
Abstract
Our previous studies demonstrated that propofol protects rat brain against focal cerebral ischemia. However, whether propofol attenuates early brain injury after subarachnoid hemorrhage in rats remains unknown until now. The present study was performed to evaluate the effect of propofol on early brain injury after subarachnoid hemorrhage in rats and further explore the potential mechanisms. Sprague-Dawley rats underwent subarachnoid hemorrhage (SAH) by endovascular perforation then received treatment with propofol (10 or 50 mg/kg) or vehicle after 2 and 12 h of SAH. SAH grading, neurological scores, brain water content, Evans blue extravasation, the myeloperoxidase activity, and malondialdehyde (MDA) content were measured 24 h after SAH. Expression of nuclear factor erythroid-related factor 2 (Nrf2), nuclear factor-kappa B (NF-κB) p65, and aquaporin 4 (AQP4) expression in rat brain were detected by Western blot. Expression of cyclooxygenase-2 (COX-2) and matrix metalloproteinase-9 (MMP-9) were determined by reverse transcription-polymerase chain reaction (RT-PCR). Expressions of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) were assessed by ELISA. Neurological scores, brain water content, Evans blue extravasation, the myeloperoxidase activity, and MDA content were significantly reduced by propofol. Furthermore, expression of Nrf2 in rat brain was upregulated by propofol, and expression of NF-κB p65, AQP4, COX-2, MMP-9, TNF-α, and IL-1β in rat brain were attenuated by propofol. Our results demonstrated that propofol improves neurological scores, reduces brain edema, blood-brain barrier (BBB) permeability, inflammatory reaction, and lipid peroxidation in rats of SAH. Propofol exerts neuroprotection against SAH-induced early brain injury, which might be associated with the inhibition of inflammation and lipid peroxidation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) is a devastating stroke with high morbidity and mortality. In the past decade, vasospasm and delayed cerebral ischemia have long been considered to be involved in the poor outcomes after SAH (Caner et al. 2012). In recent years, early brain injury (EBI), which occurs within 72 h of cerebral aneurysm rupture, has been reported to be the primary cause of high morbidity and mortality after SAH more often than delayed vasospasm (Sehba et al. 2012). EBI after SAH is a complex clinical process including increased blood-brain barrier (BBB) disruption, brain edema, inflammation, oxidative stress, and intracranial pressure (Fujii et al. 2013). Reduction of EBI through inhibiting BBB disruption, inflammation, and oxidative stress could be the therapeutic targets for SAH (Sabri et al. 2013).
Propofol, one of the widely used anesthetic agents for maintenance of anesthesia for surgical procedures, has been shown to reduce ischemic brain damage in rats of cerebral ischemia (Cui et al. 2012). In our previous research, we demonstrated that propofol attenuates the infiltration of neutrophil, downregulates the expression of NF-κB and COX-2 in ischemic brain, and decreases the release of TNF-α in blood in a rat model of permanent focal cerebral ischemia (Shi et al. 2014). To our knowledge, however, it is unclear whether also propofol attenuates EBI after SAH. In the present study, we first tried to investigate whether propofol provides a neuroprotection against EBI after SAH in rats then further explored the potential molecular mechanisms.
Materials and Methods
Animal Model
All animal experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals. Clean adult male Sprague-Dawley rats weighing 250–300 g were obtained from Shanghai Laboratory Animal Center, Chinese Academy of Sciences. Animals were housed in a colony room under controlled temperature (22 °C), and a 12:12 light-dark cycle, with food and water available. The rat SAH model was induced by endovascular perforation described in a previous study (Park et al. 2004), with minor modifications. Briefly, rats were anesthetized with an intraperitoneal injection of chloral hydrate (300 mg/kg), and the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were carefully exposed through a ventral midline neck incision, a blunted 3–0 monofilament nylon suture was inserted into the ICA until resistance was felt (18–20 mm from the common carotid bifurcation). The suture was carefully pushed approximately 3 mm further to perforate the artery wall to create a SAH. Sham operation rats were manipulated in the same way without perforation.
