
Abstract
Myocardial infarction (MI) is the most common cause of cardiac injury in the Western world. Cardiac injury activates innate immune mechanisms initiating an inflammatory reaction. Inflammatory cytokines and vascular cell adhesion molecules (VCAM) promote adhesive interactions between leukocytes and endothelial cells, resulting in the transmigration of inflammatory cells into the site of injury. Low vitamin D levels are associated with higher prevalence of cardiovascular risk factors and a higher risk of MI. In this paper, we examine the effects of short-term vitamin D supplementation on inflammatory cytokine levels after an acute coronary syndrome. We recruited patients arriving to the hospital with an acute MI. All patients received optimal medical therapy and underwent a coronary catheterization. Half of the patients were randomly selected and treated with a daily supplement of vitamin D (4,000 IU) for 5 days. A short course of treatment with vitamin D effectively attenuated the increase in circulating levels of inflammatory cytokines after an acute coronary event. Control group patients had increased cytokine and cellular adhesion molecules serum concentrations after 5 days, while the vitamin D-treated group had an attenuated elevation or a reduction of these parameters. There were significant differences in VCAM-1 levels, C-reactive protein, and interleukin-6. There were trends toward significance in interleukin-8 levels. There were no significant differences in circulating levels of intercellular adhesion molecule 1, E-selectin, vascular endothelial growth factor, and tumor necrosis factor-α. These findings provide information on the anti-inflammatory effects of vitamin D on the vascular system and suggest mechanisms that mediate some of its cardioprotective properties. There is place for further studies involving prolonged vitamin D treatment in patients suffering from ischemic heart disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myocardial infarction (MI) is the most common form of acute cardiac injury and is a leading cause of morbidity and mortality in the Western world. Advances in therapy have led to a decline in mortality during the acute phase, but this decrease in mortality is paralleled by an increase in the incidence of chronic heart failure (HF) in patients surviving with significant residual myocardial damage [1, 2]. The human heart has negligible regenerative capacity and cardiac injury poses a great challenge for the reparative mechanisms of the body, ultimately resulting in formation of a scar. The development of HF after MI is determined by the size of the necrotic area, the wound healing response that occurs in the days and weeks after the event, and the chronic remodeling of the infarct scar and the remote, non-infarcted left ventricular (LV) myocardium.
The post-MI healing process is mediated by inflammatory mechanisms. Ischemia triggers a series of events that culminates in the death of the ischemic cardiomyocytes [3]. The acute ischemic episode leads to both systemic humoral and local cellular inflammatory responses that promote the myocardial healing process. Both systemic and local responses trigger a complex myocardial inflammatory reaction. Inflammation participates in physiological myocardial scar formation. However, in the case of an exuberant inflammatory reaction, the extent of the damage to myocardial tissue may paradoxically increase [4, 5].
Vitamin D is known to have several immunomodulatory functions, including suppression of proinflammatory cytokine expression and regulation of immune cell activity [6–15]. Low levels of 25-hydroxyvitamin D are linked to the presence of cardiovascular, autoimmune, endocrine, neurological, and malignant disorders [6–8, 10, 11, 16–30]. Vitamin D and its analogs have been noted to have positive effects on fibrinolysis, blood lipids, thrombogenicity, endothelial regeneration, and smooth muscle cell growth [31]. These findings are supported by a meta-analysis showing that oral vitamin D3 treatment is associated with reduced all-cause mortality, partially by decreasing cardiovascular deaths [32]. Vitamin D deficiency has been related to higher rates of MI [31], and vitamin D supplementation has been related to reduced proinflammatory cytokines in patients with HF [27, 33].
Most of the data now known about the effects of vitamin D are drawn from observational studies and experimental models. Few prospective papers exist that examine the effects of vitamin D treatment on the acute inflammatory response associated with acute coronary syndrome (ACS). In this study, we tested the effect of treating patients with vitamin D after an acute coronary event on blood profile of inflammatory cytokines and adhesion molecules.
