
Abstract
Emerging evidence suggests a stimulating effect of parathyroid hormone (PTH) on the renin–angiotensin–aldosterone system (RAAS). In primary hyperparathyroidism, chronic-elevated PTH levels seem to stimulate the RAAS which may explain the increased risk of cardiovascular disease (CVD). In addition to increased PTH levels, low vitamin D levels may also directly increase risk of CVD, as vitamin D, itself, has been shown to inhibit the RAAS. Angiotensin II, aldosterone and cortisol all negatively impact bone health. Hyperaldosteronism is associated with a reversible secondary hyperparathyroidism due to increased renal calcium excretion. Moreover, the angiotensin II receptor is expressed by human parathyroid tissue, and angiotensin may therefore directly stimulates PTH secretion. An increased bone loss is found in patients with hyperaldosteronism. The angiotensin II receptor seems main responsible for the RAAS-initiated bone loss due to a receptor activator of NF-κB ligand-mediated activation of the osteoclasts. Available data suggest a reduced fracture rate and increased bone mineral density in patients treated with angiotensin II receptor blockers, whereas treatment with angiotensin-converting enzyme inhibitors causes the opposite effects. Mineralocorticoid receptor antagonists seem to be beneficial to bone in patients with hyperaldosteronism, but it is unknown whether this also applies to other individuals. Further long-term studies are needed to clarify the effect of RAAS inhibitors on bone health. RAAS inhibitors, are widely prescribed worldwide and beneficial as well as harmful effects may have large impact on bone health in the general population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVD) and osteoporosis are highly prevalent age-related diseases characterized by lack of symptoms until adverse outcomes occur. They represent two major public health problems and frequently coexist.
Increased activation of the RAAS plays a central role in the pathogenesis of hypertension and CVD. Emerging evidence supports a “new endocrine axis” between the RAAS and the parathyroid glands [1–3]. Patients with calcium metabolic disorders have an increased risk of CVD [4–7] and vice versa [8]. This interaction is clinical relevant because it, at least partly, may explain the association between CVD and calcium metabolic disorders.
Vitamin D has for decades been associated with a blood pressure (BP) lowering effect as well as reduced risk of CVD [9], whereas less attention has been paid to treatment with RAAS inhibitors (RAASi) in calcium metabolic disorders.
RAASi are widely used in clinical practice as antihypertensive agents and in the treatment of conditions with hyperaldosteronism. Accumulating data suggesting this “new endocrine axis” raise the question on whether treatment with RAASi improves bone mineral density (BMD) and reduces fracture risk.
The aim of this review is to summarize the evidence of a clinical important relationship between the two hormone systems and subsequently present existing data regarding the effect of ACE inhibitors (ACEi), angiotensin II receptor blockers (ARBs) and mineralocorticoid receptor antagonists (MRAs) on BMD, bone turnover markers and fracture risk. Because renin inhibitors are rarely used, they are not included in this review.
Method
For a review of literature regarding a possible interaction between RAAS and PTH, we searched PubMed for the terms “PTH,” “renin–angiotensin–aldosterone system,” “aldosterone,” “hyperaldosteronism,” “osteoporosis,” “hyperparathyroidism,” “vitamin D deficiency,” “angiotensin II,” “renin” and “cardiovascular disease.”
For the review of bone and RAAS inhibition, a systematic search in PubMed for the MeSH terms “ACE inhibitors,” “Mineralocorticoid Receptor Antagonists [Pharmacological Action]” and “Angiotensin ii receptor antagonist [Pharmacological Action]” combined with the terms, “bone mineral density,” “bone,” “fracture” and “bone turnover,” respectively.
Papers were further selected for inclusion based on our own knowledge and by searching reference lists of identified articles.
Only full-text English language articles were considered and inclusion criteria were data on fracture risk, BMD or bone turnover markers.
Effects of Calciotropic Hormones on the Renin–Angiotensin–Aldosterone System
Accumulating evidence suggests that PTH may directly and indirectly activate the RAAS. It is widely accepted that increased levels of PTH are associated with CVD (including hypertension), as well as osteoporosis [10]. Moreover, high PTH levels, even within the upper limit of the reference interval, have been associated with an increased all-cause and CVD mortality [11] suggesting an underlying precarious effect of PTH on non-skeletal adverse health outcome.
Experimental Studies on Effects of PTH Treatment
The biologically active region of PTH (the N-terminal 1–34 sequence) displays remarkable homology with that of adrenocorticotropic hormone (ACTH) [12]. A directly stimulation of aldosterone synthesis by PTH is supported by the fact that PTH type 1 receptors as well as receptors for parathyroid hormone-related protein are expressed by normal human adrenocortical zona glomerulosa cells [13]. An effect of PTH on aldosterone secretion is further supported by findings from in vitro studies showing that PTH increased the basal secretion of aldosterone and cortisol [14, 15] as well as PTH increases the angiotensin II (Ang II)-mediated aldosterone secretion [16].
In healthy men (n = 4), 12 days of continuous synthetic human PTH 1–34 (PTH1–34) infusion was investigated [15]. In addition to reversible hypercalcemia and hypertension, treatment caused a transient increased excretion of the tetrahydroaldosterone (a major metabolic product of aldosterone) and increased plasma cortisol concentration [15]. In another study including healthy men only (n = 8), a single infusion of 12.5 µg PTH1–34 doubled the plasma renin concentration [2]. Similarly, in postmenopausal women with severe osteoporosis (n = 20) treatment with PTH1–34 injections, 20 µg once-a-day, for 1 year, increased cortisol levels by approximate one-third compared to controls treated with vitamin D and calcium supplementation [12]. Unfortunately, this study did not report aldosterone levels.
Primary Hyperparathyroidism
An increased risk of CVD in patients with PHPT is well documented. In a number of studies, PHPT has been associated with an increased risk of hypertension [5, 6, 17, 18], left ventricular hypertrophy [7], and an increased pulse wave velocity [7]. Patients with PHPT are at increased risk of suffering from stroke [6], myocardial ischemia [4, 6] and have an overall increased mortality due to CVD [19–21]. Several [4–7] although not all [19, 22] studies report that CVD-related adverse outcomes are reversible following PTX.
The increased risk of CVD may be mediated by increased RAAS activity. Compared to normal parathyroid tissue, a twofold to fourfold increase in the expression of the angiotensin II type 1 receptor (AT1R) and mineralocorticoid receptors (MR) has been detected in parathyroid adenoma tissue [1]. A newer study by Brunaud et al. [23] reports a positive correlation between PTH and aldosterone levels pre-, but not postoperatively in patients with PHPT. Cortisol concentrations might as well be increased in patients with PHPT [24].