Experimental Groups
Rats were divided into four groups: (i) sham operation group, which underwent sham operation and received vehicle; (ii) SAH group, which was subjected to SAH and received vehicle; (iii) propofol 10 mg/kg group (P10), which was subjected to SAH and treated with propofol 10 mg/kg; (iv) propofol 50 mg/kg group (P50), which was subjected to SAH and treated with propofol 50 mg/kg. Rats were treated with normal saline as the vehicle control at the same volume as propofol.
SAH Severity
The severity of the SAH was quantified as reported previously (Sugawara et al. 2008). The scale was based on the amount of subarachnoid blood in six segments of basal cistern: grade 0, no subarachnoid blood; grade 1, minimal subarachnoid blood; grade 2, moderate blood clot with recognizable arteries; grade 3: blood clot obliterating all arteries within the segment. A total score ranging from 0 to 18 was obtained by adding the scores from all six segments.
Neurological Scores
Neurological scores were evaluated after 24 h of SAH according to the scoring system of Garcia (Garcia et al. 1995) with minor modification. Briefly, the neurobehavioral study consisted of six tests, including spontaneous activity, symmetry in the movement of four limbs, forepaw outstretching, climbing, body proprioception, and response to vibrissae touch. A lower score represents serious deficit of neurological function. Neurological scores were assessed by a “blinded” co-worker.
Assessment of Brain Edema
Rats were killed 24 h after SAH, and the brains were removed. The rat brains were gently blotted with filter paper and weighed on an electronic balance, as the wet weights (WW), and then dried for 24 h in 100 °C vacuum oven to obtain the dry weights (DW). Cerebral water content was calculated according to the following formulation: H2O (%) = (WW − DW) / WW × 100 %.
Measurement of BBB Permeability
BBB permeability was assessed by measurement of Evans blue (EB) extravasation. EB dye (2 % in saline) was injected into the left jugular vein 23 h after SAH. Then, the rats were transcardially perfused with PBS to remove the intravascular dye; rat brains were homogenized in a tenfold volume of 50 % trichloroacetic acid solution to precipitate protein and centrifuged for 10 min at 3000 r/min. The supernatant was diluted with ethanol (1:3), and its fluorescence was measured at 610 nm for absorbance of EB. The results were expressed as micrograms per gram tissue.
Biochemical Analysis
Neutrophil infiltration was estimated by measuring the enzymatic activity of myeloperoxidase (MPO). MPO activity in rat brain was detected according to the manufacturer’s instructions from the assay kit (Nanjing Jiancheng Bioengineering Institute, China). The results were expressed as units per gram tissue.
Lipid peroxidation was evaluated by measuring the formation of malondialdehyde (MDA). The content of MDA in rat brain was measured according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, China). The results were expressed as micromoles per gram protein.
Western Blot
Brain samples were used and total protein was extracted using protein extraction kit (Beyotime Biotech. Co., China) according to the manufacturer’s instructions. Protein samples (50 μg) were separated on 10 % SDS polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were respectively incubated at 4 °C for 2 h with a mouse monoclonal antibody against Nrf2 (1:500, Abcam), mouse monoclonal antibody against NF-κB p65 (1:200, Santa Cruz), and mouse monoclonal antibody against AQP4 (1:200, Santa Cruz). The nitrocellulose membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:3000, Zhongshan Biotechnology Co. LTD, Beijing) for 2 h at 25 °C and developed with an enhanced chemiluminescence detection system. GAPDH was used as a loading control. The optical densities of protein bands were analyzed by the Quantity One software (Bio-Rad).
Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted from brain sample using TRIzol reagents (Invitrogen, USA) and reverse-transcribed to obtain single-strand cDNA using a Reverse Transcription System (Promega, USA) according to the manufacturer’s instructions. Single-strand cDNA was amplified by polymerase chain reaction (PCR) in a 100-μl reaction mixture containing 50 mM KCl, 10 mM Tris-HCl (pH 9.0), 2 mM MgCl2, 200 μM dNTPs, 0.5 μM sense and antisense primers, and 2.5 units Taq DNA polymerase (Promega, USA). The primer sequences were as follows: COX-2 (primers, sense 5′-CTT CGG GAG CAC AAC AGA-3′; antisense 5′-GCG GAT GCC AGT GAT AGA-3′, product size 253 bp), MMP-9 (primers, sense 5′-TCC AGT AGA CAA TCC TTG CAA TGT G-3′; antisense 5′-CTC CGT GAT TCG AGA ACT TCC AAT A-3′, product size 110 bp), β-actin as an internal standard (primers, sense 5′-CCC ATC TAT GAG GGT TAC GC-3′; antisense 5′-TTT AAT GTC ACG CAC GAT TTC-3′, product size 150 bp). The reactions were initially heated at 94 °C for 4 min then at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s, totally 30 cycles, and finally stopped at 72 °C for 10 min. PCR products were electrophoresed through 2 % agarose gels containing ethidium bromide (EB, 0.5 μg/ml). The optical densities of DNA bands were analyzed using the UVP gel analysis system (Quantity One, Bio-Rad).
ELISA
Twenty-four hours after the induction of SAH, the frozen brain tissue (200 mg) was homogenized with a plastic homogenizer in cell lysis buffer containing 1 mM phenylmethanesulfonylfluoride (PMSF), 20 mM Tris pH 7.5, 150 mM NaCl, 1 % Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM EDTA, 1 % Na3VO4, and 0.5 μg/ml leupeptin and centrifuged at 14,000g for 20 min at 4 °C. The contents of cytokines (TNF-α and IL-1β) in the brain tissue were measured using a rat TNF-α or IL-1β immunoassay enzyme-linked immunosorbent assay (ELISA) kits (Boster Biological Technology, LTD, Wuhan, China) according to the manufacturer’s instructions. The results were expressed as picograms per milliliter homogenate.
Statistical Analysis
Experimental data were presented as mean ± SD. Statistical analysis was performed using ANOVA followed by Bonferroni test for individual comparisons between group means (SPSS 13.0 for Windows, USA). A value of P < 0.05 was considered statistically significant.
Results
Effect of Propofol on SAH Grade and Neurological Scores
After 24 h of SAH, the rat brains were quickly obtained; subarachnoid blood clots were found on the ipsilateral side, around the circle of Willis, and within the ventral surface of the brain stem (Fig. 1a). Propofol (10 and 50 mg/kg) administration did not alter the grade of SAH when compared with SAH + vehicle group (P > 0.05, Fig. 1b). The neurological scores were reduced after 24 h of SAH, which were significantly improved by propofol (10 and 50 mg/kg) administration (P < 0.01, Fig. 1c).

SAH grading score (a, b), effect of propofol on neurological scores (c), cerebral water content (d), and BBB permeability (e) after 24 h of SAH. The upper panels (a) show representative macroscopic pictures of brain after 24 h of SAH, and the SAH scores were summarized in (b). Subarachnoid blood clots exist equally at the ventral surface of the brain and brain stem between the SAH groups. The neurological scores were reduced 24 h after SAH, which were significantly improved by propofol. The brain water content and EB content were increased 24 h after SAH, which were attenuated by propofol. # P < 0.01 compared to sham operation group; *P < 0.05 and **P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg; NS not significant
Effect of Propofol on Brain Edema and BBB Permeability
The brain water content was elevated after 24 h of SAH, which was attenuated by propofol (10 and 50 mg/kg) administration (P < 0.01, Fig. 1d). The EB content was obviously increased after 24 h of SAH, which was attenuated by propofol (10 and 50 mg/kg) administration (P < 0.05 and P < 0.01, Fig. 1e).
Effect of Propofol on Neutrophil Infiltration and Lipid Peroxidation
Experimental results demonstrated that the enzymatic activity of MPO was increased 24 h after SAH, which was significantly attenuated by administration of propofol 10 or 50 mg/kg (P < 0.01 and P < 0.05, Fig. 2a). The MDA content was increased 24 h after SAH, which was significantly attenuated by propofol administration (10 and 50 mg/kg, P < 0.01, Fig. 2b).