Patients and Methods
Study Design
The study was designed as a prospective, randomized, open-label, single-center trial. It was conducted in accordance with the ethical principles of the Declaration of Helsinki, the Good Clinical Practice Guidelines of the International Conference of Harmonization, and local regulatory requirements; it was approved by the Ethics Committee of Meir Medical Center.
Study Population
Study patients were adult ACS patients hospitalized at the Intensive Cardiac Care Unit of Meir Medical Center in Israel between July and December 2010. Criteria used to diagnose acute myocardial infarction (AMI) were characteristic elevation and gradual decline of biochemical markers of myocardial necrosis with at least one of the following symptoms: ischemic symptoms, pathologic Q wave in the electrocardiogram (ECG), or changes in ECG waves indicating ischemia (elevation or depression of ST segment) [34].
The major exclusion criteria were renal failure (defined as creatinine level of 1.5 mg/dl or higher), known calcium homeostasis or parathyroid hormone abnormalities, febrile disorders, acute or chronic inflammatory disease upon presentation, steroid or immunosuppressive therapy, or active vitamin D supplementation within the month prior to admission. Informed consent was obtained from the patient after explanation from the researcher (Y.A). Eligible patients were randomly assigned in a 1:1 ratio to receive either daily treatment with orally administered vitamin D (4,000 IU 25(OH)-vitamin D/day) or no treatment. The study was designed to end after 5 days of supplementation.
Biochemical Analysis
For routine blood count and blood chemistry analysis, peripheral venous blood samples were collected using standard methods and sent to the institution’s clinical laboratory as part of the patient’s standard treatment. Blood tests for adhesion molecules and cytokines were drawn from all participants twice. The first test was drawn within 12 h of presentation to the hospital with an ACS. The second test was drawn after 5 days of hospitalization. All samples were centrifuged at 3,000 rpm for 10 min. The serum was stored at −40 °C for subsequent analysis. All samples were processed at the same time by technicians blinded to the origin of samples.
We used LIAISON® 25(OH)-vitamin D (DiaSorin) immunoassay to measure serum concentrations of 25(OH)-vitamin D. Interleukin (IL) levels were assayed using a high-sensitivity enzyme-linked immunosorbent assay (ELISA). Serum levels of adhesion molecules (vascular cell adhesion molecule 1 [VCAM-1], intercellular adhesion molecule 1 [ICAM-1], E-selectin, and vascular endothelial growth factor [VEGF]) were measured by commercially available ELISA kits according to the manufacturer’s instructions (R&D Systems, Inc., Minneapolis, MN, USA).
Statistical Analysis
Quantitative parameters were presented as the mean value and standard deviation. Qualitative parameters were presented as the number and percentage. Continuous variables were compared using Student’s t test. Nominal variables such as gender and other comorbidities were analyzed using Fisher’s exact test. Probability values of p < 0.05 were considered statistically significant.
Results
Patients
During the study period, 50 patients were recruited. The patients were randomized into 2 groups, with 25 participants each. The two groups were well-matched with respect to baseline characteristics (age, sex, and major cardiovascular risk factors). A total of nine patients did not complete the 5-day trial (six patients from the treatment group and three patients from the control group). Three patients progressed to cardiac bypass surgery after coronary angiography, four patients were discharged from the medical facility after 3 to 4 days of hospitalization, and 2 patients refused the second blood test. Table 1 presents the clinical characteristics and the biochemical data for the study groups.
Blood Test Analysis
The levels of cytokines and cellular adhesion molecules (CAM) closely after an acute coronary event are listed in Table 1. Vitamin D levels at arrival averaged 18.5 ± 7.1 ng/ml. After 5 days, the average vitamin D level for the control group was 22.86 ± 4.6 ng/ml and for the treated patients was 24.9 ± 7.3 ng/ml. The differences between the two groups were not significant.
