
Abstract
Our previous data indicated that hypoxic preconditioning (HPC) ameliorates transient global cerebral ischemia (tGCI)-induced neuronal death in hippocampal CA1 subregion of adult rats. However, the possible molecular mechanisms for neuroprotection of this kind are largely unknown. This study was performed to investigate the role of the mitogen-activated protein kinase/extra-cellular signal-regulated kinase kinase (MEK)/extra-cellular signal-regulated kinase (ERK) pathway in HPC-induced neuroprotection. tGCI was induced by applying the four-vessel occlusion method. Pretreatment with 30 min of hypoxia applied 1 day before 10 min tGCI significantly decreased the level of MEK1/2 and ERK1/2 phosphorylation in ischemic hippocampal CA1 subregion. Also, HPC decreased the expression of phosphorylated ERK1/2 in degenerating neurons and astrocytes. However, the administration of U0126, a MEK kinase inhibitor, partly blocked MEK1/2 and ERK1/2 phosphorylation induced by tGCI. Meanwhile, neuronal survival was improved, and glial cell activation was significantly reduced. Collectively, these data indicated that the MEK/ERK signaling pathway might be involved in HPC-induced neuroprotection following tGCI. Also, HPC resulted in a reduction of glial activation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transient episodes of nonlethal pretreatments confer profound protection on the neurons in response to a prolonged lethal episode of ischemia–reperfusion, a phenomenon that has been termed preconditioning or ischemic tolerance [1]. We recently demonstrated that hypoxic preconditioning (HPC) for 30–120 min significantly reduced cell death in the CA1 subregion after 10 min of transient global cerebral ischemia (tGCI). HPC was effective only when applied 1–4 days before ischemia. The maximum protection was observed with 30 min of hypoxia and 1-day interval between hypoxia and tGCI [2]. However, the possible molecular mechanisms by which HPC intervenes in transient global ischemia-induced cell death are not fully understood. The signaling pathways of phosphoinositide 3-kinase (PI3K)/Akt [3, 4] and extracellular signal-regulated kinase (ERK) 1/2 [5] may play important roles in regulating apoptosis after brain ischemia. Interestingly, we recently demonstrated that HPC activated phosphoinositide 3-kinase (PI3K)/Akt signaling pathway which may mediate ischemic tolerance after tGCI [2]. Nonetheless, the role of protein kinases involved in the ERK signaling pathway in HPC is unknown.
As is known, mitogen-activated protein kinase (MAPK) pathways are crucial signal transduction cascades that transmit and integrate extracellular signals to mount an appropriate response. Among the subgroups of MAPKs, the ERK1/2, phosphorylated by MAPK/ERK1 and 2 (MEK1/2) in response to growth factors, is believed to be associate with cell survival, proliferation, and differentiation [6, 7]. However, emerging evidence suggested that the activation of ERK1/2 may lead to neuronal death [8, 9], and the inhibition of MEK/ERK pathway can provide neuroprotection against oxidative stress [10, 11] and spinal cord ischemia/reperfusion (I/R) injury [12]. In vivo and in vitro studies of cerebral ischemia models had demonstrated that MEK/ERK pathway was involved in regulating brain cell death and survival after ischemia [13, 14]. Despite these findings, the role of the MEK/ERK pathway in cerebral ischemia/reperfusion (I/R) injury still needs to be explored. Besides, whether ERK1/2 is involved in the mechanisms of neuroprotection brought about by HPC is unknown.
Therefore, in this study, we sought to clarify the role of the MEK/ERK pathway in HPC-induced neuroprotection. We hypothesized that this pathway is involved in mediating astrocytes recruitment and microglia activation after transient global ischemia. We also examined whether the inhibition of this pathway with selective inhibitor U0126 could diminish glia cells activation, have a similar effect as hypoxia preconditioning, and provide neuroprotection.
Materials and Methods
Experiments were performed on adult male Wistar rats weighing 250∼300 g (Southern Medical University, Guangdong, China). Rats were treated in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23, Revised 1996). The Guangzhou Medical College Committee on Use and Care of Animals closely monitored the experiments to ensure compliance with the NIH regulations. All efforts were made to reduce the number of animals used and minimize animal suffering.
Hypoxic Preconditioning
Preconditioning was conducted by expositing rats to a 30-min period of systemic hypoxia 24 h before tGCI. Hypoxia was induced as described previously [2]. Briefly, rats were placed in a sealed plastic chamber of 9,000 cm3 through which air containing 8 % O2 and 92 % N2 flowed continuously at room temperature. Total gas flow was 200 mL/min, and no more than three rats were placed in the chamber at any given time.
Transient Global Ischemia
Transient global ischemia was induced with the four-vessel occlusion method [15]. Briefly, the animals were anesthetized with chloral hydrate (350 mg/kg, i.p.). Vertebral arteries were electrocauterized, and common carotid arteries were isolated. Forebrain ischemia was induced in awake rats 24 h after surgery by occluding both common carotid arteries for 10 min. After occlusion, rats that had lost righting reflex within 1 min and whose pupils were dilated were selected for experiments. Rectal temperature was maintained at 37∼38 °C throughout the procedure. Sham-operated rats were performed with the same surgical procedures except that the arteries were not occluded.