Propofol reduced the MPO activity and MDA content. SAH caused an increase of MPO activity and MDA content, which were significantly attenuated by propofol administration. n = 5; # P < 0.01 compared to sham operation group; *P < 0.05 and ** P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg
Effect of Propofol on Protein Expression of Nrf2, NF-κB p65, and AQP4
Experimental results showed that expression of Nrf2 was downregulated after 24 h of SAH, which was upregulated by administration of propofol 10 or 50 mg/kg (P < 0.01 and P < 0.05, Fig. 3). SAH causes an increased expression of NF-κB p65 and AQP4 in rat brain compared with sham operation group (P < 0.01), which were inhibited by propofol (10 and 50 mg/kg, P < 0.01, Figs. 4 and 5)

Propofol upregulates the protein expression of Nrf2. Representative protein bands of Nrf2 expression in sham-operated, vehicle-treated, and propofol-treated groups were detected by Western blot (a), and the data were summarized in b. n = 5; # P < 0.01 compared to sham operation group; *P < 0.05 and **P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg

Propofol downregulates the protein expression of NF-κB p65. Representative protein bands of NF-κB p65 expression in sham-operated, vehicle-treated, and propofol-treated groups were detected by Western blot (a), and the data were summarized in b. n = 5, # P < 0.01 compared to sham operation group, **P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg

Propofol downregulates the protein expression of AQP4. Representative protein bands of AQP4 expression in sham-operated, vehicle-treated, and propofol-treated groups were detected by Western blot (a), and the data were summarized in b. n = 5, #P < 0.01 compared to sham operation group, **P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg
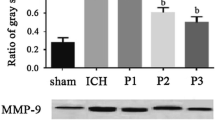
Effect of Propofol on mRNA Expression of COX-2 and MMP-9
Experimental results showed that SAH causes an increased mRNA expression of COX-2 and MMP-9 in rat brain compared with sham operation group (P < 0.01). Propofol (10 and 50 mg/kg) significantly downregulated the mRNA expression of COX-2 and MMP-9 in rats of SAH (P < 0.01, Fig. 6).

Propofol downregulates the mRNA expression of COX-2 and MMP-9. Representative bands of COX-2 and MMP-9 in rat brain from sham-operated, vehicle-treated, propofol-treated rats (a), and the data were summarized in b. n = 5, # P < 0.01 compared to sham operation group, **P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg
Effect of Propofol on Expression of Inflammatory Cytokines
Experimental results showed that SAH causes an increased expression of inflammatory cytokines (TNF-α and IL-1β) in rat brain compared with sham operation group (P < 0.01). Propofol (10 and 50 mg/kg) significantly downregulated the expression of TNF-α and IL-1β in rats of SAH (P < 0.01, Fig. 7).

Propofol downregulates the expression of TNF-α and IL-1β. SAH caused an increased expression of TNF-α and IL-1β in rat brain, which were significantly attenuated by propofol 10 and 50 mg/kg. n = 5, # P < 0.01 compared to sham operation group, **P < 0.01 compared to SAH group. P10 means propofol 10 mg/kg, P50 means propofol 50 mg/kg
Discussion
The recent study from our laboratory showed that propofol reduces ischemic brain injury following permanent cerebral ischemia in rats, and the mechanism was involved in the inhibition of pro-inflammatory NF-κB signaling pathway. In this study, we found that (a) propofol reduces SAH-induced early brain injury (EBI); (b) propofol attenuates SAH-induced brain edema and BBB breakdown; (c) propofol decreases neutrophil infiltration and lipid peroxidation; (d) propofol upregulates the low expression of Nrf2; (e) propofol downregulates the overexpression of NF-κB p65, AQP4, COX-2, MMP-9, TNF-α, and IL-1β. These findings demonstrated that propofol attenuates early brain injury after subarachnoid hemorrhage in rats, which might be associated with the inhibition of oxidative stress and inflammation through activating the Nrf2 anti-oxidative signaling and inhibiting pro-inflammatory NF-κB pathway.