The inflammatory cytokines and molecules tested were IL-6, IL-8, C-reactive protein (CRP), and tumor necrosis factor-α (TNF-α). IL-6 levels decreased by 31 % in the treated patients, compared to a 48.7 % increase in the untreated group. The difference was statistically significant, p = 0.05. IL-8 levels decreased by 24.5 % in the treated patients serum, compared to a 75.7 % increase in the control group, p = 0.1. TNF-α levels were mildly affected, showing a mild decrease in both groups after 5 days. Average CRP levels were elevated for all patients at presentation, with an average level of 1.28 ± 1.77 mg/dl. CRP concentrations continued to rise after the coronary event. The levels increased by 108 % in the treated patients, compared to an average 361 % increase for the patients without vitamin D supplementation (p = 0.03) (Table 2). The anti-inflammatory cytokine IL-10 was also tested and the levels were detectable only in 14 of 50 patients in the first blood sample and in 8 patients after 5 days (data not presented).
The vascular parameters tested were the adhesion molecules VCAM-1, ICAM-1, and E-selectin in addition to VEGF. In the untreated patients, VCAM-1, ICAM-1, and VEGF levels were increased after 5 days, representing a continuous inflammatory process. In the treated group, circulating VCAM-1 levels decreased 3.3 % (from 471.4 to 456.1 ng/ml in the study group), compared to a 23 % increase in the control group, p = 0.03. E-selectin levels decreased by 8 % among the treated patients, compared to a 0.4 % decrease in the control group, p = 0.15. ICAM-1 and VEGF levels continued to rise in both patient groups.
Discussion
Inflammation is a cornerstone of the post-MI healing process. Sudden induction of ischemia by coronary artery occlusion triggers a series of events that culminates in the death of ischemic cardiomyocytes throughout the anatomic region supplied by this artery [3]. Sudden myocardial ischemia due to coronary lumen narrowing or abrupt closure leads to both systemic humoral and local cellular inflammatory responses that promote the local myocardial healing process.
Rapid reperfusion of the infarct-related coronary artery is the most effective method of limiting tissue necrosis and improving outcome in AMI. The process of restoring blood flow to ischemic myocardium can, however, induce reperfusion injury. Reperfusion injury is partially caused by the activation of endothelial cells and recruitment of inflammatory cells to the infarct area, causing endothelial dysfunction and vascular plugging which lead to the release of degradative enzymes and reactive oxygen species [35]. The extravasation response is mediated by interactions between CAM and their cognate ligands, expressed on leukocytes, platelets, and endothelial cells. Both systemic and local responses trigger a complex myocardial inflammatory reaction. Inflammation participates in physiological myocardial scar formation. However, in the case of an exuberant inflammatory reaction, the extent of the damage to myocardial tissue may paradoxically increase [4, 36].
The Inflammatory Response and Cardiac Repair After Myocardial Infarction
Since the mammalian heart cannot produce enough energy under anaerobic conditions to maintain essential cellular processes, a constant supply of oxygen is indispensable to sustain cardiac function and viability. Ischemic myocardial injury results in decreased oxygen tension within the cell, subsequent loss of oxidative phosphorylation, and decreased generation of high-energy phosphates. Adenosine triphosphate (ATP) depletion leads to failure of the sodium pump, loss of potassium, influx of sodium and water, and cell swelling. Cessation of aerobic metabolism, ATP depletion, and accumulation of products of anoxic metabolism (such as lactic acid) occur within 10 s of occlusion. Striking loss of contractility occurs almost simultaneously and is evident within 60 s. Minutes after the onset of ischemia, reversible ultrastructural cardiomyocyte changes appear, including cellular and mitochondrial swelling and glycogen depletion. Irreversible cardiomyocyte injury, evidenced by sarcolemmal disruption and the presence of small amorphous densities in the mitochondria, develops after 20–40 min of sustained severe ischemia.
The human heart has low regenerative ability. Dying cells trigger an inflammatory reaction, activating reparative pathways that ultimately result in the formation of a scar. The inflammatory response and cytokine release from the myocardium are essential components of the host response to AMI and play a crucial role in cardiac repair. The healing process of the heart after an infraction can be divided into three phases, partially overlapping: the inflammatory phase, the proliferative phase, and wound scarring and maturation [37].
