 and blocked the activation of caspase-3. Furthermore, after p38 MAPK inhibitor SB203580 was administrated, the expression of phosphorylated p38 MAPK and MSK1 was significantly inhibited, and the activation of caspase-3 and neuronal damage was increased. Therefore, the present findings suggested that activated p38 MAPK/MSK1 pathway could add a chance of survival after HPC by keeping the activity of caspase-3 at low levels.
In summary, our study demonstrated that HPC could potentiate the survival pathway with p38 MAPK as the principal regulator to induce neuroprotection to tGCI in adult rats. To the best of our knowledge, we demonstrated that HPC mediated activation of its downstream target MSK-1 in tGCI brain for the first time. Our data also suggested that p38 MAPK/MSK1 signaling axis contributed to HPC-meditated neuroprotection, at least in part, through regulating the activation of caspase-3. To understand the mechanisms induced by hypoxia that led to neuroprotection against cerebral ischemia would be of very important for identifying potential novel therapeutic targets in cerebral ischemia/reperfusion injury.
References
Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M, Roussel S, Schumann-Bard P, Bernaudin M (2009) Delayed hypoxic postconditoning protects against cerebral ischemia in the mouse. Stroke 40:3349–3355
Zhan L, Li D, Liang D, Wu B, Zhu P, Wang Y, Sun W, Xu E (2012) Activation of Akt/FoxO and inactivation of MEK/ERK pathways contribute to induction of neuroprotection against transient global cerebral ischemia by delayed hypoxic postconditioning in adult rats. Neuropharmacology 635:873–882
Irving EA, Bamford M (2002) Role of mitogen-and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab 22:631–647
Ferrer I, Friguls B, Dalfó E, Planas AM (2003) Early modifications in the expression of mitogen-activated protein kinases (MAPK/ERK), stress-activated kinases SAPK/JNK and p38, and their phosporylated substrates following focal cerebral ischemia. Acta Neuropathol 105:425–437
Lennmyr F, Karlsson S, Gerwins P, Ata KA, Terént A (2002) Activation of mitogen-activated protein kinases in experimental cerebral ischemia. Acta Neurol Scand 106:333–340
Zarubin T, Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15:11–18
Kato N, Matsumoto M, Kogawa M, Atkins GJ, Findlay DM, Fujikawa T, Oda H, Ogata M (2013) Critical role of p38MAPK for regeneration of the sciatic nerve following crush injury in vivo. J Neuroinflammation 10:1
Deak M, Clifton AD, Lucocq LM, Alessi DR (1998) Mitogen-and stress-activated protein kinase-1(MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17:4426–4441
Arthur JS, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, Impey S (2004) Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci 24:4324–4332
Putignano E, Lonetti G, Cancedda L, Ratto G, Costa M, Maffei L, Pizzorusso T (2007) Developmental downregulation of histone pasttranslational modifications regulates visual cortical plasticity. Neuron 53:747–759
Karelina K, Hansen KF, Choi YS, DeVries AC, Arthur JS, Obrietan K (2012) MSK1 regulates environmental enrichment-induced hippocampal plasticity and cognitive enhancement. Learn Mem 19:550–560
Arthur JS (2008) MSK activation and physiological roles. Front Biosci 13:5866–5879
Nagy N, Shiroto K, Malik G, Huang CK, Gaestel M, Abdellatif M, Tosaki A, Maulik N, Das DK (2007) Ischemic preconditioning involves dual cardio-protective axes with p38MAPK as upstream target. J Mol Cell Cardiol 42:981–990
Bielewicz J, Kurzepa J, Lagowska-Lenard M, Bartosik-Psujek H (2010) The novel views on the patomechanism of ischemic stroke. Wiad Lek 63:213–220
Ferrer I (2006) Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis 21(Suppl 2):9–20
Zhan L, Peng W, Sun W, Xu E (2011) Hypoxic preconditioning induces neuroprotection against transient global ischemia in adult rats via preserving the activity of Na+/K +−ATPase. Neurochem Int 59:65–72
Slee EA, Adrain C, Martin SJ (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 276:7320–7326