Histology
Seven days after ischemia, rats were perfused intracardially with normal saline, followed by 4 % paraformaldehyde in PBS under anesthesia. The brains were removed quickly and further fixed with the same solution at 4 °C overnight. Post-fixed brains were immersed in 15 %, 30 % sucrose in the same fixative for cytoprotection, and were cut into 30-μm-thick slices using a cryotome (Thermo, Runcorn, Cheshire, UK). Sections selected from the dorsal hippocampus (between AP 4.8 and 5.8 mm, interaural or AP −3.3 to 3.4 mm, Bregma) were used for Nissl staining or Fluoro-Jade B (FJ-B) staining.
Nissl staining was performed with 0.1 % Cresyl violet (Sigma, St. Louis, MO, USA) according to the standard procedure. FJ-B staining was used to label degenerating cells [16]. Briefly, sections were immersed in 70 % ethanol, washed with distilled water, and treated with 0.06 % potassium permanganate solution for 10 min. Next, the sections were incubated with 0.004 % Fluoro-Jade B (Millipore, Bedford, MA, USA) in 0.1 % acetic acid for 20 min at 25 °C, then washed and mounted with distrene plasticizer xylene (Sigma).
The sections from Nissl staining were examined under a light microscope (×660). Survived cells showed well-stained Nissl bodies, whereas damaged cells were either swollen with loss of stainable Nissl material or necrotic with deeply staining dendrites fragmented. Meanwhile, FJ-B stained images were analyzed with a fluorescence microscope (Leica Microsystems, Wetzlar, Hessen, Germany). Cell counts were conducted by densities as described previously [17]. The cells in the CA1 pyramidal layer were quantitatively analyzed within three non-repeated rectangular areas of 0.037 mm2. Data were quantified bilaterally in sections from each brain and assessed double-blindedly. Besides, four sections for each animal were evaluated.
Immunohistochemistry
Rats were sacrificed 0, 24, 48, and 168 h after reperfusion or at 24 h after hypoxia (n = 6 in each group). Single-label, free-floating immunohistochemistry was performed as described previously [2]. The primary antibodies used in the studies include neuronal nuclei (NeuN, 1:2,000; Chemicon, Temecula, CA, USA), complement receptor type 3 (OX-42, 1:400; Chemicon), glial fibrillary acidic protein (GFAP, 1:2,000; Chemicon), phospho-MEK1/2 (Ser217/221) (1:400; Cell Signaling Technology, Beverly, MA, USA), and phospho-ERK1/2 (Thr202/Tyr204) (1:500; Cell Signaling). Immunopositive cells in which the reaction product was present within a clear and regular-shaped cytoplasmic border were quantified under a light microscope (×660). The total number of immunoreactive cells was counted by total number of four non-repeated random fields (0.037 mm2 per field × 4 = 0.148 mm2 total) in the CA1 subregions.
Western Blotting
Rats were sacrificed at 0, 24, and 48 h after reperfusion or at 24 h after hypoxia, (n = 3 in each group). The CA1 subregion protein extraction was performed as described previously [3]. Protein concentration was determined through BCA method, as recommended by the manufacturer (Beyotime, Jiangsu, China). To apply Western blotting analysis, 50 μg proteins of each sample were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10 % acrylamide gels, and then transferred to PVDF membranes (Millipore). Western blotting analyses were performed as described previously [18]. Primary antibodies included phospho-MEK1/2 (1:1,000; Cell Signaling) and phospho-ERK1/2 (1:1,000; Cell Signaling). For normalization, the membrane was stripped and reprobed with antibodies against MEK (1:1,000; Cell Signaling), ERK1/2 (1:1,000; Cell Signaling), and β-actin (1:1,000; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Densitometric analysis for the quantification of the bands was performed using image analysis software (Quantity One, Bio-Rad Laboratories, Inc., Hercules, CA, USA). Relative optical densities of protein bands were calibrated with β-actin and normalized to those in Sham-operated rats.
Drug Injection
U0126 [5 mmol/L in 25 % dimethyl sulfoxide in 0.01 M phosphate buffer saline (PBS, pH 7.4); Sigma] [19, 20] or the vehicle (25 % dimethyl sulfoxide in PBS) was injected intracerebroventricularly 24 h before tGCI (10 μL, i.c.v., Bregma: 1.5 mm lateral, 0.8 mm posterior, 4.0 mm deep).
Data Analyses
Statistical analyses were performed with the Statistical Package for Social Sciences for Windows, version 11.5 (SPSS, Inc., Chicago, IL, USA). Measurement data were summarized by mean ± SD. One-way analyses of variance (ANOVA), followed by LSD post hoc test and two-way ANOVA, followed by Student–Newman–Keuls post hoc test were used in this study. p < 0.05 was considered statistically significant.
