
Abstract
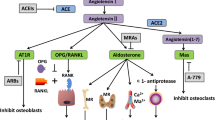
The RAS (renin–angiotensin system) plays a key role in the regulation of blood pressure, fluid and electrolyte homeostasis and cardiovascular and renal structure and function. There is evidence that in addition to the systemic RAS the components of the RAS are expressed in the local milieu of bone, where angiotensin II increases the osteoclastogenesis while inhibit the osteoblastic activity leading to a decrease in bone mineral density. Hypertension and osteoporosis are two common diseases that frequently coexist in the elderly population, and it has been hypothesized that the activation of the local RAS might be involved in the occurrence of both diseases often seen with advancing age. Epidemiological studies have found that RAS inhibitors, including angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, may exert a beneficial effect on bone mineral density, increasing the bone mass and decreasing the risk of bone fractures in patients with osteoporosis and cardiovascular diseases, and might accelerate the fracture healing process. However, both experimental and clinical studies with these RAS inhibitors led to sparse and contradictory results. Thus, in the next future a better understanding on how the components of the local RAS influence bone metabolism and remodeling will allow us to select the best therapeutic strategy for patients with osteoporosis and cardiovascular diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The circulating endocrine renin–aldosterone system (RAS) plays a key role in the regulation of arterial blood pressure, volume and electrolyte balance and cardiovascular and renal structure and function [1–5]. RAS activation has been implicated in the development of age-related cardiovascular, metabolic and kidney diseases, and elevated plasma levels of RAS peptides are associated with worse clinical outcomes [1–7]. Furthermore, RAS also exerts a much broader range of effects in other organs, as it plays an important role in tissue repair and remodeling, cognitive and autonomic functions, embryonic development and reproduction [5, 6, 9–12]. Conversely, RAS inhibitors, including angiotensin-converting enzyme inhibitors (ACEIs), which inhibit the conversion of angiotensin (Ang) I into Ang II, and angiotensin AT1 receptor (AT1R) blockers (ARBs), which block the binding and the responses of Ang II mediated via AT1R activation, are widely used for the prophylaxis and treatment of these diseases [1–10]. Interestingly, recent evidence confirmed the existence of the RAS components in the bone and activation of this local RAS stimulates the expression of osteoclastogenic mediators in the osteoblasts. Thus, it has been hypothesized that local RAS activation may play an important role in bone homeostasis, leading to imbalance between bone formation and resorption, characteristic of various bone disorders, such as osteoporosis, independent of the involvement of systemic RAS. This was the basis for the clinical use of ACEIs and ARBs to increase bone mineral density (BMD), decrease fractures and accelerate the fracture healing process in patients with osteoporosis and concomitant cardiovascular comorbidities (i.e., hypertension, heart failure, renal disease or diabetic nephropathy).
In this article, we have reviewed the systemic and local RAS, the evidence supporting the presence of a local RAS and the effects of Ang II in the bone. Then, we analyzed the role of RAS in the genesis of osteoporosis, the effects of ACEIs and ARBs in experimental models and clinical trials and the possible limitations of these studies to identify the possible gaps of knowledge that should be answered in the near future.
Bone Remodeling
Bone is a metabolically active tissue that undergoes continuous remodeling during life, a process by which old microdamaged bone is removed and replaced with newly synthesized bone to maintain bone strength and mineral homeostasis [13, 14]. Bone homeostasis depends upon the balanced function of osteoblasts, which synthesize bone matrix proteins and promote bone deposition and mineralization, and osteoclasts, which are the principal bone-resorpting cells, removing both the mineral and the organic matrix of bone (Fig. 1). Mechanical forces, proinflammatory cytokines, growth factors and hormones cause an imbalance between osteoclast and osteoblast activities and in the rate of bone remodeling which result in bone-related diseases, including postmenopausal osteoporosis, hyperparathyroidism, rheumatoid arthritis and osteopetrosis.

RANK–RANKL–OPG signaling cascades during osteoclastogenesis. Akt protein kinase B, Ang II angiotensin II, AP-1 activator protein-1, Btk/Tec Tec family tyrosine kinases, BNLK/SLP76 B cell linker/SH2 domain-containing leukocyte protein of 76 kDa, CaMKIV Ca2+/calmodulin-dependent protein kinase IV, c-fms colony-stimulating factor-1 receptor, c-fos proto-oncogene c-Fos, CREB cyclic adenosine monophosphate responsive-element-binding protein, CTR calcitonin receptor, ERK extracellular signal-regulated kinase, GRB2 growth factor receptor-bound protein 2, IKK I kappa B kinase, JNK c-Jun N-terminal kinase, M-CSF macrophage colony-stimulating factor, MAPK mitogen-activated protein kinases, MEK1/2 mitogen-activated protein kinase kinase, MEKK MAP kinase, MITF microphthalmia-associated transcription factor, MKK MAPK kinases, mTOR mammalian target of rapamycin, NFATc1 nuclear factor of activated T cells cytoplasmic 1, NF-κB nuclear factor-κB, OPG osteoprotegerin, OSCAR osteoclast-associated receptor, p38 p38 MAPK, PI3K phosphoinositide 3-kinase, PLCγ phospholipase Cγ, PU.1 transcription factor PU, Raf receptor tyrosine kinase effector, RANK receptor activator of nuclear factor-κB, RANKL receptor activator of nuclear factor-κB ligand, SLP-76 Src homology 2 (SH2) domain-containing leukocyte protein of 76 kDa, Src proto-oncogene tyrosine-protein kinase, TRAF6 tumor necrosis factor receptor-associated factor 6, TRACP tartrate-resistant acid phosphatase
Bone resorption is a multistep process initiated by the recruitment and activation of osteoclast precursors derived from mononuclear monocyte/macrophage-lineage hematopoietic cells located in the bone marrow cavity and blood stream followed by the commitment of these cells to the osteoclast phenotype [13–16]. Macrophage colony-stimulating factor (M-CSF) and RANK–RANKL–OPG signaling pathway are two major factors in osteoclast differentiation [17] (Fig. 1).
The RANK–RANKL–OPG Signaling Pathway
It is composed of three elements [18–21]:
-
(a)
The receptor activator of nuclear factor-κB or RANK (TNFRSF11A) which is strongly induced, especially on osteoclast precursor cells by M-CSF.
-
(b)
The RANK-Ligand (RANKL, TNFSF11) is expressed in osteoblastic lineage cells, synovial fibroblasts, bone marrow stromal cells and activated T cells [21, 22]. Binding of RANKL to RANK on the surface of pre-osteoclasts and mature osteoclasts provides the signal to induce osteoclast differentiation from precursors to mature osteoclasts in the presence of M-CSF, and stimulates the bone-resorbing activity and survival of mature osteoclasts [18–23] (Fig. 2). Most of the factors that induce osteoclast differentiation and stimulate osteoclast formation and activity induce RANKL expression by osteoblasts, including proinflammatory cytokines [interleukin-1 (IL-1) and IL-11 and tumor necrosis factor α (TNFα)], parathyroid hormone (PTH), prostaglandin E2, glucocorticoids, 1,25 dihydroxyvitamin D3 and Wnt ligands, while estrogens or TGFβ decrease RANKL expression [21, 24, 25]. Conversely, RANKL-deficient mice present severe osteopetrosis due to a complete lack of osteoclasts [26–28].
Fig. 2 
Schematic representation of the osteoblast–osteoclast interaction. RANKL expression is induced in osteoblasts and bone marrow stromal cells. RANKL binds to its specific membrane-bound receptor RANK located in pre-osteoclasts and mature osteoclasts promoting osteoclast differentiation, activation and survival. Proresorptive factors promote the RANK–RANKL interaction, while anti-resorptive actors increase osteoprotegerin (OPG) expression. OPG binds and neutralizes RANKL, blocks osteoclastogenesis and decreases survival of preexisting osteoclasts. Mature osteoclasts polarize, adhere to the bone surface and degrade the organic and inorganic phases of bone by secreting H+ and Cl− ions [via the activation of the carbonic anhydrase II and vacuolar ATPase proton pumps, and the Cl−/H+ antiporter ClC-7, respectively] and lytic enzymes [cathepsin K, tartrate-resistant acid phosphatase (TRAP) and matrix metalloproteinases-3 and 9] into a sealed resorption vacuole formed between the basal surface of the osteoclast and the bone surface. Ang II angiotensin II, ALP alkaline phosphatase, CLCN7 chloride channel, IL interleukin, PDGF platelet-derived growth factor, PGE2 prostaglandin E2, OPG osteoprotegerin, PTH parathyroid hormone, RANK receptor activator of nuclear factor-κB, RANKL receptor activator of nuclear factor-κB ligand, TGF-β1 transforming growth factor beta-1, TNFα tumor necrosis factor α, TRAP tartrate-resistant acid phosphatase
-
(c)
Osteoprotegerin (OPG, TNFRSF11B) is secreted by various cell types, including B lymphocytes, osteogenic stromal stem cells and osteoblasts in response to anabolic agents such as estrogens and TGFβ-related bone morphogenetic proteins [29]. OPG acts as a decoy receptor that binds to RANKL and prevents RANKL binding to RANK, exerting osteoprotective effects by inhibiting osteoclast differentiation and activation and promoting osteoclast apoptosis [30]. OPG-deficient mice develop early-onset osteoporosis, accelerated osteoclastogenesis, vascular inflammation and calcification of aorta and renal arteries, while OPG overexpression precipitates osteopetrosis [22, 31, 32]. Thus, the RANKL/OPG ratio is critical for controlling osteoclast differentiation and bone resorptive function, so that the osteoclast number and activity increased when the RANKL/OPG ratio increases due to either an increase in RANKL or a decrease in OPG [18, 22, 33]. Postmenopausal osteoporosis is associated with a high rate of bone remodeling and an increased RANKL/OPG ratio [24, 34], and OPG treatment can reverse the accelerated osteoclastogenesis and bone loss in ovariectomized (OVX) female rats [22].

Osteoclastogenic Differentiation
As already mentioned binding of RANKL to RANK commits monocytic precursor cells to the osteoclastic lineage. RANK lacks intrinsic kinase activity, and thus, it must interact with several adaptor and docking proteins [TNF receptor-associated factors (TRAFs), Gab2 and Cbl] to activate downstream signaling pathways (Fig. 1). Binding of RANKL to RANK, located on the surface of pre-osteoclasts and mature osteoclasts, leads to receptor trimerization followed by the recruitment of the cytoplasmic adaptor protein TRAF6, leading to the activation of several downstream signaling pathways, including [18–21, 25, 28, 34]:
-
(a)
The IKK (I kappa B kinase complex)-NF-κB (nuclear factor-kappa B) signaling pathway.
-
(b)
Three MAPKs, MKK6-p38-MITF (microphthalmia-associated transcription factor), MEKK7-JNK1-c-Jun and Raf-MEK1/2-ERK1/2 (extracellular receptor kinases)-c-fos AP-1 that are essential for the differentiation of monocytic precursors into osteoclasts.
-
(c)
The c-Sr-PI3K/AKt (phosphatidylinositol 3-kinase/Akt) signaling pathway through the interaction with TRAF6 and Cbl proteins, and.
-
(d)
The Tec family of cytoplasmic tyrosine kinases [Bruton’s tyrosine kinase (Btk) and Tec] that activates phospholipase C (PLC) and regulates several cellular functions including Ca2+ influx, cytoskeletal remodeling, apoptosis and proliferation or differentiation.
-
(e)
NF-κB, c-fos and AP-1 (activator protein-1 complex, which consists of c-Fos, c-Jun and ATF proteins) upregulate the expression of a key molecule for osteoclast differentiation, the nuclear factor of activated T cells cytoplasmic 1 (NFATc1) transcription factor, which translocates into the nucleus after dephosphorylation by calcineurin, where it binds to its own promoter, resulting in the autoamplification of NFATc1 expression [15–20, 35]. Additionally, activation of MAPK pathways promotes translocation and activation of other transcription factors, including c-Fos, AP-1 (activator protein-1 complex, which consists of c-Fos, c-Jun and ATF proteins), Fra-1 (Fos-related antigen 1), CREB (cAMP response element-binding), NF-κB, PU.1 [a member of the E-26 (ETS) family of transcription factors) and MIFT. The cooperation of all these factors regulates the transcription of several target genes involved in osteoclast differentiation, including cathepsin K, calcitonin receptor (CTR), TRACP (encoding tartrate-resistant acid phosphatase), β3 integrin and osteoclast-associated receptor (OSCAR).
The Classical Renin–Angiotensin System
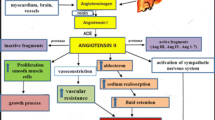
In the classical view of the RAS (Fig. 3), angiotensinogen (AGT), an α-2 macroglobulin released by the liver, is enzymatically cleaved to the inactive decapeptide angiotensin I (Ang I) by renin (EC 3.4.23.15), a highly selective protease secreted from the juxtaglomerular cells of the renal afferent arterioles in response to various physiological stimuli, including a decrease in arterial blood pressure, decreased sodium reabsorption in the distal tubule, decreased blood volume and stimulation of β1-adrenoreceptors [4–6].