In PHPT, a study by Jespersen et al. [25] reported elevated Ang II and vasopressin concentrations prior to PTX, whereas the aldosterone concentration remained unchanged compared to a control group. After PTX, the concentrations of Ang II (17 → 10 pmol/l) and vasopressin (2.9 → 1–9 pmol/l) normalized.
Beyond a vasoconstrictive and antidiuretic effect, vasopressin stimulates the ACTH concentration directly in the pituitary gland, thereby promoting the synthesis of glucocorticoids and (to a lesser extent) mineralocorticoids. Furthermore, increased calcium levels are known to increase BP and stimulate the vasopressin release from the neuroposterior pituitary [26].
Vitamin D Deficiency
Secondary hyperparathyroidism (SHPT) is often caused by vitamin D deficiency. Associations between vitamin D deficiency and risk of hypertension have been extensively investigated. Recently, a large mendelian randomization study demonstrated that low levels of 25-hydroxyvitamin D (25OHD) are associated with an increased BP [27]. Several [28–30] although not all [31] RCTs on effects of vitamin D supplementation have shown a decrease in BP in response to supplementation. Trials demonstrating an BP lowering effect are characterized by including study subjects with low vitamin D levels and an increased BP at baseline [28–30].
Vitamin D suppresses the renin biosynthesis [32, 33] contrary to PTH [2]. Lower plasma 25OHD levels are associated with higher circulating Ang II levels [33, 34] and a blunted renal plasma flow response to exogenous Ang II infusion consistent with local activation of the RAAS in the setting of lower plasma 25OHD concentrations [33]. In hypertensive patients suffering from vitamin D deficiency, high dose vitamin D3 increases the tissue sensitivity to Ang II akin to ACEis [1, 35].
In summary, several lines of evidence suggest a stimulating effect of PTH on the RAAS. In patients with abnormal high PTH levels due to an autonomously increased PTH secretion, PTH also seems to affect the RAAS which may explain the increased risk of CVD in patients with PHPT. In addition to an effect on PTH secretion, vitamin D may itself exert an inhibiting effect on the RAAS which may provide an explanation for the increased risk of CVD in vitamin D insufficiency.
The Influence of the Renin–Angiotensin–Aldosterone System (RAAS) on Calciotropic Hormones
Primary Aldosteronism
Primary aldosteronism (PA) is probably underdiagnosed. Up to 10 % of hypertensive patients might suffer from PA. Normokalemic hypertension constitutes the most common presentation, and high aldosterone and abnormal low Ang II and renin concentration characterize the disease [36].
The occurrence of cardiovascular complications is increased in patients with PA independently of BP levels. Compared to patients with essential hypertension, the odds ratio of stroke is 4.2 (95 % CI 2.0–8.6) and the odds ratio of myocardial infarction is 6.5 (95 % CI 1.5–27.4) [37]. Treatment has a high cure rate of hypertension, but evidence of reduced mortality has not been documented [36].
PA is associated with osteoporosis [38–42] and SHPT which may be attributable to an increased renal calcium and magnesium excretion [38–40, 42–47].
Studies comparing patients suffering from PA and essential hypertension have shown significantly higher PTH levels at approximate one-third in PA patients [43–46], as well as lower serum calcium levels [43, 44] and an increased [43] or a trend toward an increased [44, 45] renal calcium excretion despite similar 25OHD levels.
The two major subtypes of PA are aldosterone-producing adenomas (APA) representing 30–40 % and bilateral adrenal hyperplasia (BAH) representing 60–70 % of the cases. The aldosterone [37, 38, 48, 49] and cortisol [50] concentration is highest in APA, and presumably, patients suffering from APA have more severe calcium metabolic abnormalities and more pronounced bone involvement. This has been confirmed in one study, demonstrating that PTH was only increased in patients with PA due to aldosterone-producing adenomas (APAs) compared to patients with BAH and essential hypertension [48]. In the study by Rossi et al. [48], it was suggested that raised serum PTH levels are a feature of APA that can be used for selecting the PA patients to be submitted to adrenal vein sampling. Compared with patients with incidentaloma, PA had a decreased BMD at the lumbar spine (13 %), total (8 %) and femoral neck (11 %), an increased prevalence of osteoporosis (73 vs. 20 %) and tended to have more morphometric vertebral fractures (46 vs. 13 %) [38].
It should be emphasized that elevated BP itself increases urinary calcium causing SHPT [51]. Of all studies investigating the PTH concentration in PA patients, only the study by Rossi et al. compared PA with hypertensive patients with an almost similar BP, whereas other studies used controls with significantly lower BP [38, 43, 44]. In this study by Rossi et al. [45], the PTH concentrations were significantly higher in the PA group (50 %), suggesting that aldosterone levels rather than blood pressure per se are of importance to PTH responses. However, Brown et al. [1] did not find increased PTH levels in response to a single infusion of aldosterone despite an increase in aldosterone of 892 %. Accordingly, Ang II rather than aldosterone may mediate the acute increase in PTH, whereas aldosterone may be responsible for the chronic PTH increment [1]. This hypothesis, however, needs to be further investigated.
Secondary Aldosteronism (SA)
SA is defined as increased plasma renin, Ang II and aldosterone caused by external stimulation to the adrenal glands. Commonly known causes include CVD, cirrhosis and renal failure, as well as treatment with loop diuretics.
A small human study reports SHPT in all patients (n = 9) consecutively admitted to a medical department due to decompensated congestive heart failure [52].
Hypertensive geriatric inpatients have been reported to have higher PTH levels than normotensive inpatients despite similar 25OHD levels [53], and similar results have been reported in the general population [10].
A single infusion of Ang II increases PTH in healthy men [2] and obese; vitamin D-deficient subjects in a dose-specific manner [1]. In addition to aldosterone secretion, Ang II promotes sodium and water reabsorption, systemic arteriolar vasoconstriction, endothelial dysfunction and stimulates the sympathetic nervous system. Beyond the traditional synthesis of Ang II as a part of the RAAS, Ang II can be generated by endothelial cells [54]. AT1R and Ang II type 2 receptor (AT2R) are expressed by osteoblasts and osteoclasts [51]. Moreover, AT1R is expressed by human parathyroid tissue [1]. Emerging evidence suggests a local RAAS in the bone [55–57] as the angiotensin-converting enzyme (ACE) is expressed by bone cells [54, 58].
Ang II increases bone remodeling directly with a predominance of resorption. This is caused by an increased RANKL expression in osteoblasts [54, 56, 58], and the AT1R seems to be main responsible for activating this cascade [56–60]. In response to injections with Ang II, decreased levels of the formative bone markers alkaline phosphatase (ALP) [59, 61] and osteocalcin (OC) [59] have been found.