Results
Effects of Hypoxic Preconditioning on tGCI-Induced Delayed Neuronal Damage in the Hippocampal CA1 Subregion
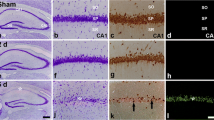
The neuronal loss in hippocampal CA1 subregion was histopathologically evaluated 7 days after 10 min of tGCI with or without HPC by Nissl staining, NeuN immunostaining, and FJ-B staining. As indicated in Fig. 1A, rats in Sham control group showed no histopathological abnormalities, while massive damage of neurons either swollen with loss of stainable Nissl material or necrotic with deeply staining dendrites fragmented were observed in CA1 subregion of tGCI rats. HPC significantly increased the number of surviving cells with palely stained nuclei and intact Nissl substance in comparison to the tGCI group. Similar results were also obtained by NeuN immunostaining. The quantitative analysis demonstrated that preconditioning with hypoxia in tGCI rats caused a marked elevation in the average number of viable CA1 pyramidal neurons compared with tGCI group (p < 0.05, Fig. 1B, C). Moreover, as expected, significant effects among groups were also observed in the number of degenerating cells detected by FJ-B staining. The increase in degenerating cells induced by tGCI was attenuated by HPC at 7 days after reperfusion (p < 0.05, Fig. 1D).

Effects of hypoxic preconditioning on neuronal damage in CA1 subregion. (A) Representative microphotographs of cresyl violet staining, NeuN staining, and Fluoro-Jade B (FJ-B) staining in the hippocampus at 7 days after transient global cerebral ischemia (tGCI) with or without hypoxic preconditioning (HPC) in rats. The pictures on the right are magnified from the square areas on the left. Scale bar: a–c, 250 μm; d–l, 25 μm. Sham group (a, d, g, j; n = 7). tGCI group: histology was performed 7 days after 10 min of tGCI (b, e, h, k; n = 7). Pre group: histology was performed 7 days after reperfusion in rats subjected to 30 min of HPC 1 day before tGCI (c, f, i, l; n = 9). Quantitative analyses of viable neurons and degenerating neurons in CA1 subregion. The bar graphs present quantitative analysis of survival cells (B), NeuN-positive cells (C), and FJ-B-positive cells (D) in CA1 subregion. (E) Representative immunoreactive cells of GFAP and OX-42 in the hippocampus at 7 days after tGCI with or without HPC in rats. Quantitative analyses of GFAP-positive cells and OX-42-positive cells in CA1 subregion. The bar graphs present quantitative analysis of GFAP-positive cells (F) and OX-42-positive cells (G) in CA1 subregion. Data are mean ± SD. *p < 0.05 vs. Sham-operated group and # p < 0.05 vs. tGCI group. Pre hypoxic preconditioning
Glial activation in hippocampal CA1 subregion was evaluated 7 days after tGCI with or without HPC by immunostaining with GFAP and OX-42. In the Sham group, microglial cells immunostained with the OX-42 antibody were scattered and had a ramified form, indicating an inactivate state (Fig. 1E(d and d′)). In tGCI group, microglial cells in the CA1 subregion were condensed and hypertrophied, indicating an active state (Fig. 1E(e and e′)). The number of activated microglial cells was significantly decreased by the pretreatment with hypoxia (Fig. 1G). The activated astrocytes, which were positively immunostained with the anti-GFAP antibody, were significantly increased in the CA1 subregion of tGCI group compared with Sham group. The pretreatment with hypoxia significantly reduced the number of activated astrocytes (Fig. 1F).
Effects of Hypoxic Preconditioning on Phosphorylation of MEK1/2
Figure 2 illustrates the changes of MEK1/2 protein levels in hippocampal CA1 subregion after 0, 4, 24, 48, and 168 h of tGCI with or without HPC. Immunohistochemistry analysis showed that most of the immunopositive neuron-like cells were seen in cortical neuronal layer (Fig. 2A(a–c)) and subcortical regions, including thalamus (Fig. 2A(d–f)), the molecular layer (Fig. 2A(g–i)), and pyramidal cell layer of hippocampus (Fig. 2A(j–l)). The phosphorylated MEK1/2 protein was expressed in neuronal cell bodies, neurofibrils, and extensive dendritic arborizations. In addition, clear immunopositive staining was observed in the axon hillocks and proximal axons of pyramidal neurons. Double-labeled immunofluorescence studies revealed that phospho-MEK1/2 was colocalized with MAP-2 in cell bodies surrounding NeuN-positive cells (Fig. 2B). No colocalization of phospho-MEK1/2 with either GFAP or OX-42 (data not shown) was found. These results showed that MEK1/2 phosphorylation occurred in neuronal cell bodies and dendritic arborizations after tGCI with or without hypoxia, instead of astrocytes or microglia. The quantitative analysis revealed that a few phosphorylated MEK1/2 immunopositive stained cells were detected in Sham-operative brain sections. MEK1/2 phosphorylation increased after 24 h of reperfusion in tGCI rats. Moreover, the levels of phosphorylated MEK1/2 expression in the HPC groups at 0, 4, 24, 48, and 168 h of reperfusion was significantly lower in comparison with the tGCI groups (Fig. 2).

