
Abstract
In this study, solid polymer blend films based on chitosan (CH) and methylcellulose (MC) were prepared in various compositions by the solution cast technique. The features of structure and complexation of the blend polymer films were studied using X-ray diffraction (XRD) and Fourier transform infrared (FTIR) analysis. The shift of FTIR peaks of the amino groups of CH and the hydroxyl groups of MC reveals the formation of interchain hydrogen-bonding between CH and MC chains in blend films. From the XRD pattern, the semi-crystalline structure of CH was depressed with the addition of MC and shows the CH:MC blend system with ratio 75:25 has the minimum degree of crystallinity. The highest room temperature conductivity was found to be \(0.05\times 10^{-6}\, \hbox {S}\,\hbox {cm}^{-1}\) for 75CH:25MC blend polymer composition. The dc conductivity exhibits Arrhenius-type behaviour with temperature. The drastic increase in conductivity up to \(37.92\times 10^{-6}\, \hbox {S}\, \hbox {cm}^{-1}\) at 373 K, can be explained by free volume model. The highest value of electrical conductivity for all prepared samples was associated with the minimum value of activation energy.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In the last few decades, much effort has been devoted to develop environmentally-friendly biodegradable polymer electrolytes based on natural polymers, such as chitosan (CH), cellulose, starch and poly(\(\upvarepsilon \)-caprolactone) (PCL) as host materials, instead of using not-eco-friendly and non-biodegradable synthetic polymers such as polyethylene oxide, polyvinyl chloride, polymethylmethacrylate, polyvinyl acetate (PVA), polyacrylonitrile and polyvinyl pyrrolidone (PVP) that impose several ecological issues [1,2,3].
It has been well reported in the literature that the ion transport in polymer electrolyte is predominant through amorphous phase rather than the crystalline phase [4]. Thus to modulate the conductivity of the polymeric system, many investigations have been focussed on increasing the amorphous phase of the polymer host material [5, 6]. For this purpose, several methods and techniques were used such as copolymerization, blending and addition of salt, plasticization or ceramic nano-fillers [7,8,9]. Among these methods, polymer blending has received ever-increasing interest [10]. The polymer blends or polymer mixture often exhibit improved physical properties and better processing characteristics compared to the properties of pure polymer components [11]. The primary points of interest of the poly-blend systems are the simplicity of preparation, low basic cost and efficient control of physical properties via compositional change [10, 12]. However, the manifestation of superior properties of blend polymers depends on the degree of miscibility of the components of blends on the molecular scale [13]. The basis of polymer–polymer miscibility may originate from any specific interaction, such as hydrogen-bonding, charge transfer complexes and dipole–dipole interactions [14, 15]. In the present study, CH and methylcellulose (MC) have been chosen to prepare polymer-blend systems due to the excellent mechanical strength and good miscibility with each other [16]. Moreover, among all popular polymer electrolyte systems, a biopolymer electrolyte based on CH and MC has special significance in view of its potential applications in solid-state electrochemical devices, due to its many advantages such as water-soluble, biodegradable, biocompatible, non-toxic, environmentally-friendly, abundant and good film-forming properties [17,18,19]. A literature search and intensive surveys reveal that a relatively small number of studies have been reported on the development of CH:MC blend-based polymer electrolyte systems [20].
Hamdan and Khiar [21] prepared a solid polymer electrolyte based on CH:MC blends with an arbitrary fixed ratio of 50:50 complexed with different amounts of ammonium triflate \((\hbox {NH}_{4}\hbox {CF}_{3}\hbox {SO}_{3})\). Whereas, Misenan et al [22] have reported the effect of adding cellulose nanocrystal on the conductivity of CH:MC blend in the ratio of 60:40. This report is concerned with the structural and electrical properties of CH and MC blends in different concentrations to find the optimal blend composition (the maximum amorphous domains). The electrical conductivity behaviour of the CH:MC poly-blend films are also reported.
2 Experimental
2.1 Preparation of the polymer blend films
In the present study, films of CH blended with MC were prepared using the solution cast technique at various weight ratios (100:0), (75:25), (50:50), (25:75) and (0:100). CH and MC were dissolved separately in 2% acetic acid and distilled water, respectively. The two solutions were stirred continuously for 2 days until the polymer powders were dissolved completely. After that, the two solutions were mixed according to desired weight ratio and the mixture was stirred at room temperature for 45 min. Finally, the obtained homogeneous solutions were poured onto cleaned plastic Petri dishes and the solvent was allowed to evaporate slowly at ambient temperature to obtain free-standing films. The prepared films were then kept in desiccators with silica gel to eliminate all traces of the solvent.