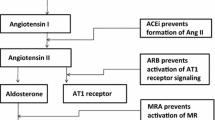
Mechanism of action of angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin AT1 receptor blockers (ARBs). AT1R angiotensin AT1 receptor, AT2R angiotensin AT2 receptor, B1/B2 bradykinin receptor B1 and B2, ET-1 endothelin-1, gK potassium conductance, NO nitric oxide, PG prostaglandin, ROS reactive oxygen species, ↑ increase
Ang I is then cleaved, mainly in the lung, by the angiotensin-converting enzyme (ACE, EC 3.4.15.1) to the octapeptide angiotensin II [Ang II or Ang (1–8)], the predominant physiologically active peptide of RAS. Ang II exerts multiple biological effects through its type 1 (AT1R, 359 amino acids, 40 kDa) and type 2 receptors (AT2R, 363 amino acids, 41 kDa), members of the 7-transmembrane-spanning G protein-coupled receptors exhibiting only ~34 % amino acid sequence identity [12, 36, 37]. The homology is mainly localized in the seven membrane-spanning domains or transmembrane helices. However, the AT1R contains three potential N-glycosylation sites [38], while the AT2R contains five N-glycosylation sites located exclusively in the extracellular N-terminal domain and almost complete divergence between AT1R and AT2R is observed in the third intracellular loop and mainly in the C-terminal tail [12, 36, 37]. Two AT1R subtypes (AT1aR and AT1bR) have been found in mouse and rat, but they show 96 % identity with each other [12, 36].
Signal Transduction from the AT1R
Following the stimulation of AT1R present in almost all tissues coupled to Gq/11 and Gi/o proteins, Ang II activates multiple second-messenger signal transduction pathways, including [7, 12, 39–41]:
-
(a)
Activation of phospholipases Cγ1 (which generates inositol 1,4,5-triphosphate and increase in intracellular Ca2+ levels), A2 and D.
-
(b)
Inhibition of adenylate cyclase leading to a decrease in the cellular levels of adenosine 3′,5′-cyclic monophosphate (cAMP).
-
(c)
Activation of various intracellular protein kinases: mitogen-activated protein kinases [MAPKs: ERK 1/2 (extracellular signal-regulated kinases), JNK (c-Jun N-terminal kinase) and p38], receptor tyrosine kinases [PDGF (platelet-derived growth factor), EGFR (epidermal growth factor receptor), PI3K (phosphoinositide 3-kinase)-Akt pathway], RhoA (ras homolog family member A), non-receptor tyrosine kinases [proto-oncogene tyrosine-protein kinase c-Src, focal adhesion kinase (FAK), protein tyrosine kinase 2 beta (PYK2), Janus kinase 2 (JAK2)] and various protein kinase C isoforms.
-
(d)
Generation of reactive oxygen species (ROS) by a mechanism that involves activation of the membrane-bound NAD(P)H oxidase.
As a result, activation of AT1R produces arterial vasoconstriction and endothelial dysfunction, increases Na+ and water reabsorption, aldosterone, endothelin-1 and vasopressin release and ROS generation, stimulates sympathetic tone and cardiovascular and renal remodeling (hypertrophy, fibrosis) and exerts proinflammatory, mitogenic, proarrhythmic and anti-apoptotic effects [7, 12, 39, 41] (Fig. 3).
Signal Transduction from the AT2R
They are highly expressed in fetal tissues, but their expression is dramatically decreased after birth, although they are upregulated in various pathological conditions associated with tissue remodeling or inflammation, including hypertension, atherosclerosis, heart failure, myocardial ischemia and diabetes mellitus [42, 43]. Stimulation of AT2R, coupled to Gi and to unknown G proteins [7, 42–44]:
-
(a)
Activates the AT2R-nitric oxide (NO)-cGMP and AT2R-PLA2 (phospholipase A2)-arachidonic acid-BKCa (large conductance Ca2+-activated potassium channels) pathways, which regulate blood pressure and natriuresis and neuronal activity, respectively.
-
(b)
Activates various protein tyrosine phosphatases [protein tyrosine phosphatase (PTP), SH2 domain-containing phosphatase 1 (SHP-1), protein phosphatase 2A (PP2A) and mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1)], which directly inhibit the activation of ERK1/2 and the growth-promoting actions associated with AT1R stimulation.
-
(c)
Inhibits AT1R-mediated tyrosine phosphorylation of signal transducers and activators of transcription (STATs) 1–3.
-
(d)
In different cell types, AT2R-induced apoptosis involves ERK inactivation, activation of JNK in a PP2A-dependent manner and/or activation of ceramide-caspase 3 pathway.
As a result, AT2R stimulation produces effects opposite to those mediated via AT1R stimulation, i.e., vasodilation (via the release of nitric oxide, NO, prostaglandins and an increase in potassium conductance), natriuresis, neuronal regeneration, cellular differentiation, growth inhibition (anti-proliferative, anti-hypertrophic and proapoptotic effects) and reduces ROS production [45] (Fig. 3). These findings confirm that the beneficial vascular effects of ARBs are, at least partially, attributed to unopposed AT2R stimulation [43, 46].
However, the finding that activation of AT2R may, in some tissues, result in parallel rather than opposite effects to AT1R activation suggests that AT1R and AT2R may share, at least in part, some common signaling pathways [43]. Indeed, there is a physiological cross-talk between AT1R and AT2R, a mechanism by which the AT2R may act as an AT1R antagonist [47, 48]. Ang II receptors can form homo- or hetero-dimers (and undergo complex associations with other G protein-coupled receptors), and it has been found that the AT2R binds directly to the AT1R and, thereby, inhibits different functions of the AT1R [43, 49]. Dimerization of the AT2R with the AT1R inhibits cell proliferation and growth effects of the latter, whereas homodimerization of the AT1R or the AT2R enhances cellular effects of these receptors [43]. Additionally, under certain pathological conditions, AT2R mimic AT1R function and exert detrimental effects including vasoconstriction and hypertrophy [46].
The RAS Much More Complex that Previously Described (Fig. 4)
Ang II (1–8) is not the only bioactive peptide as other peptides, including prorenin, renin, Ang III (2–8), Ang IV (3–8) and Ang (1–7), are also active, and additional receptors, including AT3, AT4, AT7-Mas and pro(renin), have been identified [6, 7, 50–52]. ACE2, a homolog of ACE, hydrolyses Ang I into Ang-(1–9), which is subsequently converted into Ang-(1–7), an endogenous ligand for the G protein-coupled receptor AT7-Mas through ACE and neutral endopeptidase [7, 52–54]. The biological activities of Ang-(1–7) are mediated through the release of prostaglandins and NO, and the inactivation of the PI3K-PKB/Akt, p38 MAPK and JNK signaling pathways opposes the vasopressor, proliferative, profibrotic, proinflammatory and prothrombotic actions mediated by Ang II via the AT1R [53, 55]. Thus, it has been hypothesized that at both systemic and local levels the RAS is composed of two opposite arms: the ACE/Ang II/AT1 axis and the ACE2/Ang-(1–7)/Mas axis [55].

Classic and new components of the renin–angiotensin–aldosterone system. The effects mediated via the activation of the (pro)renin and MAS receptors are also shown. Angiotensin II (Ang II) and Ang III stimulate AT1 and AT2 receptors, Ang (1–7) stimulates the Mas receptor (and possibly AT2 receptors) and Ang IV stimulates AT4 receptors or insulin-regulated membrane aminopeptidase. ACE angiotensin I-converting enzyme, ACE2 angiotensin I-converting enzyme 2, AMPA aminopeptidase A, AMPM aminopeptidase M, Ang I–IV angiotensins I, II, III and IV, Ang-(1–7) angiotensin-(1–7), AT1R angiotensin II type 1 receptor, AT2R angiotensin II type 2 receptor, IRAP insulin-regulated aminopeptidase, MASR Ang-(1-7) or MAS receptor, MMP9 matrix metallopeptidase-9, NEP neutral endopeptidase, NO nitric oxide, PAI-1 plasminogen activator inhibitor 1, PEP prolylendopeptidase, (Pro)RR (pro)renin receptor, ROS reactive oxygen species, TGF-β1 transforming growth factor-β1. Modified from Tamargo et al. [7]
Signal Transduction from the Pro(renin) Receptor
The Renin not only increased the synthesis of Ang II, but both renin and prorenin bind to a specific pro(renin) receptor (PRR) which increased the catalytic activity of renin and activated prorenin [56] (Fig. 4). The stimulation of this receptor [7, 46, 56–58]:
-
(a)
Activates ERK1/2 independently of Ang II formation and increases cell proliferation. This activation is not blocked by renin inhibitors, suggesting that binding of these inhibitors to prorenin does not prevent binding of prorenin to the PRR, although inhibitor binding prevents the generation of Ang I.
-
(b)
Increases the production of transforming growth factor-β1 (TGF-β1), resulting in the upregulation of profibrotic factors, such as the plasminogen-activated inhibitor-1 (PAI-1), fibronectin and collagen.
-
(c)
Activates the protein kinase B/Akt pathway in vascular smooth muscle cells and the p38 MAPK—heat-shock protein 27 cascade in cardiomyocytes; this latter cascade regulates actin cytoskeleton dynamics [28, 29].
Thus, activation of the PRR produces vasoconstriction, hypertrophy, fibrosis and apoptosis [27–29]. This indicates that increases in renin and prorenin levels will not only result in diminished RAS suppression, but also result in unwanted effects via PRR stimulation independently of the formation of Ang II [46, 51]. Furthermore, during RAS blockade or under pathological conditions prorenin levels in vivo are too low to stimulate the PRR. Thus, it has been proposed that the phenotype that develops in response to PRR overexpression must be the result of RAS-independent effects of PRR stimulation [46]. The PRR is colocalized with the V-type proton ATPase (V-ATPase), and PRR is indispensable for V-ATPase integrity. Additionally, PRR also acts as an adaptor between V-ATPase and Wnt signaling molecules. These findings underscore the importance of the PRR beyond renin/prorenin binding, although they do not rule out the possibility that the PRR has an important role in tissue damage, or that the beneficial effects of RAS blockade occur as a result of a reduction in PRR expression [46].
The Local Renin–Angiotensin System in Bone
In addition to the classical circulating-systemic RAS, the components of RAS are synthesized in various tissues exerting paracrine, autocrine and intracrine actions. Local RAS operates independently from systemic RAS and participates in various physiopathological processes such as inflammation, hypertension, atherosclerosis, heart failure, brain ischemia, glomerular sclerosis, insulin secretion, cardiovascular remodeling, cell growth and proliferation and apoptosis [41, 58, 59]. Under physiological conditions, the components of RAS, such as angiotensinogen, renin, ACE and AT1R and AT2R are expressed in different bone cells and chondrocytes of epiphyseal plates, which implicates local RAS in epiphyseal elongation during bone growth and healing [60–72] (Table 1). Tsukamoto et al. [73] studied the expression of local RAS components in 8-week-old C57BL/6 adult mice and in cultured bovine’s articular cartilage chondrocytes. Hypertrophic chondrocytes of epiphyseal plates in the tibia and the lamina terminals expressed local RAS components, while hyaline chondrocytes, including the knee articular cartilages, the parenchyma of nasal septums and of the tracheal walls, and cultured bovine’s articular cartilage chondrocytes, did not express the RAS components. After inducing hypertrophy with interleukin (IL)-1β or tumor necrosis factor α, (TNFα), cultured articular chondrocytes expressed AT1R and AT2R. All these findings confirmed the existence of local RAS in the bone where it can play an important role in both metabolism and remodeling independently of the systemic RAS.
Additionally, a locally active RAS affecting the growth, production, proliferation and differentiation of hematopoietic cells has been described in bone marrow cells (BMC), hematopoietic-lineage BMC and cultured marrow stromal cells (MSC) [74–80] (Table 1). Ang II stimulates AT1R present on human bone marrow CD34(+)CD38(−) cells, CD34(+)CD38(+) cells, lymphocytes and stromal cell clones increasing hematopoietic progenitor cell proliferation, an effect blocked by losartan [78, 79]. The presence of RAS components in both hematopoietic-lineage BMC and MSC, and the de novo synthesis of Ang II by MSC suggest a potential autocrine-paracrine mechanism for local RAS-mediated regulation of hematopoiesis [75]. Ang II regulates hematopoiesis by directly activating hematopoietic stem cell proliferation via the JAK/STAT pathway, or indirectly activating the synthesis of several cytokines and growth factors by stromal cells [74, 76]. Interestingly, ACE mRNA is expressed in the yolk sac endoderm in close contact with blood islands during early embryogenesis (stage HH4), at a stage when blood circulation is not yet established and systemic regulation of blood pressure by RAS is therefore not yet required [80]. All RAS components are also expressed close to these blood islands, suggesting that local RAS modulates primitive erythropoiesis. Indeed, the ACEI fosinopril decreased hematocrit by 15 %. Furthermore, while peripheral blood ACE levels increase, blast percentages in the bone marrow accumulate and migrate to the circulation. Therefore, ECA hyperfunction may lead to the degradation of negative hematopoietic regulator peptide AcSDKP, which in turn lowers its level in the bone marrow microenvironment, removing its anti-proliferative effect on the hematopoietic cells and blasts [74, 77].