The key pathological hallmark of EBI following SAH was neuronal death, BBB breakdown, and brain edema, which were mainly attributed to inflammatory reaction and oxidative stress (Uekawa et al. 2014; Ayer and Zhang 2008). SAH enhances reactive oxygen species (ROS) in the early phase, and ROS causes neuronal death and BBB breakdown in EBI after SAH; BBB breakdown and inflammatory reaction lead to vasogenic brain edema, eventually inducing exacerbation of neurological dysfunction (Sehba et al. 2012; Zhang et al. 2015). Brain edema is the main reason for increased intracranial pressure, a secondary pathophysiological change of SAH. In this study, the changes of brain water content were measured by the dry-wet method, and results showed that the brain water content was significantly increased after SAH in rats, propofol markedly reduced brain water content compared with SAH group. Further studies showed that MPO activity, MDA content, and BBB permeability were increased after SAH in rats, which were improved by propofol, indicating that propofol reduces early brain injury after SAH might be associated with the inhibition of inflammation, lipid peroxidation, and BBB breakdown. It deserves to be mentioned that propofol 10 mg/kg reduces neutrophil infiltration more obviously than propofol 50 mg/kg, demonstrating that a small dose of propofol would play an important role against inflammatory reaction following SAH.
Nuclear factor-kappa B (NF-κB), a pro-inflammatory transcription factor, has been implicated in the pathogenesis of several disease categories, including cerebral ischemia (Tu et al. 2014) and SAH (Zhang et al. 2014). Cyclooxygenase-2 (COX-2), an inflammatory enzyme that mediates the process of ischemic brain damage, was elevated after cerebral ischemia in the previous study (Tu et al. 2015). COX-2 was also over-expressed in rat brain after SAH in the present study. Tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), the pro-inflammatory cytokines regulated by NF-κB signaling, were reported to mediate the inflammatory process of brain damage after SAH (Altay et al. 2014). Our previous studies showed that propofol inhibits the expression of NF-κB and downregulates the expression of COX-2 and TNF-α in rats following focal cerebral ischemia (Shi et al. 2014). In order to provide the molecular mechanisms underlying the propofol’s neuroprotection against SAH-induced inflammatory reaction, we determined the effect of propofol on the expression of pro-inflammatory NF-κB signaling pathway. The experimental results demonstrated that propofol significantly inhibits the activation of NF-κB pathway, downregulates the mRNA expression of COX-2, and decreases the content of TNF-α and IL-1β in rats 24 h following SAH.
MDA content, an indicator of lipid peroxidation, was elevated in rat brain after SAH, which verified the oxidative damage in brain tissue (Ersahin et al. 2010). Our results demonstrated that propofol reduced MDA content in rats of SAH; therefore, the underlying neuroprotection of propofol might be in part associated with the inhibitory effect of propofol on the oxidative damage. Nuclear factor erythroid-related factor 2 (Nrf2) mediated anti-oxidative signaling in the EBI following SAH (Wang et al. 2012a). Activation of Nrf2 signaling has been clarified to play a beneficial role in EBI after SAH, possibly through inducing antioxidant to reduce cerebral oxidative stress and BBB breakdown (Chen et al. 2011; Wang et al. 2014). Propofol was demonstrated to alleviate liver oxidative stress via activation of Nrf2 signaling pathway (Ge et al. 2015). In the present study, Nrf2 expression was decreased 24 h after SAH, which was upregulated by propofol administration, demonstrating that propofol reduces lipid peroxidation and BBB breakdown might be associated with the activation of Nrf2 signaling.
Brain edema, one of the most important pathogenesis mechanisms in EBI after SAH, is mainly attributed to BBB breakdown and consequently resulted in elevation of intracranial pressure, reduction of cerebral blood flow, and suppression of cerebral perfusion (Westermaier et al. 2012). Aquaporin-4 (AQP-4) and matrix metalloproteinase-9 (MMP-9) have been demonstrated to be involved in the pathophysiological processes of BBB dysfunction and brain edema in cerebral ischemia and have become potential pharmacological targets for ischemic stroke (Li et al. 2013). However, the report is lesser about the roles of AQP-4 and MMP-9 in EBI following SAH.