2.2 Characterization techniques
Chemical modifications of the polymer due to blending were recorded using a Fourier transform infrared (FTIR) Spectrophotometer (Frontier spectrometer) in the wavelength range of 4000–400 \(\hbox {cm}^{-1}\). The X-ray diffraction (XRD) studies were conducted using (X’PERT-PRO) X-ray diffractometer with \(\hbox {CuK}\upalpha \) radiation of wavelength 1.5406 Å and a graphite monochromator, in the angle \(10^{\circ }\le 2\theta \le 70^{\circ }\) with a scan step size of \(\Delta 2\theta =0.1^{\circ }\). The electrical conductivity of the prepared films was measured using a Precision LCR Meter (Agilent/HP 4284A) in a frequency range of 100 Hz–1 MHz, and in the temperature range, 295–373 K. Here the polymer films were sandwiched between two aluminium electrodes under spring pressure (electrode–specimen contact \(\hbox {area}=4.91\, \hbox {cm}^{2}\)) and the measurements were carried out in the conduction mode. The average thicknesses of these films were determined to be in the range 126–260 \(\upmu \hbox {m}\). The real part of ac conductivity \(\left( {\sigma _{{\mathrm{ac}}} } \right) \) was calculated from the measured values of conductivity \(\left( G \right) \) using: \(\sigma _{{\mathrm{ac}}}\left( \omega \right) =Gd/A\), where d is the thickness of the sample and A is the electrode–specimen contact cross-sectional area.

FTIR plots of CH:MC polymer blend films at different weight ratios in the wavenumber ranges: (a) 400–4000 \(\hbox {cm}^{-1}\), (b) 1500–1600 \(\hbox {cm}^{-1}\) and (c) 2800–3800 \(\hbox {cm}^{-1}\).
3 Results and discussion
3.1 FTIR studies
Infrared spectral analysis monitors a wide variety of vibrational energy levels in the molecules [23]. The FTIR spectra of pure CH, MC and their blends with different compositions recorded at room temperature are given in figure 1. The FTIR spectrum of pure CH film shows a broad O–H stretching absorption band between 3419 and \(3289\, \hbox {cm}^{-1}\), which is overlapped to the stretching vibration of N–H in the same region. The C–H stretching vibration bands occur between 2931 and \(2925\, \hbox {cm}^{-1}\). More major absorption bands at 1645 and \(1564\, \hbox {cm}^{-1}\) represent the C=O stretching amino group I, and –NH bending amino group II of glucosamine, respectively [16]. The vibration modes at 1416, 1373 and \(1152\, \hbox {cm}^{-1}\) are respectively assigned to \(\hbox {CH}_{2}\) bending, –CH bending and anti-symmetric stretching of the C–O–C bridge [24].
From the FTIR spectrum of pure MC, it is observed that a strong broadband observed at 3459–3457 \(\hbox {cm}^{-1}\) is assigned to the stretching vibrations of hydroxyl groups (O–H). Moreover, the observed bands at 2934–2900 \(\hbox {cm}^{-1}\) indicate the presence of C–H stretching of \(\hbox {CH}_{2}\) and \(\hbox {CH}_{3}\) groups. The observed strong band centred at \(1647\, \hbox {cm}^{-1}\) corresponds to C=O stretching vibration and the observed band at \(1412\, \hbox {cm}^{-1}\) can be ascribed to the C–H bending in \(\hbox {CH}_{2}\) [25, 26], whereas the observed bands at \(1374\, \hbox {cm}^{-1}\) are assigned to –CH bending vibration. The band corresponding to the ether groups (C–O–C) occurs at \(1117\, \hbox {cm}^{-1}\) [17], and the asymmetric vibrations of CO appear at 1087–1055 \(\hbox {cm}^{-1}\) [27].