In summary, an expanding body of evidence supports that PA and SA are associated with a reversible SHPT due to increased renal calcium and magnesium excretion. Increased BP also causes SHPT, and both aldosterone and Ang II increases PTH.
An increased and modifiable rate of bone loss is found in hyperaldosterone subjects, and AT1R seems main responsible for the RAAS-initiated bone loss due to a RANKL-mediated activation of the osteoclasts.
Angiotensin and Aldosterone Inhibitors and Bone Health
Numerous studies investigating the link between hypertension and BMD have shown an inverse association between BP and BMD [62–64]. As hypertension is associated with an increased renal calcium excretion, a renal calcium leak has been suggested as a physiological explanation [56, 62, 63]. However, an effect of the PTH aldosterone axis may as well be of importance.
In end-stage renal failure, treatment with ACEi and ARB is associated with decreased PTH levels [65]. Moreover, Brown et al. [10] recently showed that the use of any RAASi (ACEi and ARBs) was independently associated with lower PTH levels compared to the use of non-RAASi medication (45.0 ± 19.1 vs. 47.1 ± 19.4 pg/ml), and a positive correlation between PTH and aldosterone was most pronounced in patients with a “primary aldosteronism-like” phenotype (50.2 pg/ml compared to 40.4 pg/ml in patients with “normal phenotype”).
A physiologic interaction between the parathyroid and adrenal glands as well as identification of a local RAAS in the bones raises the question on whether treatment with RAASi improves BMD and reduces fracture risk in general or in selected groups.
Effects of ACEis and ARBs on BMD, Fracture Risk, Calciotropic Hormones and Bone Turnover Markers
Findings from Animal Experimental Studies
Effects on bone density and structure: Studies reporting BMD changes following ACEi treatment in spontaneously hypertensive [66–68] ovariectomized rats have showed either a beneficial effect [67, 68] or no effect of treatment [66], whereas a study in ovariectomized normotensive rats reported a harmful effect of ACEi [69].
Another study investigating fracture healing in non-osteoporotic mice treated with ACEi reported increased BMD loss in the non-fractured femora measured by micro-CT compared to the fractured non-treated control group [55]. Further two studies investigated the effect of 4 weeks treatment with an ACEi in male [70] and female [71] normotensive, non-osteoporotic rats, showing no effects on BMD compared to non-treated controls.
In transgenic mice (expressing the human renin and angiotensinogen genes), 4 weeks treatment with ACEi increased, while ARB decreased, BMD [58]. Except from the study in transgenic mice, all rodent studies investigating ARB treatment report a beneficial [56, 57, 69, 72–75] or at least a non-harmful [76] effect on BMD.
The rat model for postmenopausal osteoporosis (ovariectomy) is also tested in ARB-treated [56, 68, 69, 72, 73], hypertensive [68, 72, 73] animals. All of these studies showed beneficial effects on BMD changes compared to ovariectomized controls.
ARB selectively blocks the AT1R which seems to be of specific importance to trabecular bone. Accordingly, in a study by Kaneko et al. [57] heterozygous (±) and homozygous (−/−) male mice, lacking the gene encoding the AT1R had increased trabecular bone volume, increased trabecular number and connectivity compared to a control group (+/+).
Fracture: A study by Garcia et al. [55] in non-hypertensive, non-osteoporotic surgical fractured mice showed an improved fracture healing in response to treatment with ACEi. Moreover, a study by Rajkumar et al. [68] showed a slower thinning of bone trabecular which prevented microfractures and increased fracture healing in hypertensive mice treated with ACEi and ARB compared to ovariectomized untreated hypertensive rats. The osteoprotective effect was most pronounced for ARB-treated mice.
Calciotropic hormones and bone turnover markers: Only one study reports on PTH levels in mice treated with ACEi, showing no difference compared to non-treated controls [55]. Effects of treatment with ACEi and ARB on urinary and plasma calcium levels have not been reported in any of the animal experimental studies. In the AT1R knockout mice, plasma and urinary calcium as well as PTH levels did not differ among the genotypes [57].
Few studies report effect of treatment on bone turnover markers. All published results support a bone formative effect of RAASi. Increased ALP levels have been reported in response to treatment with an ACEi [67], and treatment with ARBs has been shown to increase OC [74, 76]. Moreover, decreased deoxypyridinoline levels have been reported in response to treatment with both ACEi and ARB [67, 74, 76].
In addition, bone turnover is increased compared to controls in AT1R knockout mice, as assessed by measurement of OC and CTx levels [57].
In summary, AT1R seems to regulate bone turnover and bone mass. Pharmacological inhibition of AT1R by treatment with ARB seems to improve bone density, trabecula connectivity as well as fracture healing in non-osteoporotic rodents. The effect seems to be most marked in ovariectomized hypertensive animals, whereas findings from studies in non-ovariectomized and non-hypertensive, as well as male rodent, are inconclusive. ARB treatment seems superior to ACEi treatment, and results on effects of ACEi treatment are ambiguous.
Human Observational Studies
Fracture risk: In a large, Danish nationwide cohort study from 2006, patients on treatment with ACEi (a small group of ARB users was included in the ACEi group) had a significantly reduced risk of any fracture (OR 0.93; 95 % CI 0.90–0.96) as well as a 14 % reduced risk of hip fractures (OR 0.86, 95 % CI 0.80–0.92) compared to population-based controls [77].
A similar, recently published nationwide Korean cohort study investigated effects of monotherapy with an antihypertensive drug during a mean follow-up of 1.9 years [78]. Per 10.000 person-years, risk of fracture did not differ between nonusers (152.2 95 % CI 148.7–155.7) and ARB users (152.7, 95 % CI 145.4–160.4), but ACEi users had a significantly increased risk of fractures (254.0 95 % CI 225.0–286.7).
Solomon et al. [79] studied 376,061 women on low income with untreated hypertension for at least 1 year in a “new user one medicament” design and assessed relative fracture risk in the different groups of antihypertensive drugs during a median follow-up time of 70 days. Compared with patients starting treatment with a calcium channel blocker, risk of fracture was significantly reduced among patients treated with ARB (hazard ratio [HR] 0.76; 95 % CI 0.68–0.86) and thiazide diuretics (HR 0.85; 95 % CI 0.76–0.97), whereas treatment with ACEi did not affect fracture risk (HR 1.03; 95 % CI (0.90–1.18). A difference in risk of fracture between patients treated with ACEis and ARBs is, however, not supported by the finding from another population-based, retrospective cohort study by Butt et al. [80]. Although unadjusted analyses showed a significantly reduced risk of a major osteoporotic fractures in patients treated with an ARB compared to patients treated with an ACEi (HR 0.81; CI 0.70–0.93), no difference in risk of fracture was found after adjusting for dose of studied drugs [80]. Nevertheless, the study did find a statistically significant decreased risk of osteoporosis-related fractures with increasing doses of either drugs studied [80]. In contrast, the study by Solomon et al. [79] showed no dose–effect relationships.