AQP-4 may play an important role in the formation of cytotoxic brain edema by regulating water balance across brain compartments (Papadopoulos and Verkman 2007). AQP-4-associated brain edema might be modulated by the upregulation of the water channel (Yao et al. 2015). MMP-9 was demonstrated to induce the degradation of extracellular matrix and tight junction protein, leading to BBB leakage, vasogenic brain edema, and secondary brain damage (Tu et al. 2011). Wang et al. (2012b) showed that AQP4 and MMP-9 expression was significantly increased after SAH, which is consistent with increased BBB extravasation and brain edema. Ji et al. (2015) demonstrated that propofol post-conditioning protects BBB integrity and improves neurobehavioral outcome via decreasing the expression of AQP4 and MMP-9 in cerebral ischemia injury. The present study showed that expression of AQP4 protein and MMP-9 mRNA were increased after 24 h of SAH, which were inhibited by propofol administration, indicating that propofol attenuates brain edema, and BBB breakdown might be attributed to the inhibition of AQP4 and MMP-9.
In the present study, the experimental data showed that propofol 50 mg/kg was more effective than propofol 10 mg/kg in improving neurological scores and reducing brain edema and BBB breakdown, but propofol 50 mg/kg was more effective than propofol 10 mg/kg in attenuating the activities of MPO, and no difference between propofol 10 and 50 mg/kg in the expression of NF-κB, AQP4, TNF-α, and IL-1β, which might be associated with the drug’s effect specificity. Those results demonstrated that propofol protects early brain injury after subarachnoid hemorrhage not only via anti-inflammatory effect through inhibiting the expression of NF-κB, AQP4, TNF-α, and IL-1β, but also through other pathological mechanisms and molecular mechanisms, which needs further exploration in our subsequent research.
In summary, to our best knowledge, this is the first demonstration of propofol’s neuroprotection against early brain injury after SAH. Propofol reduces EBI following SAH via attenuating inflammatory reaction, oxidative stress, and BBB breakdown through inhibition of pro-inflammatory NF-κB pathway, activation of anti-oxidative Nrf2 signaling, and downregulated expression of AQP4 and MMP-9. NF-κB, Nrf2 AQP4, and MMP-9 may become the potential therapeutic targets for SAH in future.
References
Altay O, Suzuki H, Hasegawa Y, Ostrowski RP, Tang J, Zhang JH (2014) Isoflurane on brain inflammation. Neurobiol Dis 62:365–371
Ayer RE, Zhang JH (2008) Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm. Acta Neurochir Suppl 104:33–41
Caner B, Hou J, Altay O, Fujii M, Zhang JH (2012) Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. J Neurochem 123(Suppl 2):12–21
Chen G, Fang Q, Zhang J, Zhou D, Wang Z (2011) Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J Neurosci Res 89(4):515–523
Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W (2012) Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One 7(4), e35324
Ersahin M, Toklu HZ, Erzik C et al (2010) The anti-inflammatory and neuroprotective effects of ghrelin in subarachnoid hemorrhage-induced oxidative brain damage in rats. J Neurotrauma 27(6):1143–1155
Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH (2013) Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res 4(4):432–446
Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26(4):627–634, discussion 635
Ge M, Yao W, Wang Y et al (2015) Propofol alleviates liver oxidative stress via activating Nrf2 pathway. J Surg Res 196(2):373–381
Ji FT, Liang JJ, Miao LP, Wu Q, Cao MH (2015) Propofol postconditioning protects the blood brain barrier by decreasing matrix metalloproteinase9 and aquaporin4 expression and improves the neurobehavioral outcome in a rat model of focal cerebral ischemia reperfusion injury. Mol Med Rep 12(2):2049–2055