The FTIR spectra of CH:MC blends shown in figure 1 contains no distinct additional absorbance peaks compared to those for pure CH and MC. This suggests that there is no chemical interaction between the components [28], whereas the shift of some observed peaks suggests the physical interaction phenomena (i.e., ionic/hydrophobic interactions) between CH and MC chains. The shift in the position of the peak at \(1564\, \hbox {cm}^{-1}\) for CH amino groups (figure 1b) and peak at 3459–3457 \(\hbox {cm}^{-1}\) for MC hydroxyl groups (figure 1c), is due to hydrogen-bonding interaction between the positive charge of amino groups (\(-\hbox {NH}_{3}^{+}\)) of CH and negative charge of hydroxyl groups (\(-\hbox {OH}^{-}\)) of MC [29, 30]. Figure 2 shows the possible hydrogen-bonding between CH and MC. The formation of a hydrogen bond between two polymers CH and MC upon blending proves miscibility between the two polymers at the molecular-level. Ragab [31] also attributed the miscibility between the two polymers PVA/PVP blend to the hydrogen-bonding between hydroxyl groups of PVA and carbonyl groups of PVP monomeric units.

Possible mechanism for hydrogen-bonding between \(\hbox {CH}\, \{\hbox {C}_{6}\hbox {H}_{11}\hbox {NO}_{4}\}_{n}\) and \(\hbox {MC}\,\{\hbox {C}_{6}\hbox {H}_{7}\hbox {O}_{2}\hbox {(OH)}{x}(\hbox {OCH}_{3})y\}_{n}\).

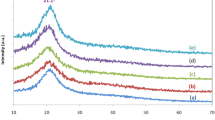
XRD patterns of CH:MC polymer blend films at different weight ratios: (a) 100:0, (b) 75:25, (c) 50:50, (d) 25:75 and (e) 0:100.
3.2 XRD studies
The XRD is a versatile, non-destructive technique that reveals detailed information about the nature of polymer blend films and their crystallographic structure [32]. The XRD patterns of pure CH, MC and their blend membranes were determined to correlate their structure with other properties. Figure 3 shows the diffractograms of CH, MC and their blends. The pure polymers films show a peak around \(2\theta =21.1^{\circ }\) which usually characterizes the semi-crystalline phase of these polymers [33, 34]. Previous studies addressed that intra- and inter-molecular hydrogen bonds are responsible for the semi-crystalline structure of both CH and MC membranes [35, 36]. The observed diffraction peak of CH:MC blend films is less intense when compared with those of pure MC films, which indicates that the blending of CH and MC causes a reduction in the degree of crystallinity and a simultaneous increase in the amorphicity of the system. It was quoted in the literature that the crystallinity fractions are directly proportional to the peak intensity and inversely related to the full-width at half-maximum of the peak [37, 38]. The intensity of XRD peaks decreases as the amorphous nature increases [39, 40], and this amorphous nature facilitates the ionic diffusivity resulting in the increase of the electrical conductivity [41, 42], as it will be seen later on.

Degree of crystallinity of CH:MC polymer blend films vs. MC wt%.
Resolution of crystalline peaks, together with an integration of the scattered intensities, provides a method for estimation of crystallinity. The degree of crystallinity (\(X_{\mathrm{c}} \)) of the samples were determined by deconvoluting peaks due to crystalline and amorphous phases, using Fityk software [43], and according to Hermans and Weidinger equation [44, 45]:
where \(A_{\mathrm{c}} \) and \(A_{\mathrm{a}} \) are the area under crystalline peaks and amorphous haloes, respectively. The calculated data of degree of crystallinity for CH and MC blend samples of different compositions are shown in figure 4. It is observed that the degree of crystallinity for blend samples lies in between the values of two pure polymers, which may be assigned to the incorporation of side branches [46]. As can be seen from figure 4, the degree of crystallinity of the 75CH:25MC polymer blend system is the lowest with a value of (20.46%), suggesting that this composition has the maximum amorphicity. The increase in the amorphous nature causes a reduction in the energy barrier to the segmental motion of the polymer blend resulting in the increase in conductivity [47].

The variation of electrical conductivity vs. MC concentration at different temperature range: (a) 295–333 K and (b) 343–373 K.

Temperature dependence dc conductivity of CH:MC polymer blend films at different weight ratios: (a) 100:0, (b) 75:25, (c) 50:50, (d) 25:75 and (e) 0:100.