Effect of hypoxic preconditioning on MEK1/2 phosphorylation in CA1 subregion. (A) Immunohistochemistry for phospho-MEK1/2 in the rat brains 24 h after 10 min of tGCI and Sham operation. Phospho-MEK1/2-positive cells were observed mainly in cortical neuronal layer (a–c) and subcortical regions, including thalamus (d–f) and the molecular layer (g–i) and pyramidal cell layer of hippocampus (j–l). Phosphorylated MEK1/2 protein was expressed in neuronal cell bodies (arrowhead), neurofibrils, and extensive dendritic arborizations (bold arrow). In addition, clear immunopositive staining was observed in axon hillocks and proximal axons of pyramidal neurons (light arrow). Scale bar: a, d, g, j, 250 μm; b, e, h, k, 150 μm; c, f, i, l, 15 μm. (B) Representative photomicrographs show fluorescent double staining of phospho-MEK1/2 (green) and MAP-2 (red), and phospho-MEK1/2 (green) and NeuN (red) in the hippocampus 24 h after 10 min of tGCI with HPC. Phospho-MEK1/2-positive cells were observed in CA1 subregion (a, d), CA3 subregion (g), and CA4 subregion (j). MAP-2-positive cells were observed in CA1 subregion (b). NeuN-positive cells were observed in CA1 subregion (e), CA3 subregion (h), and CA4 subregion (k). The overlapped images show phospho-MEK1/2 was colocalized with MAP-2 in cell bodies (c) and surrounding NeuN-positive cells (f, i, l). Scale bar, 150 μm. (C) Quantitative analysis of immunoreactive cell counting of phospho-MEK1/2 in CA1 subregion. Data are shown as mean ± SD. *p < 0.05 vs. Sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 6 in each group). (D) Representative images of Western blot using either anti-phospho-MEK1/2 (Ser217/221) or anti-MEK1/2 antibody in ischemic and hypoxic preconditioned rats. (E) Quantitative analyses of total MEK1/2 levels in CA1 subregion in ischemic and hypoxic preconditioned rats. (F) Quantitative analyses of phospho-MEK1/2 levels in CA1 subregion in ischemic and hypoxic preconditioned rats. Data are expressed as percentage of value of Sham-operated animals. Each bar represents the mean ± SD. *p < 0.05 vs. Sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 4 in each group). Is ischemia
Western blot results were almost consistent with those of immunohistochemical analysis. Phosphorylated MEK1/2 expression increased as early as 4 h after tGCI, peaked at 24 h (1.78-fold, P < 0.05), and maintained for 48 h (1.57-fold). In the HPC groups, phosphorylated MEK1/2 expression apparently decreased as early as 4 h after hypoxia and ischemia, then significantly increased during 24–48 h, and peaked at 24 h (1.48-fold, P < 0.05). Phosphorylated MEK1/2 expression was compared among groups of tGCI and HPC at various time periods of reperfusion. Two-way ANOVA revealed that the expression of phosphorylated MEK1/2 in hippocampal CA1 subregion of HPC groups at 0, 4, 24, and 48 h of reperfusion were significantly lower in comparison with the tGCI groups. However, no significant differences were observed regarding the total protein expression of MEK.
Effects of Hypoxic Preconditioning on Phosphorylation of ERK1/2
Immunohistochemistry showed faint phosphorylated ERK1/2 expression on Sham-operated hippocampal CA1 subregion sections. In Sham-operated and 0-h post-ischemic hippocampal CA1 subregion, the phosphorylated ERK1/2 expression was too faint to be detected by double-labeled immunofluorescence staining. The number of phosphorylated ERK1/2-positive cells in hippocampal CA1 subregion increased significantly 4 h after reperfusion in tGCI rats, and approximately all phosphorylated ERK1/2-positive cells in hippocampus, especially in hippocampal CA4 subregion, showed a typical neuron-like morphology, that is, had round nuclei and spindle cell body with elongated axon (Fig. 3A(a and d), B). However, double-labeled immunofluorescence studies showed that these cells were colocalized with neither MAP-2 (data not shown) nor NeuN (Fig. 3A(a–f)) which marked the survival neurons. As we previously reported [20], a marked increase in degenerating neurons, as detected by FJ-B staining, was present in hippocampus after tGCI. In the present study, in order to confirm whether phosphorylated ERK1/2 express in degenerating neurons after tGCI, FJ-B staining was performed at 4 h after ischemia. Figure 3B showed that FJ-B-positive neurons expressed phosphorylated ERK1/2 in hippocampus after tGCI. These results suggest that up-regulation of phosphorylated ERK1/2 might be an underlying cause for increased neuronal damage in tGCI rats. At 24 h after tGCI, neuronal phosphorylated ERK1/2-positive cells had grown smaller and more condensed. These cells were more pyknotic at 168 h after reperfusion. The double labeling revealed that these cells were GFAP positive, indicating a predominant astrocytes localization of phosphorylated ERK1/2 in hippocampus 168 h after tGCI (Fig. 3A(g–l)). In HPC groups, the expression pattern of phosphorylated ERK1/2-positive cells followed that of tGCI groups, but the increase was generally less extensive. The number of phosphorylated ERK1/2-positive cells returned to baseline level 48 h after reperfusion. The quantitative analysis was shown in Fig. 3C.