Effects of Angiotensin II on Bone
The identification of all the components of the RAS in bone and the finding that the transgenic activation of RAS in the transgenic Tsukuba hypertensive mouse (THM) mice [69] or the chronic infusion of subpressor doses of angiotensin II in rats [81] produced an osteopenic phenotype due to excessive bone resorption led to the hypothesis that local bone RAS might play an important role in the pathogenesis and progression of some metabolic bone diseases through two different ways: (a) an imbalance in bone remodeling, characterized by an inhibition of osteoblastic activity and an increased osteoclastic activity [70, 71, 81] (Fig. 2); and (b) changes in blood flow in bone marrow capillaries [60, 81].
Ang II had no effect on osteoclast differentiation or on bone resorption by isolated osteoclasts and did not potentiate osteoclast formation triggered by RANKL, but it stimulated osteoclastogenesis in osteoblast and osteoclast co-cultures, an effect inhibited by the ACEI captopril [63, 70, 82]. In rat calvarial osteoblastic cells, Ang II via stimulation of AT1R markedly inhibited osteoblastic differentiation and mineralization and reduced the number of mineralized nodules and in osteoblast UMR-106 cells incubated in the presence of special mineralization medium produced a dose-dependent inhibition of the mineralization [70, 71]. In bone marrow-derived mononuclear cells, which may include both osteoblasts and osteoclasts, Ang II induced cell differentiation of bone marrow mononuclear cells to multinuclear cells and of multinuclear cells to osteoclasts and increased TRAP (tartrate-resistant acid phosphatase) positive multinuclear osteoclasts. Of interest, Ang II acted on osteoblasts and not directly on osteoclast precursor cells and increased the expression of RANKL in osteoblasts, leading to the activation of osteoclasts [69, 81]. These effects were completely blocked by olmesartan and mitogen-activated protein kinase kinase inhibitors [81]. Ang II also stimulated DNA and collagen synthesis [63, 71, 82, 83], decreased the mRNA expression of osteocalcin, a protein that is specifically expressed during maturation of osteoblastic cells [70] and the activity of alkaline phosphatase (ALP, a marker of osteoblastic differentiation) in osteoblast-rich cultures obtained from fetal rat calvariae of newborn rats [39, 40, 72] and in human adult trabecular bone cells [71] (Fig. 2). Both effects were inhibited by losartan.
Role of AT1R and AT2R
The relative role of AT1R and AT2R signaling in Ang II-induced osteoclastogenesis was also studied by knocking down their expression with specific siRNAs in primary osteoblasts in culture [69]. In AT1-KO cells, the effects of Ang II on osteoclastogenesis were somewhat enhanced, while in AT2-KO cells the activity of Ang II was inhibited, which suggested that AT2 was the major transducing receptor. The same group studied the transgenic THM mice which displayed elevated serum Ang II levels, osteopenia caused by high bone turnover and hypertension [69]. Ang II had no effect on osteoclast differentiation nor exerted a stimulatory effect on osteoclastogenesis triggered by a lower concentration of RANKL, but it stimulated the formation of osteoclasts in the co-culture of calvaria-derived primary osteoblasts and bone marrow macrophages in a dose-dependent manner. These results suggested that Ang II stimulates osteoclastogenesis by acting on osteoblastic cells (i.e., ‘‘the soil cells’’) and not through a direct action on hematopoietic ‘‘seed cells.’’
Izu et al. [60] found that AT2R blockade with PD123319 increased bone volume/tissue volume (BV/TV) and trabecular number and spacing in the distal metaphyseal regions of femora in mice. AT2R-deficient mice also presented an increase in bone mass compared with wild-type mice. These effects resulted from an enhancement of osteoblastic activity and the suppression of osteoclastic activity. In organ cultures using new-born mouse, ulnae and radii PD123319 suppressed the number of TRAP-positive osteoclasts and in the proximal tibiae of 9-week-old male mice reduced the number of osteoclasts and the osteoclasts surface per bone surface. However, losartan did not affect bone mass and no gross abnormalities in bone development or osteoporosis were described in AT1aR KO mice, although they showed hypotension and hyperreninemia [84].
On the contrary, Shimizu et al. [81] reported that Ang II accelerated osteoporosis via the AT1R pathway and Kaneko et al. [61] found that mice lacking the gene encoding the AT1aR had a significantly higher BV/TV than age- and sex-matched wild-type mice. Histomorphometric analysis of the proximal tibia of AT1aR null mice showed that both bone formation (osteoid surface, osteoblast surface and bone formation rate) and resorption (number of osteoclasts, bone surface area covered by osteoclasts and eroded surface) were significantly elevated and mice presented elevated serum levels of osteocalcin and C-terminal collagen crosslink concentrations, a degradation product of type I collagen. Osteoclastogenesis and osteoblastogenesis assays in ex vivo cultures did not reveal any intrinsic alteration in the differentiation potential of AT1a-deficient cells, and serum and urinary calcium and plasma PTH levels did not differ between genotypes, which make it unlikely that the high bone mass and high bone turnover were secondary to alterations in systemic calcium metabolism. Moreover, the RANKL/OPG ratio and the expression of stromal cell-derived factor (SDF)1a increased, while the expression of the SOST gene decreased, which may account for the increased bone resorption and formation, respectively. Interestingly, 25-month-old male AT1aR KO mice maintained higher bone mass and female mice were protected against OVX-induced bone loss compared with wild-type mice. All these results suggested that AT1aR signaling negatively regulates bone turnover and mass and thus, it can be hypothesized that ARBs might represent a putative therapeutic strategy for osteoporosis.
Therefore, further studies are needed to understand the pathophysiological role of AT1 and AT2 receptors in Ang II-induced osteoclastogenesis and in bone metabolism.
Mechanism of Action of Ang II on the Bone
Local bone RAS might play an important role in the pathogenesis and progression of some bone diseases through two different ways:
-
A.
An imbalance in bone remodeling, characterized by an inhibition of osteoblastic activity and an increased osteoclastic activity, leading to increased bone resorption [70, 71, 81] (Fig. 3). This increase in osteoclastic bone resorption produced by Ang II is the result of multiple actions, among them (Table 2):
Table 2 Mechanisms involved in Ang II-induced osteoclastogenesis -
1.
An increase in RANKL expression and in the RANKL/OPG ratio. In human osteoblasts and UMR-106 cells, Ang II significantly increased RANKL (eightfold) and OPG (threefold) mRNA expression and the RANKL/OPG ratio, promoted the differentiation of mesenchymal stem cells in multinuclear cells, activated mature osteoclasts responsible for bone resorption and increased the number of TRAP-positive multinuclear osteoclasts and thereby osteoclastogenesis [69, 81, 85, 86]. These effects were abolished by the AT1R blocker olmesartan or U0126 (an extracellular signaling kinase pathway MEK/ERK inhibitor), but not by the AT2R blocker PD123329 [81]. Moreover, using mRNA isolated from the tibia and femur of AT1a-deficient mice revealed that Ang II increased the RANKL/OPG ratio and the expression of SDF1a, which may account for the increased bone resorption [61, 81]. Taken together, these results suggested that Ang II-induced osteoclast differentiation and activation may be mediated via the upregulation of RANKL following the activation of the AT1R-ERK signaling pathway. However, Asaba et al. [69] described that Ang II-increased RANKL expression via AT2R. Additionally, Ang II-induced TRAP-positive multinuclear cells were completely abolished in RANK siRNA transfected-osteoclast precursors co-cultured with human osteoblasts [81]. These results suggest that Ang II-induced osteoclast differentiation may be mediated by the RANK–RANKL system and recent evidence showed that RANKL significantly increased AT1R and ACE expression in vascular smooth muscle cells via ERK phosphorylation [87]. Thus, a cross-talk between RAS and RANKL system might work as a vicious cycle to promote bone resorption.
-
2.
An increase in cAMP levels. The differentiation of osteoblast and osteoclasts is primarily controlled by RANKL and core-binding factor subunit alpha-1 (Cbfa1/Runx2), and both are regulated by cAMP [13, 16, 88, 89]. Cbfa1/Runx2 is an essential transcription factor for the differentiation of osteoblasts from mesenchymal precursors and subsequent bone matrix mineralization [90, 91]. In cultured osteoblasts, Ang II increased dose-dependently the intracellular cAMP levels, an effect abolished by losartan, while PD123319 barely affected the stimulatory effect of Ang II [70]. In cultured rat vascular smooth muscle cells, Ang II enhances cAMP levels by facilitating the interaction between activated Gs and adenylyl cyclase [92]. The increase in cAMP levels then activates downstream signaling pathways, which, in turn, downregulates the expression of Cbfa1/Runx2, while the RANKL expression increased and subsequently reduced osteoblast number and function, leading to enhanced bone resorption and reduced bone formation [81, 88]. These effects of Ang II in rat calvarial cell [70] or in co-cultures of human osteoblast and osteoclast precursor cells were abolished by pretreatment with ARBs (losartan, olmesartan), but not by PD-123329, which suggested that Ang II inhibited osteoblast differentiation and activity throughout the AT1R-adenylyl cyclase-cAMP pathway [70, 81–83]. Of note, increased cAMP levels in plasma and urine have been found in osteoporotic and hypertensive patients [88, 93, 94].
-
3.
Upregulation of the SOST gene expression. Ang II, via the activation of AT1aR, upregulates SOST gene expression in osteocytes. SOST encodes for an osteocyte-specific secretory protein, sclerostin, which binds to LRP5/6 (low-density lipoprotein receptor-related protein) receptors on the cell membrane of osteoblasts, inhibits the Wnt/β-catenin signaling and reduces osteoblastic bone formation [95, 96]. In fact, Wnt/β-catenin signaling decreased bone resorption by inhibiting osteoclast differentiation from their precursors and increasing the expression of OPG [95, 97]. A decreased expression of SOST is responsible, at least in part, for the stimulation of bone formation in the AT1aR-KO mice [61] and the SOST-KO mice presented a high bone mass phenotype characterized by marked increases in BMD, bone volume and bone strength [98].
-
4.
An increased the expression of matrix metalloproteinase (MMP)-3 and 13. In osteoblastic ROS17/2.8 cells, Ang II stimulated the degradation process occurring during extracellular matrix turnover in the osteoid [99]. These effects were mediated via AT1R stimulation and Ang II-induced phosphorylation of ERK1/2, p38 MAPK and stress-activated protein kinases (SAP)/JNK pathways. Losartan, PD98059 (a MAPK kinase 1/2 inhibitor) and SP600125 (a JNK-specific inhibitor) suppressed Ang II-induced expression of MMP-3 and MMP-13.
-
5.
An increased expression of interleukin (IL)-6. Ang II induced IL-6 mRNA expression and protein synthesis in cultured osteoblasts through activation of the AT1R-ERK1/2 pathway, and this increase was abolished by losartan, and the ERK1/2 inhibitor U0126 [100]. IL-6 induced osteoclast formation from precursors and stimulated bone resorption [101, 102], and bone resorption promoters (such as TNFα, IL-1 and PTH) stimulated IL-6 production in cultured osteoblasts [103, 104].
-
6.
An increased expression vascular endothelial growth factor (VEGF). In the THM mice, Ang II increased VEGF mRNA levels in osteoblasts, which stimulated osteoclastogenesis through the VEGF receptor 1 (Flt-1) expressed on hematopoietic cells [69, 105].
-
7.
Inhibition of mitochondrial respiratory enzyme complexes. In primary mouse calvaria osteoblasts, Ang II exerted an inhibitory effect on mitochondrial respiratory enzyme complexes, producing membrane potential dissipation, ATP loss, generation of ROS and cell apoptosis via the JNK signaling pathway [106]. SP600125 rescued osteoblast cells from apoptosis by enhancing the anti-apoptotic protein Bcl-2 expression, suppressing the translocation of Bax from cytosol into the mitochondria and blocking cytochrome C release and caspase-3 activation.
-
8.
Changes in intracellular Ca 2+ handling. Ang II reduced Ca2+ uptake into calvarial bone disks, an effect abolished by an antiserum to TGF-β1 [107], and accumulation of calcium in the matrix layer [70], decreased ionized Ca2+ and increased PTH levels [108, 109].
-
9.
Activation of TGF-β1. Many bone cells (osteoblasts, fibroblasts and osteoclasts) produce TGF-β1, so that this is the most abundant growth factor in bone [109] and exhibits both stimulant and suppressive actions on osteoclast differentiation and bone resorption [110]. Osteoblast-derived TGFβ1 acts directly on osteoclast precursors to prime them for RANKL-induced osteoclast formation; however, following the initiation of resorption, TGF-β acts on osteoblasts to reduce the availability of RANKL and further osteoclast formation [110]. The expression of ACE, TGFβ1 and IL-11 increased in osteolytic lesions of patients with Langerhans cell histiocytosis, which is consistent with a stimulation of osteoclastogenesis by TGF-β1 and IL-11 released by the action of locally generated Ang II [111].
-
1.
-
B.