Bone mineral density: Discrepant results have been reported on effects of treatment with ACEi on BMD. In two cross-sectional studies, treatment with ACEi was associated with a significantly higher BMD in Chinese men and women [81] and in American men [82], whereas another cross-sectional study (also including Chinese patients) showed no association between ACEi treatment and BMD [83]. In two of the studies, follow-up was performed after at least 4 years of treatment, showing an increased bone loss in American men as well as in the Chinese women (but not in men) on treatment with ACEi compared with the controls [82, 83].
Only very few data are available on effects of treatment with ARB on BMD. In the study by Kwok et al. [82], BMD did not differ between American men on treatment or not on treatment with an ARB and changes in BMD during 4 years of follow-up did not differ between treated and untreated men.
Human Intervention Studies
Injection of a single dose of captopril has been shown to acutely decrease PTH as well as aldosterone levels in obese vitamin D-deficient subjects [1].
In a randomized open-label study in which 134 hypertensive patients with a non-osteoporotic BMD were treated for 1 year with either quinapril, 40 mg/d, quinapril, 40 mg/d plus hydrochlorothiazide 12.5 mg/d or enalapril 20 mg/d, changes in BMD did not differ between groups [84]. However, quinapril increased plasma calcium levels without affecting urinary calcium, levels of PTH or bone turnover markers. In the study, ACE insertion/deletion (I/D) polymorphisms were also analyzed, as polymorphism I is negatively associated with ACE activity. Compared to the DD genotype, II and ID genotypes had higher BMD at baseline and treatment caused a decrease in BMD in women but not in men [85, 86].
In summary, all human trials investigating bone outcome report a osteoprotective [79, 80] or at least a non-harmful [78, 82] effect of ARB. Treatment with ACEi, on the contrary, remains controversial. All longitudinal studies on ACEi therapy have shown an increased bone loss in treated patients [82, 83], whereas data on fracture risk are conflicting [77–79]. The picture is further complicated by the fact that two out of three cross-sectional analyses showed higher baseline BMD in ACEi-treated patients compared to controls. Further studies are needed to explain these contradictory findings.
ARB treatment seems superior to ACEi treatment in animal as well as in human studies comparing the two drug types [78, 79, 82]. It is likely that the AT1R is of importance to bone turnover and bone mass. Regarding the human studies, confounding by indication has, nevertheless, to be considered. For example, ARB treatment was previously reserved to patients developing side effects to ACEis because of a higher price and this might be a potential confounder including a so-called healthy user effect.
Effects of MRA’s on Bone and Calciotropic Hormones
Spironolactone and eplerenone are aldosterone antagonist, and they work by blocking the mineralocorticoid receptor (MR). Both drugs cause a secondary persistent increase in the secretion of renin and aldosterone. MR as well as glucocorticoid receptors are expressed by osteoblastic and osteoclastic cells [86, 87]. Eplerenone is a new drug, and it is considered more specific to the MR, as it has a lower affinity for the progesterone, androgen and glucocorticoid receptor [88]. Whether the two drugs act different on bone metabolism has not yet been investigated.
Findings from Animal Experimental Studies
A rat model on SA has been developed by continuous infusion of aldosterone (0.75 µg/h). This intervention has been shown to promote SHPT and bone loss [39, 40, 42], an effect which may be attenuated or prevented by spironolactone treatment [39, 40, 42]. However, in rats without SA, treatment with spironolactone (n = 5) was not found to affect alveolar bone loss in the setting of experimental periodontitis [89].
Beneficial effects of eplerenone on bone have been reported in a rat model on glucocorticoid induced osteopenia, in which treatment with eplerenone increased trabecular bone volume fraction as well as the connectivity, number and thickness of trabecular compared to untreated control animals [87].
Bone turnover markers have not been reported in any rodent studies.
Findings from Human Studies
Very few data are available from human studies on effects of MRA on bone. Only one case–control study has reported on risk of fracture. In this study, 167 male cases with chronic heart failure were matched by age and race to 668 controls without fractures [90]. After adjustment for potential confounders, the study showed a decreased risk of fracture in patients on treatment with spironolactone (OR 0.58; 95 % CI 0.35–0.96). The study did not allow for analyses of dose–response relationships as the majorities of patients were treated with less than 25 mg/d.
Recently, two cohort studies investigated bone outcome in PA patients following treatment [38, 43]. In both studies, medical (MRA) or surgical treatments are included, but none of the studies distinguished between the two types of treatment, as well as they did not report the proportion of patients suffering from either APA or BAH.
In the study by Salcuni et al. [38], BMD at the lumbar spine (n = 5) had increased significantly 1 year after the treatment. Similarly, Ceccoli et al. [43] found an improved BMD at the lumbar spine, femoral neck and total hip (n = 40) after a median of 2 years of follow-up. Compared with baseline values, Ceccoli et al. [43] also found an increase in serum calcium levels with a concomitant decrease in PTH levels following treatment, as well as CTX-I levels tended to be lower and bone ALP higher following treatment.
It has to be noticed that patients suffering from APA have more severe calcium and adrenal hormone abnormalities why the osteoprotective effect of treatment most likely is most pronounced in this group. Therefore, it may be that the reported improvements following treatment only apply to the adrenalectomized patients.
Due to the anti-androgen effect of spironolactone, it may also be used in the treatment of hirsutism. Three studies have reported effects on bone of treatment with spironolactone in hyperandrogenic women. In an open-label study, women with polycystic ovary syndrome (PCOS; n = 41) were randomized to 6 months of treatment with a gonadotropin-releasing hormone (GnRH) agonist alone or in combination with spironolactone 100 mg/d [91]. Compared with baseline values, women treated with a GnRH agonist alone experienced a decreased BMD which was prevented by co-treatment with spironolactone. However, in another study on women with PCOS or idiopathic hirsutism (n = 43), 1 year of treatment with an oral contraceptive in combination with spironolactone 50 mg/d did not affect BMD compared with treatment with only oral contraceptive. Finally, in a case series of 17 young women with androgen excess, 1 year of treatment with spironolactone 100 mg/d three out of 4 weeks in combination with progestogen (linestrenol 7 days per months) was associated with a significantly decreased BMD at the lumbar spine [92].
In two of the studies, plasma levels of calcium and bone turnover markers were measured and treatment did not affect measured levels [91, 93]. However, a reduced urinary calcium excretion was found in the study reporting a bone beneficial effect of co-treatment with spironolactone [91].