Effect of hypoxic preconditioning on ERK1/2 phosphorylation in CA1 subregion. a Representative photomicrographs show fluorescent double staining of phospho-ERK1/2 (green) and NeuN (red) in the hippocampus 4 and 168 h after 10 min of tGCI, and phospho-ERK1/2 (green) and GFAP (red) in the hippocampus 168 h after 10 min of tGCI. Phospho-ERK1/2-positive cells were observed in CA1 subregion (a, g, j) and CA4 subregion (d). GFAP-2-positive cells were observed in CA1 subregion (k). The overlapped images show no colocalization of phospho-ERK1/2 and NeuN in the hippocampus neither at 4 nor 168 h after tGCI (c, f, i). Nevertheless, phospho-ERK1/2 and GFAP were completely overlapped 168 h after 10 min of tGCI in CA1 subregion (l). Scale bar, 150 μm. b Immunohistochemistry for phospho-ERK1/2 and FJ-B staining in the hippocampus 4 h after 10 min of tGCI. Phospho-ERK1/2-positive cells were observed in CA1 subregion (a, b), CA3 subregion (e, f), and CA4 subregion (i, j). FJ-B-positive cells were observed in CA1 subregion (c, d), CA3 subregion (g, h), and CA4 subregion (k, l). FJ-B-positive neurons express phosphorylated ERK1/2 in hippocampus. Scale bar: a, e, i, c, g, k, 25 μm; b, f, j, d, h, l, 15 μm. c Quantitative analysis of immunoreactive cell counting of phospho-ERK1/2 in CA1 subregion. Data are shown as mean ± SD. *p < 0.05 vs. Sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 6 in each group). d Representative images of Western blot using either anti-phospho-ERK1/2 (Thr202/Tyr204) or anti-ERK1/2 antibody in ischemic and hypoxic preconditioned rats. e Quantitative analyses of total ERK1/2 levels in CA1 subregion in ischemic and hypoxic preconditioned rats. f Quantitative analyses of phospho-ERK1/2 levels in CA1 subregion in ischemic and hypoxic preconditioned rats. Data are expressed as percentage of value of Sham-operated animals. Each bar represents the mean ± SD. *p < 0.05 vs. Sham-operated animals and # p < 0.05 vs. tGCI group at the same time point (n = 4 in each group)
As shown in Fig. 3D of Western blot, low-level constitutive expression of phosphorylated ERK1/2 was observed in Sham-operated hippocampal CA1 subregion. In the tGCI groups, I/R injury resulted in a strong induction of ERK1/2 phosphorylation in hippocampal CA1 subregion 4 h after reperfusion. The peak was reached at 4 h, and the increased expression lasted for 4–48 h after tGCI. Noteworthily, HPC significantly decreased ERK1/2 phosphorylation in ischemic hippocampal CA1 subregion, which is well consistent with the results of immunohistochemical studies. Incidentally, no significant difference was observed regarding the total ERK1/2 expression. These findings indicate that tGCI induced ERK1/2 phosphorylation after reperfusion in hippocampal CA1 subregion, while HPC dramatically decreased the phosphorylation of ERK1/2 in tGCI rats.
MEK/ERK Signaling Pathway is Critical to Hypoxic Preconditioning-Induced Neuroprotection
To examine whether the MEK/ERK signaling pathway is involved in the HPC-induced neuroprotection, a MEK inhibitor U0126 or vehicle was administered into the right lateral ventricle 24 h before ischemia. Figure 4A showed the phospho-MEK1/2 expression measured after 4 h of tGCI in the U0126 pretreatment group of rats. Compared with the tGCI or vehicle group, the administration of U0126 significantly reduced the phospho-MEK1/2 level. To study whether ERK1/2 phosphorylation was reduced after its upstream kinase inhibition, we examined phospho-ERK1/2 expression in the U0126-treated rats. Western blot results showed that phospho-ERK1/2 level in hippocampal CA1 subregion was decreased after 4 h of tGCI (Fig. 4B). In addition, compared with tGCI or vehicle group, the injection of U0126 before tGCI dramatically decreased neuronal damage induced by tGCI; the results of Nissl staining and NeuN immunohistochemistry showed that the number of surviving CA1 pyramidal neurons was markedly increased in U0126 group (Fig. 4C–E). Besides, glial cell activation was completely inhibited (Fig. 4C, F, G). These data indicate that the inhibition of up-regulation of phospho-MEK1/2 and phospho-ERK1/2 significantly attenuated neuronal damage and glial activation; thus, inhibition of MEK/ERK signaling pathway may mimic the protective effect of HPC.