Changes in blood flow in bone marrow capillaries. Blood flow plays a significant role on bone remodeling and bone microvessels have just the endothelium, but they do not have muscle and connective tissue layers. Ang II can influence bone remodeling indirectly through the modulation of mean arterial blood pressure, which determines the intramedullary pressure, the driving force for transcortical interstitial fluid [66, 112, 113]. Moreover, Ang II produces endothelial dysfunction and arteriolar vasoconstriction, two effects that might reduce the flow through the bone microvasculature, thus contributing to the decrease in BMD and development of osteoporosis [112].
Osteoporosis and Hypertension: Two Common Risk Factors in the Elderly
Osteoporosis, defined as a systemic skeletal disorder characterized by a reduction in BMD, deterioration in bone microarchitecture, susceptibility to skeletal fragility and increased risk of fractures, is a major global health problem [114, 115]. In young adults, bone destruction and formation are balanced, and bone mass is maintained in a steady state. After age 40, bone resorption begins to exceed bone formation, leading to a reduction in BMD that is particularly evident in postmenopausal women. Thus, it would be expected that because of the aging of the population, the prevalence of age-related osteoporosis morbidity and mortality will increase considerably in the near future.
The estrogen loss that accompanies menopause is associated with an increased osteoclast activity and bone resorption driven by cytokines that regulate osteoclastogenesis [RANK, TNFα, IL-1, IL-6, IL-11, M-CSF (macrophage colony-stimulating factor)] and prostaglandin E [116]. In normotensive or hypertensive animal models, OVX produced a marked increase in osteoclast activity, a significant decrease in BMD and an increase in urinary deoxypyridinoline [91, 116]. OVX also induces endothelial dysfunction of microcirculation vessels in bone tissue, which reduces regional blood flow, leading to osteoporotic changes characterized by thinning of bone trabeculae, increasing intertrabecular space and microfractures [113], and these effects were prevented by enalapril and losartan.
In addition to physiological factors such as aging and estrogen deficiency, there is a potential relationship between bone and cardiovascular diseases as they present common etiological factors, including aging, postmenopausal status, diabetes and lifestyle factors (i.e., smoking, diet, sedentarism) [117–119]. Osteoporosis and hypertension are major chronic diseases that often coexist in elderly people, and almost half of the hypertensive patients are postmenopausal women at high risk of osteoporosis [120]. Clinical and epidemiological studies showed that hypertensive patients present abnormalities of calcium metabolism, including hypercalciuria, decreased BMD, vitamin D insufficiency and increased PTH levels [121–126]. The 24-h urinary calcium excretion and the lower BMD were significantly greater in female hypertensives than in female normotensive subjects [127], and the activation of the RAS was correlated with both an increase in 24-h urinary calcium excretion and a lower BMD [108]. Thus, hypertension is an independent risk factor for osteoporosis and fragility fractures [128–130] and in elderly white women is associated with increased bone loss at the femoral neck, which may contribute to the risk of hip fractures [123].
Although the mechanisms responsible for hypertension-related osteoporosis remained uncertain, it has been proposed that RAS activation participates in the progression of osteoporosis. Thus, it would be expected that RAS inhibitors (ACEIs and ARBs) may prevent the loss of BMD in hypertensive patients as a result of the normalization of RAS activity. Additionally, ACEIs and ARBs reduced other HTN-related cardiovascular risks beyond BP control and are first-choice drugs in the prophylaxis and treatment of hypertension, heart failure, coronary artery disease and diabetes mellitus [1–7]. Thus, in the next sections we shall review the role of RAS and RAS inhibitors in osteoporosis.
Role of RAS in Osteoporosis
Experimental evidence showed that excessive activation of local RAS in bone led to an osteopenic phenotype associated with an excessive bone resorption [69, 81] that is involved in the development of age-related osteoporosis [65], postmenopausal osteoporosis in OVX animals [81, 131] and glucocorticoid-induced osteoporosis (GIOP) [67, 132] and in the process of fracture healing [133], and in bone deterioration in obstructive nephropathy (unilateral ureteral obstruction) [134] and type 1 diabetes [68, 135] (see Table 1).
In aging mice (12 months old), there was a significant reduction in BMD in the proximal tibial metaphysis associated with increased cathepsin K (a protease involved in bone resorption) mRNA expression, whereas the expression of osteoblast-specific genes (Runx2 and ALP) was not modified as compared to young (2 months old) animals [62]. Renin and AGT mRNA expression in the tibia and femur of aged mice was significantly higher than in young mice (2 months old), leading to an increased production of Ang II which might play an important role in the pathology age-related osteoporosis [62].
Normotensive and spontaneously hypertensive OVX (SHR-OVX) rats showed a significant increase in osteoclast activity as assessed by an increased TRAP activity and a significant decrease in BMD [81, 136]. Moreover, OVX might accelerate Ang II-induced signaling. Ang II promoted bone resorption via the AT1R in OVX mice and rats [81, 136], while estrogens effectively downregulated Ang II production and attenuated AT1R expression and signaling [137, 138] and enhanced the improvement of vascular remodeling induced by ACEIs in female OVX-SHR through the inhibition of the ERK1/2 and STAT signaling pathways [131, 133]. Furthermore, in the tibia of OVX-SHR rats treated with a subpressor dose of Ang II (200 ng/kg/min), both ALP and TRAP activity and TRAP-positive stained area significantly increased, while BMD and the ALP/TRAP ratio decreased as compared to sham-operated SHR [81]. These effects were inhibited with imidapril and losartan. These results suggested that Ang II via the stimulation of AT1R accelerated OVX-induced osteoporosis independent of changes in blood pressure, a pattern similar to that observed in elderly postmenopausal women at risk of osteoporosis [81, 82].
Additionally, there is an association between ACE insertion/deletion polymorphisms, which determines ACE activity, and BMD in postmenopausal hypertensive women, so that individuals with the I/I genotype (associated with a decreased ACE activity) had a higher BMD than individuals with either an I/D or D/D polymorphism, which were associated with increased ACE activity and serum Ang II levels [139, 140]. All these results were the basis to hypothesize that RAS inhibitors (ACEIs and ARBs) may represent a putative therapeutic strategy to increase BMD and accelerate the fracture healing process in osteoporotic individuals with associated cardiovascular diseases.
Reduction in bone volume and strength, due to decreased osteoblast activity and increased bone resorption, is a common complication of glucocorticoid therapy for inflammatory diseases or for immunosuppression after organ transplantation [14, 141, 142]. These effects are similar to those produced by Ang II on bone metabolism [63, 81], and there is some evidence that local RAS is activated in patients with GIOP.
In adult male rabbits, dexamethasone (DXM) increased osteoclast number, osteoclast surface, and eroded surface and urinary deoxypyridinoline was upregulated, while serum osteocalcin decreased [67, 143]. Moreover, DXM stimulated AGT transcription and secretion in several tissues and upregulated bone levels of Ang II and expression of AT1R and AT2R, and ACE at mRNA and/or protein levels. However, Ang II levels and ACE activity in circulatory system remained unchanged, indicating that there is a local RAS in bone which might be involved in the pathogenesis of GIOP [67, 143]. Furthermore, using RNA isolated from the lumbar vertebrae it was found that DXM markedly increased the expression of osteocyte-related genes (SOST and DMP1—dentin matrix acidic phosphoprotein 1) and osteoclast-related genes (TRACP), RANKL and the RANKL/OPG ratio, but decreased osteoblast-related genes (Runx2, Osterix and Col1a1).
Similarly, the mRNA expression of local bone AT1R, AT2R and RANKL was higher in the trabecular bone of lumbar vertebrae in patients with lumbar disk herniation with GIOP compared with the control group, but there were no differences in the circulating levels of ACE and Ang II and in the expression of AT1R and AT2R between patients with GIOP and control patients [66]. Simple logistic regression analysis demonstrated that GIOP was significantly associated with local OPG and RANKL mRNA expression, the RANKL/OPG ratio, and local AT1R and AT2R mRNA levels. Multiple logistic regression analysis revealed that GIOP was independently associated with the circulating RANKL/OPG ratio, as well as local AT1R and AT2R mRNA levels. These results suggested that glucocorticoids might decrease BMD by activating bone RAS and modulating the RANKL/OPG ratio in bone [66].
Mechanism of Action of RAS Inhibitors
The mechanisms by which ACEI and ARBs inhibit bone resorption and may increase BMD are unknown. The therapeutic effect of ACEIs derived from lowering Ang II levels by inhibiting the conversion of Ang I to Ang II (Fig. 3). Therefore, they inhibit the effects mediated via the activation of both AT1R and AT2R [5–7, 51, 52, 59]. Furthermore, ACEIs also interfere with the kallikrein–kinin system by inhibiting the degradation of bradykinin, which increased the synthesis and release of prostaglandins and NO following the stimulation of its specific B2 receptors, and the NO pathway regulates local blood flow in bone marrow capillaries, which might increase bone formation. ARBs exert their osteoprotective effects via blockade of the deleterious effects of Ang II mediated via the activation of AT1R. Under these circumstances, the increase in Ang II levels observed during the administration of ARBs might stimulate the unblocked AT2R, which are thought to oppose the effects of AT1R activation. Thus, the final effect of ARBs results from the AT1R blockage and the stimulation of AT2R [5–7, 43, 51, 52, 59] (Fig. 3). However, the efficacy of ACEIs and ARBs despite the occurrence of Ang II and aldosterone breakthroughs suggests that their beneficial effects are not solely attributable to blockade of the Ang II. In fact, treatment with ACEIs and ARBs results in the production of high levels of Ang II metabolites, mainly Ang-(1–7), which via the Mas receptor signaling might also contribute to the efficacy of RAS blockade [46].
Regardless of the mechanism of action, the beneficial effect of ACEIs and ARBs on bone can be related to the inhibition of Ang II-induced upregulation of RANKL expression in osteoblasts and the normalization of the RANKL/OPG ratio and the other mechanisms through which Ang II enhances osteoblast differentiation and osteoclastogenesis (Table 2), in order to maintain the balance between bone formation and bone resorption and antagonize the unfavorable effects of Ang II on bone [61, 69, 70, 74, 75, 81, 88].
Alternatively, ACEIs and ARBs can indirectly modulate bone remodeling as they improved endothelial dysfunction and produced arteriolar vasodilatation increasing blood flow in bone marrow capillaries [82, 132, 144]. Indeed, in OVX rats, ACEIs and ARBs prevented the decline in regional blood flow and improved bone remodeling, pretreated animals presented a preservation of bone structure, and no microfractures were reported [113]. In this model, following the transverse osteotomy of the proximal metaphysis of the femur, enalapril and losartan increased the rate of microcirculation in the fracture zone of proximal metaphysis and the consolidation of fractures.
ACE Inhibitors in Osteoporosis
Preclinical Studies
In animal models, ACEIs have been shown to preserve BMD. In cultured newborn rat calvaria osteoblasts, captopril increased dose-dependently the secretion of ALP and the mRNA expression of collagen I [138]. In OVX rats, captopril improved the osteopenic phenotype and increased strength, mass and trabecular connections in the femurs [136]. Moreover, in aged (10 months old) OVX rats, captopril (1 or 5 mg/kg/day, for 2 months) increased trabecular area of lumbar vertebrae (L4) and improved biomechanical properties by increasing L5 break stress and elastic modulus as compared to the OVX group [138]. These findings suggested that captopril had the potential of improving lumbar vertebral bone strength in aged OVX rats and promoted osteoblast bone formation. OVX-SHR presented an increased osteoclast activation and TRAP activity in the tibia, accompanied by a significant decrease in BMD and an increase in urinary deoxypyridinoline [82]. Imidapril significantly decreased blood pressure and attenuated OVX-induced changes in BMD, TRAP activity and urinary deoxypyridinoline. In another study in OVX rats with five-sixths nephrectomy, Ang II decreased BMD and increased TRAP activity through AT1R and these effects were attenuated by imidapril [81]. Furthermore, enalapril corrected the low bone mass phenotype and hypertension of THM mice [69].
García et al. [133] studied the expression and function of ACE during fracture healing in a murine femur fracture model. ACE, AT1R and AT2R were expressed in osteoblasts and hypertrophic chondrocytes in the periosteal callus during fracture healing. Two weeks after fracture perindopril-treated animals showed some more cartilage and markedly less fibrous tissue and biomechanical analysis showed a significantly greater maximal torque at failure and a higher torsional stiffness as compared to control animals. After 5 weeks of treatment, perindopril reduced BMD in the unfractured femura but improved periosteal callus formation, bone bridging of the fracture gap and torsional stiffness. Moreover, perindopril did not affect cell proliferation, but reduced apoptotic cell death and increased the expression of AT2R, but not of AT1R, in the healing callus. These findings indicated that local RAS in bone can influence the process of fracture healing and that ACEIs accelerated bone healing and remodeling.