So far, only one randomized placebo-controlled trial has been reported the effects of 6 weeks of treatment with spironolactone 50 mg/d in a group of 32 obese normotensive subjects [1]. Compared with placebo, spironolactone increased serum calcium levels with a concomitant decrease in PTH levels. Furthermore, treatment caused a borderline significant decrease in CTX levels compared with placebo. Unfortunately, effects on urinary calcium and BMD were not measured in the study.
In summary, few human data are available, and data on BMD are only available in a highly selected group of patients suffering from either PA or hyperandrogenism.
BMD loss or osteoporosis might be a manifestation of primary aldosteronism, and MRA treatment seems to improve BMD. The renal calcium excretion is increased in patients with PA. This is followed by a decrease in plasma calcium and a compensatory increased PTH. Spironolactone reverses those changes, and this might be the mechanism for the BMD improvement following treatment.
Whether MRAs increase BMD in hyperandrogenic women is not clear. In women with hirsutism, only a minority suffers from adrenal disorders. Spironolactone binds to the androgen receptors, but in contrary to PA and SA, in which aldosterone is considered the main pathogenic mediator, spironolactone does not threat the underlying condition in most hyperandrogenic women. Accordingly, it may be that only patients with pathologically elevated concentrations of aldosterone benefit from antagonizing the effects of the hormone. Unfortunately, no comparisons of ACEi or ARB treated subjects exist, but it would be of great interest to investigate MRA treatment in other RAAS-related disorders to test this hypothesis.
Discussion
Overall, emerging evidences suggest a complex and clinical relevant interaction between the RAAS and calciotropic hormones. While a CVD protective and BP lowering effect of vitamin D has been investigated for decades, effects of RAASi on calcium homeostasis and bone metabolism represent a new scientific area.
In most studies, effects of ACEi have been investigated, as this drug is most commonly prescribed. Data on BMD as well as fracture risk in ACEi treated subjects are, however, conflicting. Contrary to previously published data, recent publications suggest a detrimental effect of ACEi treatment, and overall the findings are ambiguous. On the other hand, ARB treatment seems to be superior to ACEi treatment, as most studies report a reduced fracture risk and an increased BMD in response to treatment. To explain those findings, different mechanisms of action for ACEi and ARB must be considered.
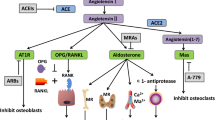
The AT1R is likely to play a central role as demonstrated in a number of experimental studies. ARBs solely block AT1R, whereas ACEis diminish the activity of AT1 as well as the less well characterized AT2R, see Fig. 1.

Mechanisms of ACEi, ARB and MRA on the RAAS
The aldosterone concentration might also be of importance. Although ACEs and ARBs reduce plasma aldosterone levels initially, an aldosterone (or Ang II) breakthrough may occur during long-term therapy in up to 53 % of treated subjects, probably due to non-ACE enzymes capable of cleaving Ang I to Ang II or increased renin concentration [94]. A lower prevalence of aldosterone (or Ang II) breakthrough in ARB treated subjects may provide an explanation for the apparent different effects of ACEi and ARB on bone [95] (Table 1).
Finally, an increased inflammatory effect of ACEi treatment must be considered, because ACE is a kinase, and inhibition leads to increased kinins, known as inflammatory mediators [96].
In MRA treated subjects, levels of renin, Ang II and aldosterone are increased, but the MR is antagonized. MRA seems to improve BMD in hyperaldosterone subjects, but whether this also applies to other patient categories is unclear and needs further investigation.
Whether a bone-protecting effect of RAASi, or at least MRA and ARB treatment, is due to a reduced renal calcium excretion because of a blood pressure lowering effect or a reduced local RAAS activity per se has not been fully clarified, but reviewing the literature suggests that both mechanisms may be of importance.
Further long-term studies are needed to clarify the effect of RAASi on bone health. This is of particular importance, as RAASi, are widely prescribed worldwide why beneficial as well as harmful effects may have large impact on bone health in the general population. Osteoporosis and osteoporotic fractures represent a major health problem, and the overlapping pathogenesis and commonly existing comorbidity make RAASi and bone outcome an interesting further research area.
In conclusion, considerable data support a physiologic clinical important association between PTH and the adrenal glands. Ang II, aldosterone and cortisol all negatively impact bone health. It is likely that the AT1R is of importance to bone turnover and bone mass. Available data suggest a reduced fracture rate and increased BMD in patients treated with ARB, whereas treatment with ACEi may cause opposite effects.
MRA treatment seems to be beneficial to bone in patients with adrenal abnormalities, but it is unknown whether this also applies to other individuals.
Abbreviations
- ACEi:
-
Angiotensin-converting enzyme inhibitor
- ACTH:
-
Adrenocorticotropic hormone
- ALP:
-
Alkaline phosphatase
- Ang II:
-
Angiotensin II
- APA:
-
Aldosterone-producing adenoma
- ARB:
-
Angiotensin II receptor blocker
- AT1R:
-
Angiotensin II type 1 receptor
- AT2R:
-
Angiotensin II type 2 receptor
- BAH:
-
Bilateral adrenal hyperplasia
- BMD:
-
Bone mineral density
- BP:
-
Blood pressure
- Ca:
-
Calcium
- CVD:
-
Cardiovascular disease
- CTx:
-
C-terminal telopeptide of type 1 collagen
- GnRH:
-
Gonadotropin-releasing hormone
- MR:
-
Mineralocorticoid receptor
- MRA:
-
Mineralocorticoid receptor antagonist
- OC:
-
Osteocalcin
- PA:
-
Primary aldosteronism
- PCOS:
-
Polycystic ovary syndrome
- PTH:
-
Parathyroid hormone
- PPHT:
-
Primary hyperparathyroidism
- PTX:
-
Parathyroidectomy
- SA:
-
Secondary aldosteronism
- SHPT:
-
Secondary hyperparathyroidism
- RAAS:
-
Renin–angiotensin–aldosterone system
- RAASi:
-
RAAS inhibitors
- SA:
-
Secondary aldosteronism
- RANKL:
-
Receptor activator of NF-κB ligand
References
Brown JM, Williams JS, Luther JM, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension. 2014;63:273–80. doi:10.1161/HYPERTENSIONAHA.113.01910.
Grant FD, Mandel SJ, Brown EM, et al. Interrelationships between the renin-angiotensin-aldosterone and calcium homeostatic systems. J Clin Endocrinol Metab. 1992;75:988–92.
Tomaschitz A, Pilz S. Interplay between sodium and calcium regulatory hormones: a clinically relevant research field. Hypertension. 2014;63:212–4. doi:10.1161/HYPERTENSIONAHA.113.02253.
Nilsson IL, Aberg J, Rastad J, Lind L. Left ventricular systolic and diastolic function and exercise testing in primary hyperparathyroidism-effects of parathyroidectomy. Surgery. 2000;128:895–902.