Effect of U0126 treatment on phosphorylation of protein kinases and neuronal cells damage in CA1 subregion after tGCI. MEK1/2 inhibitor, U0126 inhibited phospho-MEK1/2 (a) and phospho-ERK1/2 (b) in CA1 subregion 4 h after tGCI. Each bar represents the mean ± SD. *p < 0.05 vs. Sham animals and # p < 0.05 vs. tGCI group (n = 6 in each group). c Representative microphotographs of cresyl violet staining and immunohistochemistry for NeuN, GFAP, and OX-42 in the hippocampus at 7 days after tGCI with or without U0126 treatment. The pictures on the right are magnified from the square areas on the left. Scale bar: a, f, k, p, u, 250 μm; b–e, g–j, l–o, q–t, v–y, 25 μm. Sham group (a–e; n = 7). Vehicle + tGCI group: 10 min of tGCI with vehicle infusion 24 h before tGCI (f–j; n = 6). Pre group: histology was performed 7 days after reperfusion in rats subjected to 30 min of HPC 1 day before tGCI (k–o; n = 9). tGCI group: histology was performed 7 days after 10 min of tGCI (p–t; n = 7). U0126 + tGCI group: 10 min of tGCI with U0126 infusion 24 h before tGCI (u–y; n = 7). Quantitative analyses of neurons survival and glia activation in CA1 subregion. The bar graphs present quantitative analysis of survival cells (d), NeuN-positive cells (e), GFAP-positive cells (f), and OX-42-positive cells (g) in CA1 subregion. Each bar represents the mean ± SD. *p < 0.05 vs. Sham-operated animals, # p < 0.05 vs. tGCI group, and & p < 0.05 vs. Pre group
Discussion
The major findings in this study focused on the effect of the HPC on cerebral I/R injury, and our study showed that HPC protects against tGCI-induced neuronal injury in adult rats through mechanisms involved in the deactivation of MEK/ERK signaling pathway.
The current study demonstrates that phosphorylation of MEK1/2 and ERK1/2 was enhanced significantly in hippocampal CA1 subregion after reperfusion, suggesting that MEK1/2 and ERK1/2 were activated after tGCI. Our previous study showed that DNA fragmentation increased evidently in hippocampal CA1 subregion 24–48 h after tGCI [21]. Because the activation of MEK1/2 and ERK1/2 was followed by DNA fragmentation, the enhancement of phosphorylation in these kinases is therefore likely to be a forewarning of apoptosis. Several studies showed that ERK activity was associated with DNA-damaging agents and antitumor compound-induced apoptosis. Also, ERK activity has been shown to directly affect mitochondrial function by decreasing mitochondrial respiration and mitochondrial membrane potential, which could lead to mitochondrial membrane disruption and cytochrome c release [22, 23]. Whether these mechanisms are involved in neuronal damage after tGCI is unclear, and future studies are required to clarify. Also, the activation of ERK during reperfusion is likely to be a response to the generation of reactive oxygen species that was supposed to contribute to neuronal injury. Phosphorylated ERK1/2 colocalizes in areas of increased superoxide anion production after transient focal cerebral ischemia in mice [24]. Furthermore, transgenic mice overexpressed any of the three superoxide dismutase genes exhibited resistance to cerebral ischemia, whereas superoxide dismutase knockout mice displayed larger infarcts [25].
Previous studies have revealed that ERK1/2 was phosphorylated after tGCI [26, 27], whereas the current study demonstrated MEK1/2 phosphorylation after tGCI as well as ERK1/2 phosphorylation. As ERK1/2 is activated by phospho-MEK, the inhibition of MEK activation can be effective to prevent ERK1/2 activation. A previous study using a MEK inhibitor showed that ERK1/2 activation was regulated by MEK after focal cerebral ischemia [28]. Furthermore, ERK1/2 activation seems to have a deleterious effect on focal cerebral ischemia, for ERK1/2 inhibition by the MEK inhibitor reduced infarct volume [28, 29], which is consistent with our results. Our current results showed that the inhibition of MEK and ERK phosphorylation after tGCI significantly attenuated neuronal damage as well as glial activation. However, the mechanism by which kinase inhibition reduces ischemic injury is not fully understood. Many studies with in vitro models predicted that the activation of the MEK/ERK pathway should promote cell survival. Also, ERK activation can attenuate the mitochondrial cell death signaling pathway by inactivating Bad, a proapoptotic protein of Bcl-2 family [25]. In addition, ERK2 is reported to phosphorylate and inhibit the initiator caspase-9, thus blocking subsequent activation of the effector caspase-3 [30, 31]. Nonetheless, the MEK inhibitor SL327 diminished caspase-3 activation but did not prevent mitochondrial cytochrome c release as expected [32]. Alternatively, ERK activation, and specifically the MEK2/ERK2 pathway, is proposed to promote a form of nonapoptotic programmed cell death [33]. Also, ERK activation promoted cerebellar granule neurons degeneration predominantly through plasma membrane damage and independently of casepase-3 [34]. In several in vitro and in vivo models of neuronal injury, neurotrophic factors reduce apoptotic cell death, but potentiate oxidative damage, which can be blocked by MEK inhibitors—PD098059 and U0126 [10, 35]. Inhibition of the MEK/ERK pathway can also attenuate an inflammatory response after cerebral ischemia by preventing the induction of the cytokine interleukin-1β [36]. In the present study, MEK/ERK inhibition with U0126 dramatically reduced astrocytes recruitment and microglia activation, and increased neuronal survival. Taken together, these data suggested a role of the MEK/ERK pathway in the inflammatory responses after brain ischemia, and that neuroprotection provided by U0126 is at least in part mediated by its inhibitory effects on glial cell activation.