3.3 Electrical conductivity studies
The electrical conductivity of CH:MC bio-polymer blend has been investigated, with an aim to understand the origin and nature of the charge transport prevalent in this material system. The low-frequency plateau region of electrical conductivity describes the electrode–specimen interface phenomena is attributed to the space charge polarization at the blocking electrode and is associated with the dc conductivity \(\left( {\sigma _{{\mathrm{dc}}} } \right) \) of the polymer system [48]. Thus the frequency dependent electrical conductivity measurements can be used to obtain the dc conductivity \(\left( {\sigma _{{\mathrm{dc}}} } \right) \) of the polymer blend films at different temperatures by extrapolating the plateau region on the electrical conductivity axis to zero frequency (not shown here). The variation of the dc conductivity \(\left( {\sigma _{{\mathrm{dc}}} } \right) \) vs. MC concentration at different temperatures is shown in figure 5. The obtained conductivity values for pure CH (\(1.2\times 10^{-8}\, \hbox {S}\, \hbox {cm}^{-1}\)), and pure MC (\(2.6\times 10^{-8}\, \hbox {S}\, \hbox {cm}^{-1}\)) are respectively in accordance with the results reported by Ng and Mohamad [49] for CH, and the results reported by Shuhaimi et al [50] for MC. The maximum conductivity of CH:MC bio-poly blends was obtained for a composition ratio 75:25, which is ascribed to the increase in the amorphous nature of the polymer blend at this concentration, which is well consistent with the XRD results. Also, it is evident that the electrical conductivity of polymer blend systems increases with increasing temperature. The value of dc conductivity for the 75CH:25MC membrane was found to increase with increasing temperature from \(0.05\times 10^{-6}\,\hbox {S}\, \hbox {cm}^{-1}\) at ambient temperature, to \(37.92\times 10^{-6} \,\hbox {S}\, \hbox {cm}^{-1}\) at 373 K. The significant increase in dc conductivity with temperature can be explained by means of the free-volume model. According to this model, the increase in temperature results in an increase in the fraction of free volume [51]. This will facilitate the segmental motion of polymer chains, which helps the migration of charge carriers by hopping from one site to another and sequentially increase the electrical conductivity [52, 53].
The temperature dependence dc conductivity followed an Arrhenius behaviour in the studied temperature range. The dc conductivity (\(\sigma _{{\mathrm{dc}}} \)) as per Arrhenius relation can be expressed as [54]:
where \(\sigma _0 \) is a proportionality constant related to the number of charge carriers in the films, \(E_{\mathrm{a}} \) is the activation energy, \(k_{\mathrm{B}} \) is the Boltzmann constant and T is the temperature in Kelvin [55].

The variation of dc conductivity and activation energy as a function of MC wt% concentration.
The activation energy (combination of defect formation energy and ion migration energy) corresponds to the energy required for conduction from one site to another [56,57,58]. For calculating \(E_{\mathrm{a}} \) for dc conductivity, the effect of temperature on the \(\sigma _{{\mathrm{dc}}} \) was studied. Figure 6 elucidates the linear dependence of logarithm of dc conductivity with inverse absolute temperature for CH:MC poly-blend films. The regression values (\(R^{2}\)) of the plots using linear fit have been found to be close to unity suggesting that the temperature dependent dc conductivity for all prepared films obey Arrhenius relationship. The activation energies were evaluated for individual pure polymers and their blend by considering the slope after linear fitting the data, using the relation [59]:
The variation of activation energy \(\left( {E_{\mathrm{a}} } \right) \) and room temperature dc conductivity \(\left( {\sigma _{{\mathrm{dc}}} } \right) \) as a function of MC concentration is shown in figure 7. It is clear that the minimum value of \(E_{\mathrm{a}} \) (in the order of 0.81 eV for 75CH:25MC poly-blend sample) is associated with the highest dc conductivity. The decrease in activation energy for polymer blend samples is attributed to the reduction of the energy barrier to the segmental motion of polymer matrix. It is worth mentioning that the polymer electrolyte with a low value of activation energy is desirable for practical applications [60]. The inverse relationship between conductivity and activation energy was also reported earlier by Yousf et al [61] for starch–CH blend biopolymer films, and by Hafiza et al [62] for carboxyl MC–CH blend polymer electrolyte.
Generally, the electrical conductivity is controlled by the concentration of mobile charge carriers and its mobility. In the present system, the increase in conductivity with increasing temperature should be mainly ascribed to the increase in the carrier mobility, due to the increment in the amorphous nature of the blend system (as confirmed by XRD analysis), which arises from the coordination and interaction between the amino groups of CH with hydroxyl groups of MC (as proved by FTIR analysis).