Piovesan A, Molineri N, Casasso F, et al. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin Endocrinol (Oxf). 1999;50:321–8.
Vestergaard P, Mollerup CL, Frøkjaer VG, et al. Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg. 2003;27:216–22.
Rosa J, Raska I, Wichterle D, et al. Pulse wave velocity in primary hyperparathyroidism and effect of surgical therapy. Hypertens Res. 2011;34:296–300. doi:10.1038/hr.2010.232.
Sprini D, Rini GB, Di Stefano L, et al. Correlation between osteoporosis and cardiovascular disease. Clin cases Miner bone Metab. 2014;1:117–9.
Bolland MJ, Grey A, Gamble GD, Reid IR. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014;2:307–20. doi:10.1016/S2213-8587(13)70212-2.
Brown J, de Boer IH, Robinson-Cohen C, et al. Aldosterone, parathyroid hormone, and the use of renin-angiotensin-aldosterone system inhibitors: the multi-ethnic study of atherosclerosis. J Clin Endocrinol Metab. 2014;. doi:10.1210/jc.2014-3949.
Pilz S, Tomaschitz A, Drechsler C, et al. Parathyroid hormone level is associated with mortality and cardiovascular events in patients undergoing coronary angiography. Eur Heart J. 2010;31:1591–8. doi:10.1093/eurheartj/ehq109.
Lasco A, Catalano A, Morabito N, et al. Adrenal effects of teriparatide in the treatment of severe postmenopausal osteoporosis. Osteoporos Int. 2011;22:299–303. doi:10.1007/s00198-010-1222-5.
Maniero C, Fassina A, Guzzardo V, et al. Primary hyperparathyroidism with concurrent primary aldosteronism. Hypertension. 2011;58:341–6. doi:10.1161/HYPERTENSIONAHA.111.173948.
Mazzocchi G, Aragona F, Malendowicz LK, Nussdorfer GG. PTH and PTH-related peptide enhance steroid secretion from human adrenocortical cells. Am J Physiol Endocrinol Metab. 2001;280:E209–13.
Hulter H, Melby JC, Peterson JC, Cooke CR. Chronic continuous PTH infusion results in hypertension in normal subjects. J Clin Hypertens. 1986;2:360–70.
Isales CM, Barrett PQ, Brines M, et al. Parathyroid hormone modulates angiotensin II-induced aldosterone secretion from adrenal glomerulosa cell. Endocr Soc. 1991;129:489–95.
Nainby-Luxmoore JC, Langford HG, Nelson NC, et al. A case-comparison study of hypertension and hyperparathyroidism. J Clin Endocrinol Metab. 1982;55:303–6.
Letizia C, Ferrari P, Cotesta D, et al. Ambulatory monitoring of blood pressure (AMBP) in patients with primary hyperparathyroidism. J Hum Hypertens. 2005;19:901–6.
Nilsson I-L, Yin L, Lundgren E, et al. Clinical presentation of primary hyperparathyroidism in Europe–nationwide cohort analysis on mortality from nonmalignant causes. J Bone Miner Res. 2002;17(Suppl 2):N68–74.
Yu N, Donnan PT, Leese GP. A record linkage study of outcomes in patients with mild primary hyperparathyroidism: the Parathyroid Epidemiology and Audit Research Study (PEARS). Clin Endocrinol (Oxf). 2011;75:169–76. doi:10.1111/j.1365-2265.2010.03958.x.
Hedbäck G, Odén A. Increased risk of death from primary hyperparathyroidism–an update. Eur J Clin Invest. 1998;28:271–6.
Silverberg SJ, Clarke BL, Peacock M, et al. Current issues in the presentation of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. 2015;99:3580–3594. doi:10.1210/jc.2014-1415.
Brunaud L, Germain A, Zarnegar R, et al. Serum aldosterone is correlated positively to parathyroid hormone (PTH) levels in patients with primary hyperparathyroidism. Surgery. 2009;146:1035–41. doi:10.1016/j.surg.2009.09.041.
Gianotti L, Tassone F, Pia A, et al. May an altered hypothalamo-pituitary-adrenal axis contribute to cortical bone damage in primary hyperparathyroidism? Calcif Tissue Int. 2009;84:425–9. doi:10.1007/s00223-009-9245-7.
Jespersen B, Pedersen EB, Charles P, et al. Elevated angiotensin II and vasopressin in primary hyperparathyroidism. Angiotensin II infusion studies before and after removal of the parathyroid adenoma. Acta Endocrinol (Copenh). 1989;120:362–8.
Gavras I, Hatinoglou S, Benetos A, Gavras H. Calcium stimulates vasopressin release. J Hypertens. 1986;4:451–4.
Vimaleswaran KS, Cavadino A, Berry DJ, et al. Association of vitamin D status with arterial blood pressure and hypertension risk: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2014;2:719–29. doi:10.1016/S2213-8587(14)70113-5.
Krause R, Bühring M, Hopfenmüller W, et al. Ultraviolet B and blood pressure. Lancet. 1998;352:709–10.
Pfeifer M, Begerow B, Minne HW, et al. Effects of a short-term vitamin D(3) and calcium supplementation on blood pressure and parathyroid hormone levels in elderly women. J Clin Endocrinol Metab. 2001;86:1633–7.
Sugden JA, Davies JI, Witham MD, et al. Vitamin D improves endothelial function in patients with Type 2 diabetes mellitus and low vitamin D levels. Diabet Med. 2008;25:320–5. doi:10.1111/j.1464-5491.2007.02360.x.
Pittas AG, Chung M, Trikalinos T, et al. Systematic review: vitamin D and cardiometabolic outcomes. Ann Intern Med. 2010;152:307–14. doi:10.7326/0003-4819-152-5-201003020-00009.
Li YC, Qiao G, Uskokovic M, et al. Vitamin D: a negative endocrine regulator of the renin-angiotensin system and blood pressure. J Steroid Biochem Mol Biol. 2004;89–90:387–92.
Tomaschitz A, Pilz S, Ritz E, et al. Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system. The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin Chim Acta. 2010;411:1354–60. doi:10.1016/j.cca.2010.05.037.
Forman JP, Williams JS, Fisher NDL. Plasma 25-hydroxyvitamin D and regulation of the renin-angiotensin system in humans. Hypertension. 2010;55:1283–8. doi:10.1161/HYPERTENSIONAHA.109.148619.
Vaidya A, Sun B, Larson C, et al. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin ii akin to the action of a converting enzyme inhibitor in obese hypertensives: an interventional study. J Clin Endocrinol Metab. 2012;97:2456–65. doi:10.1210/jc.2012-1156.