HPC is described as an endogenous strategy in which a sublethal hypoxic exposure protects tissues from further damage caused by a severe ischemic/hypoxic insult. The protection mechanism induced by HPC makes it an attractive target for potential clinical therapeutic approaches. In this study, we found a significant decrease in the phosphorylation of MEK1/2 and ERK1/2 in hippocampal CA1 subregion of rat followed tGCI with previous hypoxic exposure. Our result is consistent with previous findings. For example, in a study of forebrain ischemia, significant decreased ERK2 activity was observed and thought to be related to the protective mechanism of hippocampus injuries [28]. The inhibition of ERK1/2 activation was reported as an effective protection strategy in studies of brain ischemia and in vitro cultured neurons following oxygen deprivation [28, 37]. In our study, the decreased MER1/2 and ERK1/2 phosphorylation in tGCI with previous hypoxic exposure was believed to be a mechanism in the self-inhibiting intracellular MEK/ERK pathway in response to HPC stimulation.
Because the MEK/ERK pathway interacts with other elements that determine cell survival or cell death, it is useful to investigate the change in such elements after ischemia to understand the role of HPC on the MEK/ERK pathway. Akt is known to play a critical role in controlling the balance between survival and apoptosis [38]. One study showed that inhibition of Akt induced phosphorylation of Raf at serine-259 and inactivated Raf, which resulted in ERK1/2 activation in cultured cells stimulated with insulin-like growth factor [39]. Another study showed that phosphatidylinositol 3-kinase inhibitor, which inhibited Akt activation, prevented nuclear translocation of protein kinase C zeta, resulting in ERK1/2 activation after ischemic hypoxia and reoxygenation in cultured cells [40]. Furthermore, we have shown that Akt is inactivated in hippocampal CA1 subregion after tGCI [2]. All of these evidences together suggest that Akt inactivation after ischemia may interact with activation of the MEK/ERK pathway. Nonetheless, future studies are needed to clarify the relationship between Akt inactivation and ERK1/2 activation after ischemia.
As we described previously, massive neuronal degeneration was observed in hippocampus after tGCI [20]. Here, we found phosphorylated ERK1/2 increases after tGCI. This temporal relationship represents the first line of evidence for a causal relationship between phosphorylated ERK1/2 and neuronal damage. Besides, pretreatment with hypoxia decreased the level of ERK1/2 phosphorylation and the number of FJ-B-positive neurons. In addition, FJ-B-positive neurons express phosphorylated ERK1/2. The results suggested that up-regulation of phosphorylated ERK1/2 might be an underlying cause for increased neuronal damage in tGCI rats. Finally, the inhibition of phosphorylated ERK1/2 by U0126 significantly increased neuronal survival and decreased glial cell activation in hippocampal CA1 subregion after tGCI.
Taken together, these data clearly demonstrate that the MEK/ERK pathway plays a critical role in promoting cell death in response to tGCI. Also, the inhibition of the MEK/ERK pathway diminished glia cells activation, mimic the hypoxia preconditioning, and provide neuroprotection to tGCI. HPC attenuates neuronal cell death by preventing MEK/ERK signaling pathways activation after tGCI in adult rats.
References
Kapinya KJ (2005) Ischemic tolerance in the brain. Acta Physiol Hung 92:67–92
Zhan L, Wang T, Li W, Xu ZC, Sun W, Xu E (2010) Activation of Akt/FoxO signaling pathway contributes to induction of neuroprotection against transient global ischemia by hypoxic preconditioning in adult rats. J Neurochem 114:897–908
Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y (2001) Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab 21:351–360
Kitano H, Young JM, Cheng J, Wang L, Hum PD, Murphy SJ (2007) Gender-specific response to isoflurane preconditioning in focal cerebral ischemia. J Cereb Blood Flow Metab 27:1377–1386
Lee SR, Lo EH (2003) Interactions between p38 mitogen-activated protein kinase and caspase-3 in cerebral endothelial cell death after hypoxia-reoxygenation. Stroke 34:2704–2709
Mebratu Y, Tesfaigzi Y (2009) How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle 8:1168–1175
Subramaniam S, Unsicker K (2010) ERK and cell death: ERK1/2 in neuronal death. FEBS J 277:22–29
Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ (2006) ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem 281:20315–20325
Zhuang S, Schnellmann RG (2006) A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther 319:991–997
Numakawa Y, Matsumoto T, Yokomaku D, Taguchi T, Niki E, Hatanaka H, Kunugi H, Numakawa T (2007) 17beta-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology 148:627–637
Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S (2000) Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett 288:163–166
Lu K, Liang CL, Liliang PC, Yang CH, Cho CL, Weng HC, Tsai YD, Wang KW, Chen HJ (2010) Inhibition of extracellular signal-regulated kinases 1/2 provides neuroprotection in spinal cord ischemia/reperfusion injury in rats: relationship with the nuclear factor-kappaB-regulated anti-apoptotic mechanisms. J Neurochem 114:237–246