4 Conclusions
The perfect composition of MC:CH bio-poly-blend system for electrical conductivity was depicted using FTIR and XRD. The FTIR study indicates that CH can be effectively blended with MC due to the formation of hydrogen bond between functional groups of two polymers. The XRD pattern of blend samples reveal that the semi-crystalline structure of both CH and MC is reduced upon blending and the poly-blend samples with compositions of 75CH:25MC showed the maximum amorphous content. The dc conductivity of this membrane increased with increase in sample temperature and followed Arrhenius equation. The highest electrical conductivity at 395 K has been found to be \(0.05\times 10^{-6}\, \hbox {S}\, \hbox {cm}^{-1}\) for the 75CH:25MC sample; the activation energy was found to be inversely proportional to the dc conductivity and was in the range 0.81–1.07 eV.
References
Muchakayala R, Song S, Gao S, Wang X and Fan Y 2017 Polym. Test. 58 116
Kulshrestha N and Gupta P N 2016 Ionics 22 671
Khiar A S A and Arof A K 2010 Ionics 16 123
Aziz S B, Abdullah O G and Hussein S A 2018 J. Electron. Mater. 47 3800
Aziz S B, Abdullah O G, Saber D R, Rasheed M A and Ahmed H M 2017 Int. J. Electrochem. Sci. 12 363
Abdullah O G 2016 J. Mater. Sci. Mater. Electron. 27 12106
Abdulwahid R T, Abdullah O G, Aziz S B, Hussein S A, Muhammad F F and Yahya M Y 2016 J. Mater. Sci. Mater. Electron. 27 12112
Abdullah O G, Aziz S B and Rasheed M A 2017 J. Mater. Sci. Mater. Electron. 28 4513
Aziz S B, Abdullah O G and Rasheed M A 2017 J. Appl. Polym. Sci. 134 44847
Abdullah O G, Aziz S B and Rasheed M A 2018 Ionics 24 777
Ramamohan K, Achari V B S, Sharma A K and Xiuyang L 2015 Ionics 21 1333
Kesavan K, Mathew C M, Rajendran S, Subbu C and Ulaganathan M 2015 Braz. J. Phys. 45 19
Reddeppa N, Reddy T J R, Achari V B S, Rao V V R N and Sharma A K 2009 Ionics 15 255
Lewandowska K 2009 Thermochim. Acta 493 42
Marsano E, Vicini S, Skopinska J, Wisniewski M and Sionkowska A 2004 Macromol. Symp. 218 251
Liu P, Wei X and Liu Z 2013 Adv. Mater. Res. 750–752 802
Aziz N A N, Idris N K and Isa M I N 2010 Int. J. Polym. Anal. Charact. 15 319
Gomes A M M, Silva P L, Moura C L, Silva C E M and Ricardo N M P S 2011 . Macromol. Symp. 299/300 220
Basavaraju K C, Damappa T and Rai S K 2006 Carbohyd. Polym. 66 357
Synytsya A, Grafova M, Slepicka P, Gedeon O and Synytsya A 2012 Biomacromolecules 13 489
Hamdan K Z and Khiar A S A 2014 Key Eng. Mater. 594–595 818
Misenan M S M, Ali E S and Khiar A S A 2018 AIP Conf. Proc. 1972 030010
Praveena S D, Ravindrachary V, Bhajantri R F and Ismayi 2014 Polym. Composit. 27 987
Silva S S, Goodfellow B J, Benesch J, Rocha J, Mano J F and Reis R L 2007 Carbohyd. Polym. 70 25
Rangelova N, Radev L, Nenkova S, Salvado I M M, Fernandes M H V and Herzog M 2011 Cent. Eur. J. Chem. 9 112
Filho G R, Assuncao R M N, Vieira J G, Meireles C S, Cerqueira D A, Barud H S et al 2007 Polym. Degrad. Stabil. 92 205
Ragab H S and El-Kader M F H A 2013 Phys. Scr. 87 025602
Tang Y, Wang X, Li Y, Lei M, Du Y, Kennedy J F et al 2010 Carbohyd. Polym. 82 833
Hema M, Selvasekarapandian S, Arunkumar D, Sakunthala A and Nithya H 2009 J. Non-Cryst. Solids 355 84