Additionally, perindopril prevented DXM-induced bone loss mainly by blocking the activated local RAS rather than the systemic RAS and reversed the DMX-induced changes in SOST and Runx2 expression and in RANKL/OPG ratio. In rabbits with methylprednisolone acetate (MPA)-induced osteonecrosis, the levels of Ang II and the mRNA and protein expression of ACE were highest 1 week and the expression of AT1R and AT2R 2 weeks following the administration of MPA [132]. However, osteonecrosis occurred most significantly 3 weeks after the administration of MPA, so that osteonecrosis was preceded by the activation of bone RAS.
On the contrary, some studies showed that ACEIs did not improve, or even accelerated, bone loss. In a comparative study of six antihypertensive agents on BMD and microarchitectural changes in OVX female C57/BL6 mice, enalapril increased BMD loss as compared to vehicle-treated mice [145]. Moreover, at doses recommended for treatment of hypertension enalapril did not modify BMD, mineral content or morphometric parameters of the femur in 14-week-old female rats [146]. Similarly, moexipril had no effect on the cancellous bone site in OVX rats and did not hamper the osteoprotective effects of 17beta-estradiol [147], and in OVX-SHR rats it reduced blood pressure but had no effect on the proximal tibial metaphysis or the tibial shaft [148].
Mice with streptozotocin-induced type 1 diabetes displayed osteoporosis and the mRNA expression of ACE and PRR, the protein expression of renin and Ang II levels were markedly upregulated in the bone of vehicle-treated diabetic mice compared to that of non-diabetic mice [135]. In this model, captopril inhibited the changes in RAS components but did not exhibit osteoprotective effects as reflected by a reduction of BMD, trabecular thickness and BV/TV in the tibial proximal and femoral distal metaphysis. Moreover, captopril significantly increased TRAP5b levels, reduced OPG/RANK ratio and increased carbonic anhydrase II mRNA expression and the number of matured osteoclasts and decreased TGF-β and osteocalcin mRNA expression in the tibia as compared to those of diabetic mice. These results indicated that captopril suppressed osteogenesis and stimulated osteoclastogenesis, even though it effectively inhibited the high activity of local RAS in the diabetic mice.
Clinical Studies
Several studies found that ACEIs could be effective in increasing BMD and reducing the risk of fractures in patients with osteoporosis which frequently present other cardiovascular and renal diseases, such as hypertension, heart failure, diabetes mellitus and chronic kidney disease [128, 129, 144, 149, 150]. In a prospective cohort study of 50 postmenopausal hypertensive women, fosinopril prevented BMD loss in lumbar spine and femoral neck as compared to untreated women [149]. In an open, prospective study including 134 patients with low-to-moderate hypertension and stable BMD, quinapril and enalapril reduced calciuria and serum 1,25-hydroxyvitamin D levels, but urinary deoxypyridinoline, a marker of bone resorption, was not significantly changed [144]. The same group found that ACEIs significantly increased BMD of lumbar spine in hypertensive women with the ACE D/D polymorphism who present an increased risk of hypertension, diabetic renal disease and cardiovascular complications, whereas women presenting the ACE I/I + I/D polymorphism had a poor response to ACEI treatment [139, 144].
A large case–control analysis using the UK General Practice Research Database which included 30,601 cases aged 30–79 years with an incident fracture diagnosis and 120,819 controls suggested a decreased fracture risk associated with longer-term use of ACEIs (OR 0.81; 95 % CI 0.73–0.89, P < 0.001) [151]. Another population-based pharmacoepidemiological case–control study investigated the association between fracture risk and treatment with commonly used cardiovascular drugs, including 124,655 cases that sustained a fracture and 373,962 age- and gender-matched controls [129]. After adjustment for potential confounders, the relative risk of any fracture was reduced by 7 % (OR 0.93; 95 % CI 0.90–0.96) in postmenopausal women on ACEIs compared with nonusers. There were no differences related to sex and age in these results.
A cross-sectional study of 3887 Chinese patients (1929 women, 1958 men) aged ≥65 years studied the association between ACEI use and BMD [128]. In multiple regression analyses, ACEI use was independently associated with higher femoral neck BMD (+0.015 g/cm2, P = 0.035) in women, and higher femoral neck (+0.015 g/cm2, P = 0.017), total hip (+0.016 g/cm2, P = 0.021) and lumbar spine (+0.043 g/cm2, P < 0.001) BMD in men, after adjusting for many potential confounders including other antihypertensive agents, osteoporosis, cardiovascular risk factors and lifestyle measures. Similarly, the sub-analysis of a clinical study showed that the usage of ACEIs significantly reduced the fracture risk [150].
However, some clinical studies showed that ACEIs did not change the rate and risk of fractures, or even accelerated, bone loss. Solomon et al. [152] examined the relative risk of fracture in a large cohort of Medicare beneficiaries (376,061 subjects) with a diagnosis of hypertension initiating single-drug therapy for treatment. Fracture rate in the total cohort was 35.2 per 1000 person-years. After models adjusting for relevant comorbidities and co-medications accessible in health care utilization data, the risk of fracture was not significantly different from the reference for loop diuretics, beta blockers, and ACEIs. A recent prospective cohort study in 5229 American men (87.2 %) aged ≥65 years from the Osteoporotic Fractures in Men Study (MrOS) found that when compared with nonusers, continuous use of ACEIs was associated with a small (0.004 g/cm2) but significant increase in the average rate of BMD loss at total hip and trochanter over 4.6 years after adjustment for confounders. Use of ARBs, however, was not associated with bone loss at any site [153]. Thus, ACEIs may marginally increase bone loss in older men. The association between ACEI use and increased bone loss was confirmed in a cohort study in 2111 middle-aged Japanese subjects (67 % women) from the Adult Health Study followed through biennial medical examinations since 1958. Patients presented a mean annual percentage change in BMD at the femoral neck of −0.38 % for men and of −1.14 % for women [154]. After adjustment for sex, age, weight, alcohol consumption and smoking status, the annual percentage change in BMD decreased by 0.61 % among ACEI users in comparison with non-ACEI users (P = 0.002). Furthermore, in 2161 patients from two cohort studies which investigated the risk factors of osteoporotic fractures in Hong Kong-dwelling elderly Chinese, the annualized percentage bone loss of male ACEI users was not different from nonusers; however, after adjusting for significant confounders, female continuous ACEI users over 4 years had significantly greater bone loss both in total hip and femoral neck than nonusers [155].
Angiotensin Receptor Blockers in Osteoporosis
Preclinical Studies
Several experimental studies showed that ARBs antagonized the effects of Ang II and preserved bone mass and AT1a-deficient mice exhibited a high bone mass phenotype and an increased trabecular bone volume, trabecular number and connectivity [61].
In osteoblast UMR-106 cells, Ang II produced a dose-dependent inhibition of mineralization and this effect was inhibited by losartan [64]. OVX rats develop endothelial dysfunction in the microcirculation vessels of osteal tissue, resulting in decreased regional blood flow, thinning of bone trabeculae, osteoporosis, presence of microfractures and delayed consolidation of fractures. In this model, enalapril (0.5 mg/kg) and losartan (6 mg/kg) prevented the reduction of microcirculation in bone, slowed the thinning of bone trabeculae, prevented the occurrence of microfractures and improved the fracture healing process, although losartan had a more pronounced osteoprotective effect [113]. In OVX rats and mice, losartan, olmesartan and telmisartan improved the osteopenic phenotype and increased BMD, bone strength and trabecular connections of rats femurs, making the bone stronger [81, 113, 136, 145]. Olmesartan also downregulated RANKL expression, decreased osteoclastogenesis and improved BMD in OVX-SHR [156]. In this model telmisartan significantly improved rosiglitazone-induced decrease in BMD of femur and lumbar vertebrae and bone formation indices such as bone volume fraction, mineralizing surface/bone surface, mineral apposition and bone formation rates [157]. Telmisartan also promoted femur fracture healing in mature male BALB/c mice [158]. At 2 weeks post-fracture, the diameter of the callus in telmisartan-treated animals was significantly increased, the biomechanical analysis showed a greater torque to failure and a higher torsional stiffness, and the histomorphometric analysis showed that the callus of these animals showed some more cartilage and markedly less fibrous tissue. These effects were most probably due to an increase of cell proliferation in the periosteal callus, as indicated by an increased fraction of VEGF- and PCNA (proliferating cell nuclear antigen)-positive cells within the callus in telmisartan-treated animals compared with vehicle-treated controls. After 2–5 weeks, telmisartan treatment resulted in a greater periosteal callus formation and an accelerated healing process with an earlier histological bridging of the fracture gap.
Peroxisome proliferator-activated receptor gamma (PPARγ) controls bone mass by regulating commitment and differentiation of mesenchymal stem cells (MSCs) toward osteoblasts and adipocytes [159]. When activated with thiazolidinediones, PPARγ suppresses osteoblasts, promotes adipocytes development, and enhances osteoclast development [160, 161]. In aged animals, rosiglitazone-induced bone loss was related to an increased osteoclastogenesis in part due to diversion of marrow mesenchymal stem cells differentiation from osteoblastic toward adipocyte lineage [162], and the prolonged use of rosiglitazone led to bone loss and increased occurrence of fractures in elderly women [163]. Telmisartan, an ARB that also exhibits partial PPARγ agonist properties [164], did not affect bone mass or osteoblast phenotype and actively blocked rosiglitazone-induced anti-osteoblastic activity and dephosphorylation of S112pPPARγ in two murine models of type 2 diabetic [165].
As previously described for ACEIs, some studies found that ARBs produced no significant effects in rat calvarial osteoblasts [70] or in co-cultures of human osteoblast and osteoclast precursor cells [81] and did not affect the bone mass, pattern of trabecular bone and BV/TV in C57BL/6 J male mice [60]. ARBs also did not cause significant changes of bone properties in normal [146] or OVX female rats [145, 166], type 2 diabetic mice [165], orchiectomized male hypertensive and normotensive rats [167]. Moreover, in AT1aR-deficient mice losartan improved hypertension but exacerbated the osteopenic phenotype [61].
More importantly, in some animal models ARBs may lead to more bone injuries. In mice with type 1 diabetes induced by streptozotocin, RNA expressions of AGT, ACE and renin receptor in the tibia and protein expressions of AGT and AT1R in the femur were markedly upregulated in the diabetic osteoporotic group, suggesting that high local bone RAS activity contributes to the development of type 1 diabetic osteoporosis [68, 135]. Losartan did not exert osteoprotective effects in this model as shown by the reduction of BMD and microarchitectural parameters at the proximal metaphysis of the tibia. This result can be explained because losartan induced the upregulation of renin and Ang II protein expression and downregulated AT2R protein expression, i.e., because losartan enhanced RAS activity. In the chimeric THM mice, activation of RAS induced a high turnover osteoporosis with accelerated bone resorption [69]. In this model, enalapril improved osteoporosis and hypertension, whereas losartan improved hypertension but exacerbated the osteopenic phenotype. It was hypothesized that the blockade of AT1R alone somehow activated signaling through the unblocked AT2R by the high levels of circulating Ang II. If so, the blockade of the synthesis of Ang II may be a more effective therapeutic strategy in patients with osteoporosis and hypertension. Interestingly, the finding that losartan and enalapril had opposite effects on bone mass confirmed that local RAS contributed to the development of osteoporosis independently of systemic effects on blood pressure. Finally, in the case–control analysis of the UK General Practice Research Database, the use of ARBs was not associated with an altered fracture risk [151].
Clinical Studies
A population-based, retrospective cohort study using administrative databases in Ontario examined the risk of osteoporosis-related fractures in 87,625 hypertensive elderly patients newly treated with ARBs versus ACEIs. The primary outcome was hip fracture, and secondary outcomes were non-hip major osteoporotic fractures (other femoral, clinical vertebral, forearm, wrist, humerus) and other osteoporotic fractures [168]. No significant differences in hip fracture risk were found between new users of ARBs relative to ACEIs. However, there was a statistically significant 19 % risk reduction in other major osteoporotic fractures among new users of ARBs compared with ACE inhibitors (HR 0.81; CI 0.70–0.93). In a post hoc dose-dependency analysis, after adjusting for ARB and ACEI dose, there was no statistically significant difference between the effects of ARBs and ACEIs on risk of hip fractures (HR 0.99; CI 0.78–1.25), other major osteoporotic fractures (HR 0.87; CI 0.75–1.01) and other osteoporotic fractures (HR 0.90; CI 0.74–1.08). However, in this analysis there was a statistically significant decreased risk of osteoporosis-related fractures with increasing doses of either study drug.
In the previously mentioned large cohort of Medicare beneficiaries with a diagnosis of hypertension who had not filled a prescription for an antihypertensive medication in the prior 365 days after model adjusting for relevant comorbidities and comedications accessible in health care utilization data, the risk of fracture was reduced in users of ARBs (HR 0.76; 95 % CI 0.68–0.86) and thiazide diuretics (HR 0.85; 95 % CI 0.76–0.97) compared with calcium channel blockers [152]. The adjusted fracture risk was not significantly different from the reference for loop diuretics, beta blockers and ACEIs, which confirmed that the risk of fracture differs across users of different antihypertensive medications.
Possible Explanations for the Contradictory Results Observed with ACEIs and ARBs
The experimental and clinical studies that analyzed the effects of ACEIs and ARBs on bone homeostasis led to contradictory results. There are some possible explanations for these discrepancies:
-
1.