Funder JW, Carey RM, Fardella C, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:3266–81. doi:10.1210/jc.2008-0104.
Milliez P, Girerd X, Plouin P-F, et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45:1243–8.
Salcuni AS, Palmieri S, Carnevale V, et al. Bone involvement in aldosteronism. J Bone Miner Res. 2012;27:2217–22. doi:10.1002/jbmr.1660.
Chhokar VS, Sun Y, Bhattacharya SK, et al. Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation. 2005;111:871–8.
Law PH, Sun Y, Bhattacharya SK, et al. Diuretics and bone loss in rats with aldosteronism. J Am Coll Cardiol. 2005;46:142–6.
Chhokar VS, Sun Y, Bhattacharya SK, et al. Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol. 2004;287:H2023–6.
Runyan AL, Chhokar VS, Sun Y, et al. Bone loss in rats with aldosteronism. Am J Med Sci. 2005;330:1–7.
Ceccoli L, Ronconi V, Giovannini L, et al. Bone health and aldosterone excess. Osteoporos Int. 2013;24:2801–7.
Pilz S, Kienreich K, Drechsler C, et al. Hyperparathyroidism in patients with primary aldosteronism: cross-sectional and interventional data from the GECOH study. J Clin Endocrinol Metab. 2012;97:75–9. doi:10.1210/jc.2011-2183.
Rossi E, Sani C, Perazzoli F, et al. Alterations of the calcium metabolism and of parathyroid function in primary aldosteronism and their revertal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am J Hypertens. 1995;7061:884–93.
Maniero C, Fassina A, Seccia TM, et al. Mild hyperparathyroidism: a novel surgically correctable feature of primary aldosteronism. J Hypertens. 2012;30:390–5. doi:10.1097/HJH.0b013e32834f0451.
Resnick LM, Laragh JH. Calcium metabolism and parathyroid function in primary aldosteronism. Am J Med. 1985;78:385–90.
Rossi GP, Ragazzo F, Seccia TM, et al. Hyperparathyroidism can be useful in the identification of primary aldosteronism due to aldosterone-producing adenoma. Hypertension. 2012;60:431–6. doi:10.1161/HYPERTENSIONAHA.112.195891.
Giacchetti G, Ronconi V, Lucarelli G, et al. Analysis of screening and confirmatory tests in the diagnosis of primary aldosteronism: need for a standardized protocol. J Hypertens. 2006;24:737–45.
Blumenfeld JD, Sealey JE, Schlussel Y, et al. Diagnosis and treatment of primary hyperaldosteronism. Ann Intern Med. 1994;121:877–85.
Afghani A. Hypertension and bone loss. Nova Biomedica/Nova Science Publishers; 2011. http://site.ebrary.com.ez.statsbiblioteket.dk:2048/lib/stats/reader.action?docID=10671131.
Khouzam RN, Dishmon DA, Farah V, et al. Secondary hyperparathyroidism in patients with untreated and treated congestive heart failure. Am J Med Sci. 2006;331:30–4.
Mateus-Hamdan L, Beauchet O, Bouvard B, et al. High parathyroid hormone, but not low vitamin D concentrations, expose elderly inpatients to hypertension. Geriatr Gerontol Int. 2013;13:783–91. doi:10.1111/j.1447-0594.2012.00945.x.
Hatton R, Stimpel M, Chambers TJ. Angiotensin II is generated from angiotensin I by bone cells and stimulates osteoclastic bone resorption in vitro. J Endocrinol. 1997;152:5–10.
Garcia P, Schwenzer S, Slotta JE, et al. Inhibition of angiotensin-converting enzyme stimulates fracture healing and periosteal callus formation—role of a local renin-angiotensin system. Br J Pharmacol. 2010;159:1672–80. doi:10.1111/j.1476-5381.2010.00651.x.
Shimizu H, Nakagami H, Osako MK, et al. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008;22:2465–75. doi:10.1096/fj.07-098954.
Kaneko K, Ito M, Fumoto T, et al. Physiological function of the angiotensin AT1a receptor in bone remodeling. J Bone Miner Res. 2011;26:2959–66. doi:10.1002/jbmr.501.
Asaba Y, Ito M, Fumoto T, et al. Activation of renin-angiotensin system induces osteoporosis independently of hypertension. J Bone Miner Res. 2009;24:241–50. doi:10.1359/jbmr.081006.
Hagiwara H, Hiruma Y, Inoue A, et al. Deceleration by angiotensin II of the differentiation and bone formation of rat calvarial osteoblastic cells. J Endocrinol. 1998;156:543–50.
Hiruma Y, Inoue A, Hirose S, Hagiwara H. Angiotensin II stimulates the proliferation of osteoblast-rich populations of cells from rat calvariae. Biochem Biophys Res Commun. 1997;230:176–8.
Lamparter S, Kling L, Schrader M, et al. Effects of angiotensin II on bone cells in vitro. J Cell Physiol. 1998;175:89–98.
Vestergaard P, Rejnmark L, Mosekilde L. Hypertension is a risk factor for fractures. Calcif Tissue Int. 2009;84:103–11.
Cappuccio FP, Meilahn E, Zmuda JM, Cauley JA. High blood pressure and bone-mineral loss in elderly white women: a prospective study. Study of Osteoporotic Fractures Research Group. Lancet. 1999;354:971–5.
Tsuda K, Nishio I, Masuyama Y. Bone mineral density in women with essential hypertension. Am J Hypertens. 2001;14:704–7.
Koiwa F, Komukai D, Hirose M, et al. Influence of renin-angiotensin system on serum parathyroid hormone levels in uremic patients. Clin Exp Nephrol. 2012;16:130–5. doi:10.1007/s10157-011-0534-x.
Ma YF, Stimpel M, Liang H, et al. Impact of antihypertensive therapy on the skeleton: effects of moexipril and hydrochlorothiazide on osteopenia in spontaneously hypertensive ovariectomized rats. J Endocrinol. 1997;154:467–74.
Shimizu H, Nakagami H, Osako MK, et al. Prevention of osteoporosis by angiotensin-converting enzyme inhibitor in spontaneous hypertensive rats. Hypertens Res. 2009;32:786–90. doi:10.1038/hr.2009.99.
Rajkumar DSR, Faitelson AV, Gudyrev OS, et al. Comparative evaluation of enalapril and losartan in pharmacological correction of experimental osteoporosis and fractures of its background. J Osteoporos. 2013. doi:10.1155/2013/325693.
Kang KY, Kang Y, Kim M, et al. The effects of antihypertensive drugs on bone mineral density in ovariectomized mice. J Korean Med Sci. 2013;28:1139–44. doi:10.3346/jkms.2013.28.8.1139.