The acute repair process is mediated by cytokines and inflammatory cells in the infarcted myocardium. The induction of proinflammatory mediators and leukocyte infiltration play a crucial role in phagocytosis and removal of necrotic cells and matrix debris from infarcted myocardium. Moreover, they promote tissue repair and scar formation [38]. During the inflammatory phase, activation of chemokine and cytokine cascades results in the recruitment of leukocytes into the infarcted area. Chemokine upregulation is a noted feature of the postinfarction inflammatory response (Table 3). Neutrophils and macrophages clear the wound from dead cells and matrix debris. Activated macrophages release cytokines and growth factors, leading to the formation of highly vascularized granulation tissue. At this stage, the expression of proinflammatory mediators is suppressed, while fibroblasts and endothelial cells proliferate. During the proliferative phase of healing, activated myofibroblasts produce extracellular matrix proteins and an extensive microvascular network is formed. Maturation of the scar follows: fibroblasts and vascular cells undergo apoptosis and a collagen-based scar is formed.
Reperfusion of the coronary vessel accentuates the inflammatory reaction and greatly accelerates the healing response. The acute localized inflammatory response is transient and is followed by resolution of the inflammatory infiltrate and fibrous tissue deposition. A crucial commitment is made during the late stages of the inflammatory phase to convert the response from phagocytosis and clearance of dead cells and debris to a mode that promotes tissue repair and scar formation [38]. The inhibition of chemokine and cytokine synthesis after a dramatic early peak is crucial for the repair process, preventing prolonged expression of inflammatory mediators in the healing infarct and suppressing continuous leukocyte recruitment and injury. Thus, optimal healing requires mechanisms inhibiting chemokine and cytokine synthesis, resulting in the resolution of the inflammatory infiltrate and transition to fibrous tissue deposition. These mechanisms involve (a) clearance of the neutrophilic infiltrate and removal of matrix debris, (b) inhibition of cytokine and chemokine synthesis, (c) removal of the fibrin-based provisional matrix, and (d) activation of fibroblasts and collagen deposition. Although very few studies have dealt with the process of resolution of inflammation in the healing infarct, understanding these concepts is crucial for planning strategies targeting the reparative response [39–41].
Vitamin D as an Effector of Systemic Inflammatory Reaction
1,25-Dihydroxyvitamin D3 (1,25(OH)2D3), originally described as an essential hormone for bone and mineral homeostasis, is the biologically active form of the secosteroid vitamin D3. Most of the known biological effects of 1,25(OH)2D3 are mediated through the vitamin D3 receptor, a member of the superfamily of nuclear hormone receptors [42].
The awareness of the role of vitamin D in the regulation of immune responses was triggered by the discovery of vitamin D receptors in almost all immune cells [7, 14]. These include activated CD4+ and CD8+ T cells, B cells, neutrophils, and antigen-presenting cells (APCs), such as macrophages and dendritic cells (DCs) [43]. Moreover, the enzyme responsible for the final and rate-limiting hydroxylation step in the synthesis of 1,25(OH)2D3, the 25(OH)D3-1α-hydroxylase enzyme, is expressed by activated macrophages, making them able to synthesize and secrete 1,25(OH)2D3 in a regulated fashion. Although the enzyme found in macrophages is identical to the known renal form, its expression is regulated differently. While renal 1α-hydroxylase is mainly regulated by mediators of calcium and bone homeostasis (e.g., parathyroid hormone and 1,25(OH)2D3 itself), its macrophage version is predominantly under the control of immune signals, such as interferon-γ (IFN-γ) [44].