Irving EA, Bamford M (2002) Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab 22:631–647
Maddahi A, Edvinsson L (2010) Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation 7:14
Pulsinelli WA, Brierley JB (1979) A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 10:267–272
Schmued LC, Stowers CC, Scallet AC, Xu (2005) Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res 1035:24–31
Wang Y, Zhan L, Zeng W, Li K, Sun W, Xu ZC, Xu E (2011) The effect of GABA on the hypoxia-induced increase of epilepsy susceptibility in neonate rat. Neurochem Res 36:2409–2416
Endo H, Nito C, Kamada H, Nishi T, Chan PH (2006) Activation of the Akt/GSK3β signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J Cereb Blood Flow Metab 26:1479–1489
Chan SH, Wu CA, Wu KL, Ho YH, Chang AY, Chan JY (2009) Transcriptional upregulation of mitochondrial uncoupling protein 2 protects against oxidative stress-associated neurogenic hypertension. Circ Res 105:886–896
Zhan L, Li D, Liang D, Wu B, Zhu P, Wang Y, Sun W, Xu E (2012) Activation of Akt/FoxO and inactivation of MEK/ERK pathways contribute to induction of neuroprotection against transient global cerebral ischemia by delayed hypoxic postconditioning in adult rats. Neuropharmacology 63:873–882
Zhan L, Peng W, Sun W, Xu E (2011) Hypoxic preconditioning induces neuroprotection against transient global ischemia in adult rats via preserving the activity of Na+/K + −ATPase. Neurochem Int 59:65–72
Kim GS, Hong JS, Kim SW, Koh JM, An CS, Choi JY, Cheng SL (2003) Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem 278:21920–21929
Nowak G, Clifton GL, Godwin ML, Bakajsova D (2006) Activation of ERK1⁄ 2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol 291:F840–F855
Noshita N, Sugawara T, Hayashi T, Lewen A, Omar G, Chan PH (2002) Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J Neurosci 22:7923–7930
Sugawara T, Fujimura M, Noshita N, Kim GW, Saito A, Hayashi T, Narasimham P, Maier CM, Chan P (2004) Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx 1:17–25
Zhang F, Signore AP, Zhou Z, Wang S, Cao G, Chen J (2006) Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: potential signaling mechanisms. J Neurosci Res 83:1241–1251
Zheng YQ, Liu JX, Wang JN, Xu L (2007) Effects of crocin on reperfusion-induced oxidative/nitrative injury to cerebral microvessels after global cerebral ischemia. Brain Res 1138:86–94
Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A (2001) Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA 98:11569–11574
Farrokhnia N, Ericsson A, Terént A, Lennmyr F (2008) MEK-inhibitor U0126 in hyperglycaemic focal ischaemic brain injury in the rat. Eur J Clin Invest 38:679–685
Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR (2003) Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol 5:647–654
Kitazumi I, Tsukahara M (2011) Regulation of DNA fragmentation: the role of caspases and phosphorylation. FEBS J 278:427–441
Wang X, Wang H, Xu L, Rozanski J, Sugawara T, Chan PH, Trazskos JM, Feuerstein GZ (2003) Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther 304:172–178
Castro-Obregon S, Rao R, del Rio G, Chen S, Poksay K, Rabizadeh S, Vesce S, Zhang X, Swanson RA, Bredesen DE (2004) Alternative, nonapoptotic programmed cell death. J Biol Chem 279:17543–17553
Subramaniam S, Zirrgiebel U, von Bohlen Und Halbach O, Strelau J, Laliberté C, Kaplan DR, Unsicker K (2004) ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol 165:357–369
Lobner D, Liot G (2004) Role of MAPK/ERK neurotropin-4 potentiation of necrotic neuronal death. Neurochem Res 29:2303–2309
Wang ZQ, Wu DC, Huang FP, Yang GY (2004) Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res 996:55–66
Zhang F, Wang S, Signore AP, Chen J (2007) Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke 38:2329–2336
Franke TF, Kaplan DR, Cantley LC (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88:435–437
Zimmermann S, Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286:1741–1744
Mizukami Y, Kobayashi S, Uberall F, Hellbert K, Kobayashi N, Yoshida K (2000) Nuclear mitogen-activated protein kinase activation by protein kinase czeta during reoxygenation after ischemic hypoxia. J Biol Chem 275:19921–19927
Acknowledgments
This work was supported by National Research Foundation for the Doctoral Program of Higher Education of China (Grant No. 20124423110002), and partly supported by the National Natural Science Foundation of China (Grant No. 81100800). Our sincere thanks go to Mr. Peifeng DU (Institute for Standardization of Nuclear Industry) and Donghai Liang (Environmental Health Sciences, Yale School of Public Health) for editing this paper.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhan, L., Yan, H., Zhou, H. et al. Hypoxic Preconditioning Attenuates Neuronal Cell Death by Preventing MEK/ERK Signaling Pathway Activation after Transient Global Cerebral Ischemia in Adult Rats. Mol Neurobiol 48, 109–119 (2013). https://doi.org/10.1007/s12035-013-8436-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8436-4