Differences in animal species, experimental models (in vitro vs in vivo, normotensive rats vs SHR), phenotype of the cultured cells or age of animals/individuals (older populations with normal RAS activity vs elderly hypertensive and/or diabetic patients) [64, 153]. Thus, ACEIs and ARBs did not affect ALP activity or cellular DNA content in osteoblastic MC3T3-E1 cells [143] but inhibited the effects of Ang II in osteoblasts and osteoclasts derived from newborn mouse calvaria as well in the bones (tibia/femur) of adult mice [60, 62, 69], and enalapril and losartan had no effect on BMD, mineral content or morphometric parameters in OVX rats [148], while imidapril or olmesartan attenuated osteoporosis in OVX-SHRs [81, 82].
-
2.
The effects of Ang II via AT1R and/or AT2R may vary depending on the background of animals or the levels of Ang II. Shimizu et al. [81] showed that Ang II induced RANKL expression directly in osteoblasts by activating the AT1R, which might explain why ARBs reduce the fracture risk in older adults, whereas Asaba et al. [69] found that Ang II induced RANKL expression via the AT2R. Although the reason for this discrepancy is currently unknown, these findings are against the general idea that the effects mediated via the AT1R and AT2R are counter-regulatory to each other or that the AT2R-mediated signaling is the protective arm of the RAS [7, 169]. Additionally, under certain pathological conditions, AT2R mimic AT1R function and exert detrimental effects including vasoconstriction and hypertrophy [46]. Furthermore, ARBs and ACEIs can modify the expression of AT1R and AT2R, although the results are inconsistent, so that an increase in both AT1R and AT2R expression [67], an increase in AT1R but not in AT2R [133] or no change [170] has been reported. In the type 1 diabetic mice model, losartan exerted opposite effects on the expression of these receptors, upregulating AT1R and downregulating AT2R.
In many in vitro and in vivo studies, supraphysiological concentrations of Ang II were tested, so that the extrapolation of the observed results to clinical practice is doubtful. Furthermore, AT1R can be downregulated dose-dependently by Ang II [171], which may explain the bimodal dose responses to progressively increasing concentrations of Ang II in UMR-106 cells or in human primary bone cells from trabecular explants [45, 71, 172].
-
3.
The clinical benefit of ACEIs and ARBs can be partially counteracted by the reactive increase in pre-renin and renin mRNA levels which not only stimulate the conversion of Ang I to Ang II, leading to a reduction in the efficacy of these RAS inhibitors, but stimulate the PRR, producing tissue damage in an Ang II-independent manner insensitive to ACEIs or ARBs [46, 68, 173–175] (Fig. 4). This can be the explanation why losartan promoted the loss of bone mass and the deterioration of trabecular bone microarchitecture in type 1 diabetic mice [68]. Furthermore, Ang II can be synthesized via non-ACE enzymatic pathways (chymase, carboxypeptidase, cathepsin G, or tonin) which might explain the lack of effect of ACEIs [4–7, 51, 176].
-
4.
ACEIs decreased free testosterone and dehydroepiandrosterone (DHEA) plasma levels in men [177] and increased sex hormone-binding globulin (SHBG) in women [178]. This latter effect correlated with greater bone loss and vertebral and peripheral fractures [179], particularly in elderly patients [154].
-
5.
Drugs from the same family class can exhibit different pharmacological properties. Indeed, ARBs exhibit important differences in their binding affinity for the AT1R, which translates into differences in potency and duration of their effects (candesartan and olmesartan vs losartan) and in off-target effects (i.e., on PPARγ, oxidative stress, expression of adhesion molecules and proinflammatory cytokines) [156, 180, 181]. Thus, it should not be a surprise that the results described with one ARB cannot be replicated by another ARB even though they produce a similar reduction in blood pressure.
-
6.
Even when short-term therapy with ACEIs and ARBs decreased Ang II and aldosterone levels, long-term RAS inhibition resulted in a return of Ang II and aldosterone levels toward baseline values. Unfortunately, the local bone effects of aldosterone remain unknown.
-
7.
The RAS plays a key role in regulating mean arterial blood pressure (MAP) which determines the intramedullary pressure that is a driving force for transcortical interstitial fluid flow and is positively correlated with BMD, so that an increase in intramedullary pressure increases BMD, whereas decreased intramedullary pressure is associated with a decreased BMD [182]. Both ACEIs and ARBs decreased MAP and therefore, it would be expected that they can reduce the intramedullary pressure and BMD. In fact, perindopril significantly reduced blood pressure and decreased BMD in the distal femoral metaphysis of unfractured femora [133]. Further studies are needed to analyze the bone microvascular effects of RAS inhibitors.
Conclusions and Future Areas of Research
There is evidence that both local and systemic RAS may play an important role in bone metabolism and remodeling, but present evidence is sparse and sometimes contradictory. Therefore, there is an unmet need to understand the pathophysiological role of local and circulating RAS in human bone homeostasis and remodeling and the mechanisms involved in the possible therapeutic and/or adverse effects of ACEIs and ARBs observed in preclinical and, particularly, in clinical trials. Further studies are also needed to identify the signaling pathways downstream AT1R and AT2R stimulation, the interaction between RAS and sex steroids and glucocorticoids and the possible cross-talk between AT1R and AT2R in bone. Furthermore, as ACEIs and ARBs are first-choice drugs in the treatment of patients with hypertension, heart failure, diabetes mellitus or chronic kidney disease, situations frequently associated with osteoporosis, particularly in postmenopausal women, a key question to be answered is: Which is the most effective way to inhibit the local RAS? Interestingly, there is no information on the role of other RAS components on bone function: What is the role of the ACE2-Ang (1–7)-MasR pathway? What is the role of prorenin and renin? Or what role has aldosterone in bone homeostasis? We need also to understand the role of local RAS in patients with osteoporosis, arthritis and with metastases of primary tumors into bone. Finally, we need validated animal models to study the role of circulating and local RAS in bone homeostasis and in the pathophysiology of the most prevalent degenerative bone disorders which frequently coexist in aging populations with cardiovascular diseases where RAS inhibitors are the first-choice drugs.
References
Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Böhm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2013;34:2159–219.
Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren M, et al. European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. 2012;33(13):1635–701.
Casas JP, Chua W, Loukogeorgakis S, Vallance P, Smeeth L, Hingorani AD, et al. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet. 2005;366(9502):2026–33.
Ma TK, Kam KK, Yan BP, Lam YY. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br J Pharmacol. 2010;160(6):1273–92.
Farag E, Maheshwari K, Morgan J, Esa WAS, Doyle DJ. An update of the role of renin angiotensin in cardiovascular homeostasis. Anesth Analg. 2015;120(2):275–92.
Dendorfer A, Dominiak P, Schunkert H. ACE inhibitors and angiotensin II receptor antagonists. Handb Exp Pharmacol. 2005;170:407–42.
Tamargo J, Duarte J, Ruilope LM. New antihypertensive drugs under development. Curr Med Chem. 2015;22(3):305–42.
Wu HY, Huang JW, Lin HJ, Liao WC, Peng YS, Hung KY, et al. Comparative effectiveness of renin-angiotensin system blockers and other antihypertensive drugs in patients with diabetes: systematic review and bayesian network meta-analysis. BMJ. 2013;347:f6008.
Leung PS, Sernia C. The renin-angiotensin system and male reproduction: new functions for old hormones. Mol Cell Endocrinol. 2003;30(3):263–70.
Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: a target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2012;302(6):H1219–30.
Gironacci MM, Cerniello FM, Carbajosa NAL, Goldstein J, Cerrato BD. Protective axis of the renin-angiotensin system in the brain. Clin Sci. 2014;127(5):295–306 (Lond).
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52(3):415–72.
Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289(5484):1504–8.
Rodan GA, Martin TJ. Therapeutic approaches to bone disease. Science. 2000;289(5484):1508–14.
Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci USA. 1990;87(18):7260–4.
Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170(2):427–35.
Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202(9):1261–9.
Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473(2):139–46.
Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012;23(11):582–90.
Boyce BF. Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts. J Bone Miner Res. 2013;28(4):711–22.
Walsh CW, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone and beyond. Frontiers Inmmunol. 2014;5(5):1–8.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–19.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95(7):3597–602.
Syed F, Khosla S. Mechanisms of sex steroid effects on bone. Biochem Biophys Res Commun. 2005;328(3):688–96.
Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12(1):17–25.
Blair JM, Zhou H, Seibel MJ, Dunstan CR. Mechanisms of disease: roles of OPG, RANKL and RANK in the pathophysiology of skeletal metastasis. Nat Clin Pract Oncol. 2006;3(1):41–9.
Lee S-K, Lorenzo J. Cyrokine regulating osteoclast formation and function. Curr Opin Rheumatol. 2006;18(4):411–8.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–23.
Kikuta J, Ishii M. Osteoclast migration, differentiation and function: novel therapeutic targets for rheumatic diseases. Rheumatology. 2013;52(2):226–34 (Oxford).
Tat SK, Padrines M, Theoleyre S, Couillaud-Battaglia S, Heymann D, Redini F, Fortun Y. OPG/membranous—RANKL complex is internalized via the clathrin pathway before a lysosomal and a proteasomal degradation. Bone. 2006;39(4):706–15.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12(9):1260–8.
Indridason OS, Franzson L, Sigurdsson G. Serum osteoprotegerin and its relationship with bone mineral density and markers of bone turnover. Osteoporos Int. 2005;16(4):417–23.
Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15(6):457–75.
Lewiecki EM. New targets for intervention in the treatment of postmenopausal osteoporosis. Nat Rev Rheumatol. 2011;7(11):631–8.
Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. Segregation of TRAF6-mediated signalling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001;20(6):1271–80.
Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. CGTP Collaborators. the concise guide to pharmacology 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170(8):1459–581.
Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, Dzau VJ. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem. 1993;268(33):24539–42.
Murphy TJ, Alexander RW, Griendling KK, Runge MS, Beinstein KE. Isolation of a cDNA encoding the vascular type 1 angiotensin II receptor. Nature. 1991;351(6323):233–6.
Haendeler J, Berk BC. Angiotensin II mediated signal transduction. Important role of tyrosine kinases. Regul Pept. 2000;95(1–3):1–7.
Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci. 2007;112(8):417–28 (Lond).
Paul M, Poyan Mehr A, Kreuz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86(3):747–803.
Nouet S, Nahmias C. Signal transduction from the angiotensin II AT2 receptor. Trends Endocrinol Metab. 2000;11(1):1–6.
Lemarié CA, Schiffrin EL. The angiotensin II type 2 receptor in cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2010;11(1):19–31.
Steckelings UM, Rompe F, Kaschina E, Namsolleck P, Grzesiak A, Funke-Kaiser H, et al. The past, present and future of angiotensin II type 2 receptor stimulation. J Renin Angiotensin Aldosterone Syst. 2010;11(1):67–73.
Levy BI. How to explain the differences between renin angiotensin system modulators. Am J Hypertens. 2005;18(9 Pt 2):134S–41S.
Pessôa BS, van der Lubbe N, Verdonk K, Roks AJ, Hoorn EJ, Danser AJ. Key developments in renin–angiotensin—aldosterone system inhibition. Nat Rev Nephrol. 2013;9(1):26–36.
Horiuchi M, Akishita M, Dzau VJ. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension. 1999;33(2):613–21.
Akishita M, Yamada H, Dzau VJ, Horiuchi M. Increased vasoconstrictor response of the mouse lacking angiotensin II type 2 receptor. Biochem Biophys Res Commun. 1999;261(2):345–9.
Abdalla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276(43):39721–6.
Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216(2):R1–17.
Speth RC, Giese MJ. Update on the renin-angiotensin system. J Pharmacol Clin Toxicol. 2013;1(1):1–13.
Carey RM. Newly discovered components and actions of the renin-angiotensin system. Hypertension. 2013;62(5):818–22.
Santos RA, e Silva ACS, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100(14):8258–63.
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–43.
Ferreira AJ, Santos RA, Bradford CN, Mecca AP, Sumners C, Katovich MJ, et al. Therapeutic implications of the vasoprotective axis of the renin–angiotensin system in cardiovascular diseases. Hypertension. 2010;55(2):207–13.
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109(11):1417–27.
Funke-Kaiser H, Zollmann FS, Schefe JH, Unger T. Signal transduction of the (pro)renin receptor as a novel therapeutic target for preventing end-organ damage. Hypertens Res. 2010;33(2):98–104.
Krop M, Lu X, Danser AH, Meima ME. The (pro)renin receptor. a decade of research: what have we learned? Pflugers Arch. 2013;465(1):87–97.
Hoogwerf BJ. Renin-angiotensin system blockade and cardiovascular and renal protection. Am J Cardiol. 2010;105(1 Suppl):30A–5A.
Izu Y, Mizoguchi F, Kawamata A, Hayata T, Nakamoto T, Nakashima K, et al. Angiotensin II type 2 receptor blockade increases bone mass. J Biol Chem. 2009;284(8):4857–64.
Kaneko K, Ito M, Fumoto T, Fukuhara R, Ishida J, Fukamizu A, et al. Physiological function of the angiotensin AT1a receptor in bone remodeling. J Bone Miner Res. 2011;26(12):2959–66.