Pfeilschifter W, Czech B, Hoffmann BP, Sujak M, Kahles T, Steinmetz H, Neumann-Haefelin T, Pfeilschifter J (2010) Pyrrolidine dithiocarbamate activates p38 MAPK and protects brain endothelial cells from apoptosis: a mechanism for the protective effect in stroke? Neurochem Res 35:1391–1401
Pulsinelli WA, Brierley JB (1979) A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 10:267–272
Zhan L, Wang T, Li W, Xu ZC, Sun W, Xu E (2010) Activation of Akt/FoxO signaling pathway contributes to induction of neuroprotection against transient global cerebral ischemia by hypoxic pre-conditioning in adult rats. J Neurochem 114:897–908
Wang Y, Zhan L, Zeng W, Li K, Sun W, Xu ZC, Xu E (2011) The effect of GABA on the hypoxia-induced increase of epilepsy susceptibility in neonate rat. Neurochem Res 36:2409–2416
Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y (2001) Activation of Akt/protein kinase B contribute to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab 21:351–360
Endo H, Nito C, Kamada H, Nishi T, Chan PH (2006) Activation of the Akt/GSK 3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rat. J Cereb Blood Flow Metab 26:1479–1489
Wu DC, Ye W, Che XM, Yang GY (2000) Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab 20:1320–1330
Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL, Parsons AA (2001) Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev 21:129–145
Lennmyr F, Ericsson A, Gerwins P, Ahlström H, Terént A (2003) Increased brain injury and vascular leakage after pretreatment with p38-inhibitor SB203580 in transient ischemia. Acta Neurol Scand 108:339–345
Nishimura M, Sugino T, Nozaki K, Takagi Y, Hattori I, Hayashi J, Hashimoto N, Moriguchi T, Nishida E (2003) Activation of p38 kinase in the gerbil hippocampus showing ischemic tolerance. J Cereb Blood Flow Metab 23:1052–1059
Zhan L, Yan H, Zhou H, Sun W, Hou Q, Xu E (2013) Hypoxic preconditioning attenuates neuronal cell death by preventing MEK/ERK signaling pathway activation after transient global cerebral ischemia in adult rats. Mol Neurobiol 48:109–119
Walton KM, DiRocco R, Bartlett BA, Koury E, Marcy VR, Jarvis B, Schaefer EM, Bhat RV (1998) Activation of p38MAPK in microglia after ischemia. J Neurochem 70:1764–1767
Bhat NR, Zhang P, Lee JC, Hogan EL (1998) Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci 18:1633–1641
Cuadrado A, Nebreda AR (2010) Mechanisms and functions of p38 MAPK signalling. Biochem J 429:403–417
Lee SH, Park J, Che Y, Han PL, Lee JK (2000) Constitutive activity and differential localization of p38alpha and p38beta MAPKs in adult mouse brain. J Neurosci Res 60:623–631
Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA (2009) Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest 119:886–898
Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-Johnson D (1999) Selective activation of p38alpha and p38gamma by hypoxia. Role in regulation of cyclin D1 by hypoxia in PC12 cells. J Biol Chem 274:23570–23576
Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344
Lee HT, Chang YC, Wang LY, Wang ST, Huang CC, Ho CJ (2004) cAMP response element-binding protein activation in ligation preconditioning in neonatal brain. Ann Neurol 56:611–623
Meller R, Minami M, Cameron A, Impey S, Chen D, Simon J-QP (2005) CREB-mediated Bcl-2 protein expression after ischaemic preconditioning. J Cerebr Blood flow Metab 25:234–246
Alvarado-Kristensson M, Melander F, Leandersson K, Rönnstrand L, Wernstedt C, Andersson T (2004) p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med 199:449–458
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81371303 and 81100880) and National Research Foundation for the Doctoral Program of Higher Education of China (Grant No. 20124423110002). Our sincere thanks go to Peifeng DU (Institute for Standardization of Nuclear Industry) for editing this paper.
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhu P and Zhan L contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhu, P., Zhan, L., Zhu, T. et al. The Roles of p38 MAPK/MSK1 Signaling Pathway in the Neuroprotection of Hypoxic Postconditioning Against Transient Global Cerebral Ischemia in Adult Rats. Mol Neurobiol 49, 1338–1349 (2014). https://doi.org/10.1007/s12035-013-8611-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8611-7