Abdullah O G, Aziz S B and Rasheed M A 2016 Results Phys. 6 1103
Ragab H M 2011 Physica B 406 3759
Rajeswari N, Selvasekarapandian S, Karthikeyan S, Prabu M, Hirankumar G, Nithya H et al 2011 J. Non-Cryst. Solids 357 3751
Abdullah O G, Salman Y A K and Saleem S A 2016 J. Mater. Sci. Mater. Electron. 27 3591
Abdullah O G and Saleem S A 2016 J. Electron. Mater. 45 5910
Aziz S B, Abdulwahid R T, Rasheed M A, Abdullah O G and Ahmed H M 2017 Polymers 9 486
Aziz S B, Abdullah O G, Hussein S A and Ahmed H M 2017 Polymers 9 622
Wang J, Song S, Muchakayala R, Hu X and Liu R 2017 Ionics 23 1759
Aziz S B, Abdullah O G and Rasheed M A 2017 J. Mater. Sci. Mater. Electron. 28 12873
Bdewi S F, Abdullah O G, Aziz B K and Mutar A A R 2016 J. Inorg. Organomet. Polym. Mater. 26 326
Abdullah O G, Tahir D A and Kadir K 2015 J. Mater. Sci. Mater. Electron. 26 6939
Achari V B, Reddy T J R, Sharma A K and Rao V V R N 2007 Ionics 13 349
Abdullah O G, Aziz S B, Saber D R, Abdullah R M, Hanna R R and Saeed S R 2017 J. Mater. Sci. Mater. Electron. 28 8928
Wojdyr M 2010 J. Appl. Crystallogr. 43 1126
Sownthari K and Suthanthiraraj S A 2013 Express Polym. Lett. 7 495
Rani N S, Sannappa J, Demappa T and Mahadevaiah 2015 Ionics 21 133
El-Kader M F H and Ragab H S 2013 Ionics 19 361
Baskaran R, Selvasekarapandian S, Kuwata N, Kawamura J and Hattori T 2006 Solid State Ion. 177 2679
Abazine K, Anakiou H, El Hasnaoui M, Graca M P F, Fonseca M A, Costa L C et al 2016 J. Compos. Mater. 50 3283
Ng L S and Mohamad A A 2008 J. Membrane Sci. 325 653
Shuhaimi N E A, Teo L P, Woo H J, Majid S R and Arof A K 2012 Polym. Bull. 69 807
Elashmawi I S, Elsayed N H and Altalhi F A 2014 J. Alloy. Compd. 617 877
Dam T, Karan N K, Thomas R, Pradhan D K and Katiyar R S 2015 Ionics 21 401
Aziz S B, Rasheed M A, Saeed S R and Abdullah O G 2017 Int. J. Electrochem. Sci. 12 3263
Hebbar V, Bhajantri R F and Naik J 2017 J. Mater. Sci. Mater. Electron. 28 5827
Shukur M F, Ithnin R, Illias H A and Kadir M F Z 2013 Opt. Mater. 35 1834
Mohan V M, Raja V, Bhargav P B, Sharma A K and Rao V V R N 2007 J. Polym. Res. 14 283
Badr S, Sheha E, Bayomi R M and El-Shaarawy M G 2010 Ionics 16 269
Raja V, Mohan V M, Sharma A K and Rao V V R N 2009 Ionics 15 519
Sugumaran S and Bellan C S 2014 Optik 125 5128
Ravi M, Bhavani S, Kumar K K and Rao V V R N 2013 Solid State Sci. 19 85
Yusof Y M, Shukur M F, Illias H A and Kadir M F Z 2014 Phys. Scr. 89 035701
Hafiza M N, Bashirah A N A, Bakar N Y and Isa M I N 2014 Int. J. Polym. Anal. Charact. 19 151
Acknowledgements
The authors would like to thank the Ministry of Higher Education and Scientific Research, University of Sulaimani, and Komar University of Science and Technology, for the financial support of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Abdullah, O.G.H., Hanna, R.R. & Salman, Y.A.K. Structural and electrical conductivity of CH:MC bio-poly-blend films: optimize the perfect composition of the blend system. Bull Mater Sci 42, 64 (2019). https://doi.org/10.1007/s12034-019-1742-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12034-019-1742-3