Bandow K, Nishikawa Y, Ohnishi T, Kakimoto K, Soejima K, Iwabuchi S, et al. Low-intensity pulsed ultrasound (LIPUS) induces RANKL, MCP-1, and MIP-1beta expression in osteoblasts through the angiotensin II type 1 receptor. J Cell Physiol. 2007;211(2):392–8.
Hatton R, Stimpel M, Chambers TJ. Angiotensin II is generated from angiotensin I by bone cells and stimulates osteoclastic bone resorption in vitro. J Endocrinol. 1997;152(1):5–10.
Sernia C, Li L, Huang H, Nguyuen K, Li YH, Hsu S, et al. Bone homeostasis: an emerging role for the renin-angiotensin system. In: Leung PS, editor. Frontiers in research of the renin-angiotensin system on human disease. New York: Springer; 2007. p. 179–95.
Gu SS, Zhang Y, Li XL, Wu SY, Diao TY, Hai R, et al. Involvement of the skeletal renin-angiotensin system in age-related osteoporosis of ageing mice. Biosci Biotechnol Biochem. 2012;76(7):1367–71.
Shuai B, Yang YP, Shen L, Zhu R, Xu XJ, Ma C, et al. Local renin-angiotensin system is associated with bone mineral density of glucocorticoid-induced osteoporosis patients. Osteoporos Int. 2015;26(3):1063–71.
Yongtao Z, Kunzheng W, Jingjing Z, Hu S, Jianqiang K, Ruiyu L, Chunsheng W. Glucocorticoids activate the local renin-angiotensin system in bone: possible mechanism for glucocorticoid-induced osteoporosis. Endocrine. 2014;47(2):598–608.
Zhang Y, Diao TY, Gu SS, Wu SY, Gebru YA, Chen X, et al. Effects of angiotensin II type 1 receptor blocker on bones in mice with type 1 diabetes induced by streptozotocin. J Renin Angiotensin Aldosterone Syst. 2014;15(3):218–27.
Asaba Y, Ito M, Fumoto T, Watanabe K, Fukuhara R, Takeshita S, et al. Activation of renin-angiotensin system induces osteoporosis independently of hypertension. J Bone Miner Res. 2009;24(2):241–50.
Hagiwara H, Hiruma Y, Inoue A, Yamaguchi A, Hirose S. Deceleration by angiotensin II of the differentiation and bone formation of rat calvarial osteoblastic cells. J Endocrinol. 1998;156(3):543–50.
Lamparter S, Kling L, Schrader M, Ziegler R, Pfeilschifter J. Effects of angiotensin II on bone cells in vitro. J Cell Physiol. 1998;175(1):89–98.
Bowler WB, Gallagher JA, Bilbe G. G-protein coupled receptors in bone. Front Biosci. 1998;1:d769–80.
Tsukamoto I, Akagi M, Inoue S, Yamagishi K, Mori S, Asada S. Expressions of local renin-angiotensin system components in chondrocytes. Eur J Histochem. 2014;58(2):2387.
Haznedaroglu IC, Beyazit Y. Pathobiological aspects of the local bone marrow renin-angiotensin system: a review. J Renin Angiotensin Aldosterone Syst. 2010;11(4):205–13.
Strawn WB, Richmond RS, Ann Tallant E, Gallagher PE, Ferrario CM. Renin–angiotensin system expression in rat bone marrow haematopoietic and stromal cells. Br J Haematol. 2004;126(1):120–6.
Haznedaroglu IC, Beyazit Y. Local bone marrow renin-angiotensin system in primitive, definitive and neoplastic haematopoiesis. Clin Sci. 2013;124(5):307–23 (Lond).
Beyazit Y, Purnak T, Guven GS, Haznedaroglu IC. Local bone marrow renin-angiotensin system and atherosclerosis. Cardiol Res Pract. 2011;1(1):714515.
Mrug M, Stopka T, Julian BA, Prchal JF, Prchal JT. Angiotensin II stimulates proliferation of normal early erythroid progenitors. J Clin Invest. 1997;100(9):2310–4.
Rodgers KE, Xiong S, Steer R, diZerega GS. Effect of angiotensin II on hematopoietic progenitor cell proliferation. Stem Cells. 2000;18(4):287–94.
Savary K, Michaud A, Favier J, Larger E, Corvol P, Gasc JM. Role of the renin-angiotensin system in primitive erythropoiesis in the chick embryo. Blood. 2005;105(1):103–10.
Shimizu H, Nakagami H, Osako MK, Hanayama R, Kunugiza Y, Kizawa T, et al. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008;22(7):2465–75.
Shimizu H, Nakagami H, Osako MK, Nakagami F, Kunugiza Y, Tomita T, et al. Prevention of osteoporosis by angiotensin-converting enzyme inhibitor in spontaneous hypertensive rats. Hypertens Res. 2009;32(9):786–90.
Hiruma Y, Inoue A, Hirose S, Hagiwara H. Angiotensin II stimulates the proliferation of osteoblast-rich populations of cells from rat calvariae. Biochem Biophys Res Commun. 1997;230(1):176–8.
Sugaya T, Nishimatsu S, Tanimoto K, Takimoto E, Yamagishi T, Imamura K, et al. Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem. 1995;270(32):18719–22.
Nakagami H, Morishita R. Hormones and osteoporosis update. Effect of angiotensin II on bone metabolism. Clin Calcium. 2009;19(7):997–1002.
Dossing DA, Stern PH. Receptor activator of NF-kappa B ligand protein expression in UMR-106 cells is differentially regulated by parathyroid hormone and calcitriol. J Cell Biochem. 2005;95(5):1029–41.
Osako MK, Nakagami H, Shimamura M, Koriyama H, Nakagami F, Shimizu H, et al. Cross-talk of receptor activator of nuclear factor-κB ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler Thromb Vasc Biol. 2013;33(6):1287–96.
Guan XX, Zhou Y, Li JY. Reciprocal roles of angiotensin II and Angiotensin II Receptors Blockade (ARB) in regulating Cbfa1/RANKL via cAMP signaling pathway: possible mechanism for hypertension-related osteoporosis and antagonistic effect of ARB on hypertension-related osteoporosis. Int J Mol Sci. 2011;12(7):4206–13.
Tintut Y, Parhami F, Le V, Karsenty G, Demer LL. Inhibition of osteoblast-specific transcription factor Cbfa1 by the cAMP pathway in osteoblastic cells. J Biol Chem. 1999;274(41):28875–9.
Thaler R, Spitzer S, Rumpler M, Fratzl-Zelman N, Klaushofer K, Paschalis EP, et al. Differential effects of homocysteine and beta aminopropionitrile on preosteoblastic MC3T3-E1 cells. Bone. 2010;46(3):703–9.
Cola C, Almeida M, Li D, Romeo F, Mehta JL. Regulatory role of endothelium in the expression of genes affecting arterial calcification. Biochem Biophys Res Commun. 2004;320(2):424–7.
Kubalak SW, Webb JG. Angiotensin II enhancement of hormone-stimulated cAMP formation in cultured vascular smooth muscle cells. Am J Physiol. 1993;264(1 Pt 2):H86–96.
MacGregor GA, Cappuccio FP. The kidney and essential hypertension: a link to osteoporosis? J Hypertens. 1993;11(8):781–5.
Neelon FA, Birch BM, Drezner M, Lebovitz HE. Urinary cyclic adenosine monophosphate as an aid in the diagnosis of hyperparathyroidism. Lancet. 1973;1(7804):631–3.
Paszty C, Turner CH, Robinson MK. Sclerostin: a gem from the genome leads to bone-building antibodies. J Bone Miner Res. 2010;25(9):1897–904.
Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–64.
Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, et al. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283(10):6509–18.
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–9.
Nakai K, Kawato T, Morita T, Iinuma T, Kamio N, Zhao N, et al. Angiotensin II induces the production of MMP-3 and MMP-13 through the MAPK signaling pathways via the AT(1) receptor in osteoblasts. Biochimie. 2013;95(4):922–33.
Guo L, Wang M, Zhang ZY, Hao L, Lou BY, Li XY, et al. Angiotensin II induces interleukin-6 synthesis in osteoblasts through ERK1/2 pathway via AT1 receptor. Arch Oral Biol. 2011;56(3):205–11.
Algan SM, Purdon M, Horowitz SM. Role of tumor necrosis factor alpha in particulate-induced bone resorption. J Orthop Res. 1996;14(1):30–5.
Seck T, Diel I, Bismar H, Ziegler R, Pfeilschifter J. Expression of interleukin-6 (IL-6) and IL-6 receptor mRNA in human bone samples from pre- and postmenopausal women. Bone. 2002;30(1):217–22.
Dai JC, He P, Chen X, Greenfield EM. TNF alpha and PTH utilize distinct mechanisms to induce IL-6 and RANKL expression with markedly different kinetics. Bone. 2006;38(4):509–20.
Patil C, Zhu X, Rossa C Jr, Kim YJ, Kirkwood KL. p38 MAPK regulates IL-1 beta induced IL-6 expression through mRNA stability in osteoblasts. Immunol Invest. 2004;33(2):213–33.
Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, et al. VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci USA. 2005;102(39):14016–21.
Li G, Wang M, Hao L, Loo WT, Jin L, Cheung MN, et al. Angiotensin II induces mitochondrial dysfunction and promotes apoptosis via JNK signalling pathway in primary mouse calvaria osteoblast. Arch Oral Biol. 2014;59(5):513–23.
Schurman SJ, Bergstrom WH, Shoemaker LR, Welch TR. Angiotensin II reduces calcium uptake into bone. Pediatr Nephrol. 2004;19(1):33–5.
Grant FD, Mandel SJ, Brown EM, Williams GH, Seely EW. Interrelationships between the renin-angiotensin-aldosterone and calcium homeostatic systems. J Clin Endocrinol Metab. 1992;75(4):988–92.
Kanaan RA, Kanaan LA. Transforming growth factor beta1, bone connection. Med Sci Monit. 2006;12(8):164–9.
Fox SW, Lovibond AC. Current insights into the role of transforming growth factor-β in bone resorption. Molec Cell Endocrinol. 2005;243(1–2):19–26.
Brown RE. Angiotensin-converting enzyme, transforming growth factor beta(1), and interleukin 11 in the osteolytic lesions of Langerhans cell Histiocytosis. Arch Pathol Lab Med. 2000;124(9):1287–90.
Alagiakrishnan K, Juby A, Hanley D, Tymchak W, Sclater A. Role of vascular factors in osteoporosis. J Gerontol. 2003;58(4):362–6.
Rajkumar DS, Faitelson AV, Gudyrev OS, Dubrovin GM, Pokrovski MV, Ivanov AV. Comparative evaluation of enalapril and losartan in pharmacological correction of experimental osteoporosis and fractures of its background. J Osteoporos. 2013;2013:325693.
Keen R. Osteoporosis: strategies for prevention and management. Best Pract Res Clin Rheumatol. 2007;21(1):109–22.
NIH Consensus Development Panel on Osteoporosis Prevention. Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285(6):785–95.
Flieger J, Karachalios T, Khaldi L, Raptou P, Lyritis G. Mechanical stimulation in the form of vibration prevents postmenopausal bone loss in ovariectomized rats. Calcif Tissue Int. 1998;63(6):510–4.
von der Recke P, Hansen MA, Hassager C. The association between low bone mass at the menopause and cardiovascular mortality. Am J Med. 1999;106(3):273–8.
Choi SH, An JH, Lim S, Koo BK, Park SE, Chang HJ, et al. Lower bone mineral density is associated with higher coronary calcification and coronary plaque burdens by multidetector row coronary computed tomography in pre- and postmenopausal women. Clin Endocrinol. 2009;71(5):644–51 (Oxf).
Nishio K, Mukae S, Aoki S, Itoh S, Konno N, Ozawa K, et al. Congestive heart failure is associated with the rate of bone loss. J Intern Med. 2003;253(4):439–46.
Vasan RS, Beiser A, Seshadri S, Larson MG, Kannel WB, D’Agostino RB, et al. Residual lifetime risk for developing hypertension in middle-aged women, men: the Framingham Heart Study. JAMA. 2002;287(8):1003–10.
Resnick LM, Laragh JH, Sealey JE, Alderman MH. Divalent cations in essential hypertension: relations between serum ionized calcium, magnesium, and plasma renin activity. N Engl J Med. 1983;309(15):888–91.
Strazzullo P, Nunziata V, Cirillo M, Giannattasio R, Ferrara LA, Mattioli PL, et al. Abnormalities of calcium metabolism in essential hypertension. Clin Sci. 1983;65(2):137–41 (Lond).
Cappuccio FP, Meilahn E, Zmuda JM, Cauley JA. High blood pressure and bone mineral loss in elderly white women: a prospective study. Study of Osteoporotic Fractures Research Group. Lancet. 1999;354(9183):971–5.
McCarron DA, Pingree PA, Rubin RJ, Gaucher SM, Molitch M, Krutzik S. Enhanced parathyroid function in essential hypertension: a homeostatic response to a urinary calcium leak. Hypertension. 1980;2(2):162–8.