Li M, Ma RN, Li LH, Qu YZ, Gao GD (2013) Astragaloside IV reduces cerebral edema post-ischemia/reperfusion correlating the suppression of MMP-9 and AQP4. Eur J Pharmacol 715(1-3):189–195
Papadopoulos MC, Verkman AS (2007) Aquaporin-4 and brain edema. Pediatr Nephrol 22(6):778–784
Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH (2004) Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke 35(10):2412–2417
Sabri M, Lass E, Macdonald RL (2013) Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013:394036
Sehba FA, Hou J, Pluta RM, Zhang JH (2012) The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 97(1):14–37
Shi SS, Yang WZ, Chen Y, Chen JP, Tu XK (2014) Propofol reduces inflammatory reaction and ischemic brain damage in cerebral ischemia in rats. Neurochem Res 39(5):793–799
Sugawara T, Ayer R, Jadhav V, Zhang JH (2008) A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods 167(2):327–334
Tu XK, Yang WZ, Liang RS et al (2011) Effect of baicalin on matrix metalloproteinase-9 expression and blood–brain barrier permeability following focal cerebral ischemia in rats. Neurochem Res 36(11):2022–2028
Tu XK, Yang WZ, Chen JP et al (2014) Curcumin inhibits TLR2/4-NF-kappaB signaling pathway and attenuates brain damage in permanent focal cerebral ischemia in rats. Inflammation 37(5):1544–1551
Tu XK, Yang WZ, Chen JP et al (2015) Repetitive ischemic preconditioning attenuates inflammatory reaction and brain damage after focal cerebral ischemia in rats: involvement of PI3K/Akt and ERK1/2 signaling pathway. J Mol Neurosci 55(4):912–922
Uekawa K, Hasegawa Y, Ma M et al (2014) Rosuvastatin ameliorates early brain injury after subarachnoid hemorrhage via suppression of superoxide formation and nuclear factor-kappa B activation in rats. J Stroke Cerebrovasc Dis 23(6):1429–1439
Wang Z, Ma C, Meng CJ et al (2012a) Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J Pineal Res 53(2):129–137
Wang Z, Meng CJ, Shen XM et al (2012b) Potential contribution of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 to blood–brain barrier disruption and brain edema after experimental subarachnoid hemorrhage. J Mol Neurosci 48(1):273–280
Wang Z, Ji C, Wu L et al (2014) Tert-butylhydroquinone alleviates early brain injury and cognitive dysfunction after experimental subarachnoid hemorrhage: role of Keap1/Nrf2/ARE pathway. PLoS One 9(5), e97685
Westermaier T, Stetter C, Raslan F, Vince GH, Ernestus RI (2012) Brain edema formation correlates with perfusion deficit during the first six hours after experimental subarachnoid hemorrhage in rats. Exp Transl Stroke Med 4(1):8
Yao X, Derugin N, Manley GT, Verkman AS (2015) Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci Lett 584:368–372
Zhang XS, Zhang X, Wu Q et al (2014) Astaxanthin offers neuroprotection and reduces neuroinflammation in experimental subarachnoid hemorrhage. J Surg Res 192(1):206–213
Zhang T, Su J, Guo B, Wang K, Li X, Liang G (2015) Apigenin protects blood–brain barrier and ameliorates early brain injury by inhibiting TLR4-mediated inflammatory pathway in subarachnoid hemorrhage rats. Int Immunopharmacol 28(1):79–87
Acknowledgments
The National Natural Science Foundation of China (81100987), the Doctoral Program Foundation of Institutions of Higher Education of China (20113518120005), the Natural Science Foundation of Fujian Province of China (2011 J05066), the Clinical Key Subject (Neurosurgery) Funding of Fujian Medical University, and the Key Laboratory (Neurosurgical Department) Funding from the Affiliated Union Hospital of Fujian Medical University, supported this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Song-sheng Shi and Hua-bin Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Shi, Ss., Zhang, Hb., Wang, Ch. et al. Propofol Attenuates Early Brain Injury After Subarachnoid Hemorrhage in Rats. J Mol Neurosci 57, 538–545 (2015). https://doi.org/10.1007/s12031-015-0634-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0634-2