Mathai ML, Naik S, Sinclair AJ, et al. Selective reduction in body fat mass and plasma leptin induced by angiotensin-converting enzyme inhibition in rats. Int J Obes (Lond). 2008;32:1576–84. doi:10.1038/ijo.2008.126.
Broulík PD, Tesař V, Zima T, Jirsa M. Impact of antihypertensive therapy on the skeleton: effects of enalapril and AT1 receptor antagonist losartan in female rats. Physiol Res. 2001;50:353–8.
Ma L, Ji JL, Ji H, et al. Telmisartan alleviates rosiglitazone-induced bone loss in ovariectomized spontaneous hypertensive rats. Bone. 2010;47:5–11. doi:10.1016/j.bone.2010.03.016.
Donmez BO, Ozdemir S, Sarikanat M, et al. Effect of angiotensin II type 1 receptor blocker on osteoporotic rat femurs. Pharmacol Rep. 2012;64:878–88.
Araújo AA, Souza TO, Moura LM, et al. Effect of telmisartan on levels of IL-1, TNF-α, down-regulated COX-2, MMP-2, MMP-9 and RANKL/RANK in an experimental periodontitis model. J Clin Periodontol. 2013;40:1104–11. doi:10.1111/jcpe.12160.
Izu Y, Mizoguchi F, Kawamata A, et al. Angiotensin II type 2 receptor blockade increases bone mass. J Biol Chem. 2009;284:4857–64. doi:10.1074/jbc.M807610200.
Araújo AA, Lopes De Souza G, Souza TO, et al. Olmesartan decreases IL-1β and TNF-α levels; Downregulates MMP-2, MMP-9, COX-2, and RANKL; and upregulates OPG in experimental periodontitis. Naunyn Schmiedebergs Arch Pharmacol. 2013;386:875–84. doi:10.1007/s00210-013-0886-8.
Rejnmark L, Vestergaard P, Mosekilde L. Treatment with beta-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: a nationwide case-control study. J Hypertens. 2006;24:581–9.
Choi HJ, Park C, Lee Y-K, et al. Risk of fractures in subjects with antihypertensive medications: a nationwide claim study. Int J Cardiol. 2015;184:62–7. doi:10.1016/j.ijcard.2015.01.072.
Solomon DH, Mogun H, Garneau K, Fischer MA. Risk of fractures in older adults using antihypertensive medications. J Bone Miner Res. 2011;26:1561–7. doi:10.1002/jbmr.356.
Butt DA, Mamdani M, Gomes T, et al. Risk of osteoporotic fractures with angiotensin ii receptor blockers versus angiotensin-converting enzyme inhibitors in hypertensive community-dwelling elderly. J Bone Miner Res. 2014;29:2483–8. doi:10.1002/jbmr.2271.
Lynn H, Kwok T, Wong SYS, et al. Angiotensin converting enzyme inhibitor use is associated with higher bone mineral density in elderly Chinese. Bone. 2006;38:584–8.
Kwok T, Leung J, Zhang YF, et al. Does the use of ACE inhibitors or angiotensin receptor blockers affect bone loss in older men? Osteoporos Int. 2012;23:2159–67. doi:10.1007/s00198-011-1831-7.
Zhang Y-F, Qin L, Leung P-C, Kwok TCY. The effect of angiotensin-converting enzyme inhibitor use on bone loss in elderly Chinese. J Bone Miner Metab. 2012;30:666–73. doi:10.1007/s00774-012-0363-3.
Pérez-Castrillón JL, Silva JJ, Justo I, et al. Effect of quinapril, quinapril-hydrochlorothiazide, and enalapril on the bone mass of hypertensive subjects: relationship with angiotensin converting enzyme polymorphisms. Am J Hypertens. 2003;16:453–9.
Pérez-Castrillón JL, Justo I, Silva J, et al. Relationship between bone mineral density and angiotensin converting enzyme polymorphism in hypertensive postmenopausal women. Am J Hypertens. 2003;16:233–5.
Beavan S, Horner A, Bord S, et al. Colocalization of glucocorticoid and mineralocorticoid receptors in human bone. J Bone Miner Res. 2001;16:1496–504.
Fumoto T, Ishii K-A, Ito M, et al. Mineralocorticoid receptor function in bone metabolism and its role in glucocorticoid-induced osteopenia. Biochem Biophys Res Commun. 2014;447:407–12. doi:10.1016/j.bbrc.2014.03.149.
Struthers A, Krum H, Williams GH. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clin Cardiol. 2008;31:153–8. doi:10.1002/clc.20324.
Grauballe MCB, Bentzen BH, Björnsson M, et al. The effect of spironolactone on experimental periodontitis in rats. J Periodontal Res. 2005;40:212–7.
Carbone LD, Cross JD, Raza SH, et al. Fracture risk in men with congestive heart failure risk reduction with spironolactone. J Am Coll Cardiol. 2008;52:135–8. doi:10.1016/j.jacc.2008.03.039.
Moghetti P, Castello R, Zamberlan N, et al. Spironolactone, but not flutamide, administration prevents bone loss in hyperandrogenic women treated with gonadotropin-releasing hormone agonist. J Clin Endocrinol Metab. 1998;84:1250–4.
Prezelj J, Kocijancic A. Antiandrogen treatment with spironolactone and linestrenol decreases bone mineral density in eumenorrhoeic women with androgen excess. Horm Metab Res. 1994;26:46–8.
Gregoriou O, Bakas P, Konidaris S, et al. The effect of combined oral contraception with or without spironolactone on bone mineral density of hyperandrogenic women. Gynecol Endocrinol. 2000;14:369–73.
Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3:486–92.
Cohn JN, Anand IS, Latini R, et al. Sustained reduction of aldosterone in response to the angiotensin receptor blocker valsartan in patients with chronic heart failure: results from the Valsartan Heart Failure Trial. Circulation. 2003;108:1306–9.
Gonçalves-Zillo TO, Pugliese LS, Sales VMT, et al. Increased bone loss and amount of osteoclasts in kinin B1 receptor knockout mice. J Clin Periodontol. 2013;40:653–60. doi:10.1111/jcpe.12097.
Disclosures
Conflict of interest
Lise Sofie Bislev, Tanja Sikjaer, Lars Rolighed and Lars Rejnmark declare that they have no conflict of interest.
Animal/Human Studies
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Because this article does not contain any studies with human participants or animals performed by any of the authors, informed consent was not obtained.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bislev, L.S., Sikjær, T., Rolighed, L. et al. Relationship Between Aldosterone and Parathyroid Hormone, and the Effect of Angiotensin and Aldosterone Inhibition on Bone Health. Clinic Rev Bone Miner Metab 13, 194–205 (2015). https://doi.org/10.1007/s12018-015-9182-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-015-9182-0