McCarron DA. Low serum concentrations of ionized calcium in patients with hypertension. N Engl J Med. 1982;307(4):226–8.
Ilic K, Obradovic N, Vujasinovic-Stupar N. The relationship among hypertension, antihypertensive medications, and osteoporosis: a narrative review. Calcif Tissue Int. 2013;92(3):217–27.
Tsuda K, Nishio I, Masuyama Y. Bone Mineral Density in Women With Essential Hypertension. Am J Hypertens. 2001;14(7 Pt 1):704–7.
Lynn H, Kwok T, Wong SY, Woo J, Leung PC. Angiotensin converting enzyme inhibitor use is associated with higher bone mineral density in elderly Chinese. Bone. 2006;38(4):584–8.
Rejnmark L, Vestergaard P, Mosekilde L. Treatment with beta-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: a nationwide case-control study. J Hypertens. 2006;24(3):581–9.
Yang S, Nguyen ND, Center JR, Eisman JA, Nguyen TV. Association between hypertension and fragility fracture: a longitudinal study. Osteoporos Int. 2014;25(1):97–103.
Liu HW, Iwai M, Takeda-Matsubara Y, Wu L, Li JM, Okumura M, et al. Effect of estrogen and AT1 receptor blocker on neointima formation. Hypertension. 2002;40(4):451–7.
Zhang Y, Wang K, Song Q, Liu R, Ji W, Ji L, et al. Role of the local bone renin-angiotensin system in steroid-induced osteonecrosis in rabbits. Mol Med Rep. 2014;9(4):1128–34.
Garcia P, Schwenzer S, Slotta JE, Scheuer C, Tami AE, Holstein JH, et al. Inhibition of angiotensin-converting enzyme stimulates fracture healing and periosteal callus formation-role of a local renin-angiotensin system. Br J Pharmacol. 2010;159(8):1672–80.
Gu SS, Zhang Y, Wu SY, Diao TY, Gebru YA, Deng H. Early molecular responses of bone to obstructive nephropathy induced by unilateral ureteral obstruction in mice. Nephrology. 2012;17(8):767–73.
Diao TY, Pan H, Gu SS, Chen X, Zhang FY, Wong MS, et al. Effects of angiotensin-converting enzyme inhibitor, captopril, on bone of mice with streptozotocin-induced type 1 diabetes. J Bone Miner Metab. 2014;32(3):261–70.
Donmez BO, Ozdemir S, Sarikanat M, Yaras N, Koc P, Demir N, et al. Effect of angiotensin II type 1 receptor blocker on osteoporotic rat femurs. Pharmacol Rep. 2012;64(4):878–88.
Takeda-Matsubara Y, Nakagami H, Iwai M, Cui TX, Shiuchi T, Akishita M, et al. Estrogen activates phosphatases and antagonizes growth-promoting effect of angiotensin II. Hypertension. 2002;39(1):41–5.
Liu YY, Yao WM, Wu T, Xu BL, Chen F, Cui L. Captopril improves osteopenia in ovariectomized rats and promotes bone formation in osteoblasts. J Bone Miner Metab. 2011;29(2):149–58.
Pérez-Castrillón JL, Justo I, Silva J, Sanz A, Martín-Escudero JC, Igea R, et al. Relationship between bone mineral density and angiotensin converting enzyme polymorphism in hypertensive postmenopausal women. Am J Hypertens. 2003;16(3):233–5.
Woods D, Onambele G, Woledge R, Skelton D, Bruce S, Humphries SE, et al. Angiotensin-I converting enzyme genotype-dependent benefit from hormone replacement therapy in isometric muscle strength and bone mineral density. J Clin Endocrinol Metab. 2001;86(5):2200–4.
Steinbuch M, Youket TE, Cohen S. Oral glucocorticoid use is associated with an increased risk of fracture. Osteoporos Int. 2004;15(4):323–8.
Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;48(11):3224–9.
Skov J, Persson F, Frøkiær J, Christiansen JS. Tissue renin-angiotensin systems: a unifying hypothesis of metabolic disease. Front Endocrinol. 2014;5:23 (Lausanne).
Pérez-Castrillón JL, Silva J, Justo I, Sanz A, Martín-Luquero M, Igea R, et al. Effect of quinapril, quinapril-hydrochlorothiazide, and enalapril on the bone mass of hypertensive subjects: relationship with angiotensin converting enzyme polymorphisms. Am J Hypertens. 2003;16(6):453–9.
Kang KY, Kang Y, Kim M, Kim Y, Yi H, Kim J, et al. The effects of antihypertensive drugs on bone mineral density in ovariectomized mice. J Korean Med Sci. 2013;28(8):1139–44.
Broulík PD, Tesar V, Zima T, Jirsa M. Impact of antihypertensive therapy on the skeleton: effects of enalapril and AT1 receptor antagonist losartan in female rats. Physiol Res. 2001;50(4):353–8.
Stimpel M, Jee WS, Ma Y, Yamamoto N, Chen Y. Impact of antihypertensive therapy on postmenopausal osteoporosis: effects of the angiotensin converting enzyme inhibitor moexipril, 17beta-estradiol and their combination on the ovariectomy-induced cancellous bone loss in young rats. J Hypertens. 1995;13(12 Pt 2):1852–6.
Ma YF, Stimpel M, Liang H, Pun S, Jee WS. Impact of antihypertensive therapy on the skeleton: effects of moexipril and hydrochlorothiazide on osteopenia in spontaneously hypertensive ovariectomized rats. J Endocrinol. 1997;154(3):467–74.
García-Testal A, Monzó A, Rabanaque G, González A, Romeu A. Evolution of the bone mass of hypertense menopausal women in treatment with fosinopril. Med Clin. 2006;127(18):692–4 (Barc).
Nakagami H, Kiomy O, Shimizu H, Hanayama R, Morishita R. Potential contribution of action of renin angiotensin system to bone metabolism. Curr Hypertens Rev. 2007;3(2):129–32.
Schlienger RG, Kraenzlin ME, Jick SS, Meier CR. Use of beta-blockers and risk of fractures. JAMA. 2004;292(11):1326–32.
Solomon DH, Mogun H, Garneau K, Fischer MA. Risk of fractures in older adults using antihypertensive medications. J Bone Miner Res. 2011;26(7):1561–7.
Kwok T, Leung J, Zhang YF, Bauer D, Ensrud KE, Barrett-Connor E, et al. Osteoporotic Fractures inMen(MrOS) Research Group. Does the use of ACE inhibitors or angiotensin receptor blockers affect bone loss in older men? Osteoporos Int. 2012;23(8):2159–67.
Masunari N, Fujiwara S, Nakata Y, Furukawa K, Kasagi F. Effect of angiotensin converting enzyme inhibitor and benzodiazepine intake on bone loss in older Japanese. Hiroshima J Med Sci. 2008;57(1):17–25.
Zhang YF, Qin L, Leung PC, Kwok TC. The effect of angiotensin-converting enzyme inhibitor use on bone loss in elderly Chinese. J Bone Miner Metab. 2012;30(6):666–73.
Daikuhara H, Fukunaga K, Ohshima T. Difference in the effects of switching from candesartan to olmesartan or telmisartan to olmesartan in hypertensive patients with type 2 diabetes: the COTO study. Drug Des Devel Ther. 2014;8:219–26.
Ma L, Ji JL, Ji H, Yu X, Ding LJ, Liu K, et al. Telmisartan alleviates rosiglitazone-induced bone loss in ovariectomized spontaneous hypertensive rats. Bone. 2010;47(1):5–11.
Zhao X, Wang JX, Feng YF, Wu ZX, Zhang Y, Shi L, et al. Systemic treatment with telmisartan improves femur fracture healing in mice. PLoS One. 2014;9(3):e92085.
Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113(6):846–55.
Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13(12):1496–503.
Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARγ2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 2009;106(2):232–46.
Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148(6):2669–80.
Lecka-Czernik B. Bone loss in diabetes: use of anti-diabetic thiazolidinediones and secondary osteoporosis. Curr Osteoporos Rep. 2010;8(4):178–84.
Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-γ activity. Circulation. 2004;109(17):2054–7.
Kolli V, Stechschulte LA, Dowling AR, Rahman S, Czernik PJ, Lecka-Czernik B. Partial agonist, telmisartan, maintains PPARγ serine 112 phosphorylation, and does not affect osteoblast differentiation and bone mass. PLoS One. 2014;9(5):e96323.
Li YQ, Ji H, Shen Y, Ding LJ, Zhuang P, Yang YL, et al. Chronic treatment with angiotensin AT1 receptor antagonists reduced serum but not bone TGF-beta1 levels in ovariectomized rats. Can J Physiol Pharmacol. 2009;87(1):51–5.
Zhang YF, Qin L, Kwok TC, Yeung BH, Li GD, Liu F. Effect of angiotensin II type I receptor blocker losartan on bone deterioration in orchiectomized male hypertensive and normotensive rats. Chin Med J. 2013;126(14):2661–5 (Engl).
Butt DA, Mamdani M, Gomes T, Lix L, Lu H, Tu K. Tu K; Hypertension Outcome, Surveillance Team. Risk of osteoporotic fractures with angiotensin II receptor blockers versus angiotensin converting-enzyme inhibitors in hypertensive community-dwelling elderly. J Bone Miner Res. 2014;29(11):2483–8.
Nishiya Y, Sugimoto S. Effects of various antihypertensive drugs on the function of osteoblast. Biol Pharm Bull. 2001;24(6):628–33.
Zhu YC, Zhu YZ, Li J, Schäfer H, Schmidt WE, Unger T, et al. Effects of ramipril on cardiac gene transcription levels of angiotensin II receptors after myocardial infarction. Zhongguo yao lixue bao. 1999;20(6):481–5.
Bouscarel B, Wilson PB, Blackmore PF, Lynch CJ, Exton JH. Agonist-induced down-regulation of the angiotensin II receptor in primary cultures of rat hepatocytes. J Biol Chem. 1988;263(29):14920–4.
Wilms H, Rosenstiel P, Unger T, Deuschl G, Lucius R. Neuroprotection with angiotensin receptor antagonists: a review of the evidence and potential mechanisms. Am J Cardiovasc Drugs. 2005;5(4):245–53.
Zhang Z, Zhang Y, Ning G, Kong J, Deb DK, Li YC. Combination therapy with AT1 receptor blocker and vitamin D analog markedly ameliorates diabetic nephropathy. Proc Natl Acad Sci U S A. 2008;105(41):15896–901.
Danser AH. The increase in renin during renin inhibition: does it result in harmful effects by the (pro)renin receptor? Hypertens Res. 2010;33(1):4–10.
Pimenta E, Oparil S. Role of aliskiren in cardio-renal protection and use in hypertensives with multiple risk factors. Vasc Health Risk Manag. 2009;5(1):453–63.
Azizi M, Chatellier G, Guyene TT, Murieta-Geoffroy D, Ménard J. Additive effects of combined angiotensin-converting enzyme inhibition and angiotensin II antagonism on blood pressure and renin release in sodium-depleted normotensives. Circulation. 1995;92(4):825–34.
Kwok T, Ohlsson C, Vandenput L, Tang N, Zhang YF, Tomlinson B, et al. ACE inhibitor use was associated with lower serum dehydroepiandrosterone concentrations in older men. Clin Chim Acta. 2010;411(15–16):1122–5.
Koshida H, Takeda R, Miyamori I. Lisinopril decreases plasma free testosterone in male hypertensive patients and increases sex hormone binding globulin in female hypertensive patients. Hypertens Res. 1998;21(4):279–82.
Hoppé E, Bouvard B, Royer M, Audran M, Legrand E. Sex hormone-binding globulin in osteoporosis. Joint Bone Spine. 2010;77(4):306–12.
Schiling P, Löffler G. Effects of angiotensin II on adipose conversion and expression of genes of the renin-angiotensinogen system in human preadipocytes. Horm Metab Res. 2001;33(4):189–95.
Dandona P, Kumar V, Aljada A, Ghanim H, Syed T, Hofmayer D, et al. Angiotensin 2 receptor blocker valsartan suppresses reactive oxygen species generation in leucocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: evidence of an anti-inflammatory action. J Clin Endocrinol Metab. 2003;88(9):4496–501.
Bergula A, Huang W, Frangos J. Femoral vein ligation increases bone mass in the hindlimb suspended rat. Bone. 1999;24(3):171–7.
Acknowledgments
To Paloma Vaquero for her technical assistance.
Funding
This review was supported by Instituto de Salud Carlos III (Red RIC, and PI11/01030) and Comunidad de Madrid (S2010/BMD-2374).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Juan Tamargo, Ricardo Caballero, and Eva Delpón declare that they do not have any conflict of interest.
Animal/Human studies
The article does not contain any studies with human or animal subjects performed by the any of the authors.
Rights and permissions
About this article

Cite this article
Tamargo, J., Caballero, R. & Delpón, E. The Renin–Angiotensin System and Bone. Clinic Rev Bone Miner Metab 13, 125–148 (2015). https://doi.org/10.1007/s12018-015-9189-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-015-9189-6