1,25(OH)2D3 has several immunomodulatory functions and pleiotropic activities in the immune system, including suppression of proinflammatory cytokine expression and regulation of immune cell activity. 1,25(OH)2D3 can affect the differentiation and function of cells in the immune system. In experimental models, the addition of 1,25(OH)2D3 results in the inhibition of T lymphocyte proliferation, cytokine secretion, and cell cycle progression from G1a to G1b. By inhibiting IFN-γ transcription, the major positive feedback signal for APCs, 1,25(OH)2D3 prevents further antigen presentation and recruitment of T lymphocytes [45]. Strikingly, 1,25(OH)2D3 uses several different molecular mechanisms to regulate cytokine expression, either directly targeting transcription initiation and regulation or indirectly interfering with other intracellular signaling pathways. Moreover, APCs as well as T lymphocytes can be direct targets of the immunomodulatory effects of 1,25(OH)2D3, leading to the inhibition of pathogenic effector T lymphocytes and the enhanced frequency of regulatory T lymphocytes, largely via the induction of DCs with tolerogenic properties.
The fact that 1,25(OH)2D3 influences the immune system, not merely by suppression, but by immune modulation through induction of immune shifts and regulator cells, makes this compound very appealing for clinical use, especially in the treatment and prevention of autoimmune diseases and regulation of inflammatory reactions. Vitamin D supplementation poses a minor limitation that we should be aware of, namely, its rare dose-dependent side effects, including hypercalcemia, hypercalciuria, renal calcification, and increased bone resorption. Indeed, the in vitro observed immunomodulatory effects of 1,25(OH)2D3 only occur at concentrations of 10−10 M and higher.
Vitamin D and the Cardiovascular System
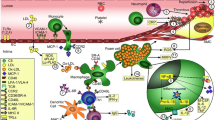
Low vitamin D levels have been linked to diseases not traditionally associated with vitamin D and calcium homeostasis, including cardiovascular diseases (CVDs) [12]. There are several mechanisms by which vitamin D may be associated with atherosclerosis and CVD events (Fig. 1). The role of vitamin D in CVDs has gained increasing interest in recent years. The mechanisms mediating a potential association with CVDs are not fully understood. Because many cell types involved in cardiovascular function like cardiomyocytes, endothelial cells, or vascular smooth muscle cells express vitamin D receptors, a direct influence of vitamin D on the cardiovascular system can be assumed. On one hand, vitamin D has been suggested to stimulate differentiation of vascular smooth muscle cells in the vessel wall to a calcifying osteoblast-like phenotype (vascular calcification) [46]. This would actually suggest a positive association of vitamin D levels with CVD risk. On the other hand, vitamin D deficiency might exert an unfavorable influence through its association with important CVD risk factors, such as diabetes [47] and hypertension [48], as well as with inflammation and cell proliferation.

Potential mechanisms for cardiovascular effects of vitamin D deficiency
Several observational studies have shown that vitamin D deficiency is a common finding in patients with CVD [49]. In a retrospective cohort study recently published [31], men with circulating 25(OH)D levels of at least 30 ng/ml had approximately half the risk of MI, independent of other CVD factors. The association was suggestively stronger for fatal coronary heart disease, but the number of cases was too small to make definitive conclusions. Two case–control studies [22, 28] and a small prospective study [30] found that individuals with low 25(OH)D levels were at higher risk for ischemic heart disease.
In a UK study of 2,686 men and women aged 65 years or more, the participants were randomized to receive 830 IU of vitamin D daily (administered once every 4 months) or placebo for 5 years. While the in-study 25(OH)D levels were 29.7 ng/ml in the vitamin D group and 21.4 ng/ml in the placebo group, only a nonsignificant decrease in CVD incidence (RR, 0.90; 95 % CI, 0.77–1.06) and CVD mortality (RR, 0.84; 95 % CI, 0.65–1.10) was observed in the intervention group [50]. A recent meta-analysis [32] of total mortality as a secondary end point of randomized controlled trials with varying levels of vitamin D vs. placebo controls found a statistically significant 8 % reduction in risk of total mortality in individuals who had received vitamin D. Although the authors could not evaluate cause-specific mortality, the relatively immediate effect of a large enough magnitude to affect total mortality would suggest a benefit on CVD risk. In an animal study conducted with mice [51], oral administration of the active form of vitamin D3 inhibited atherosclerosis development by inducing tolerogenic DCs and T regulatory cells. These cells also functioned as antiatherogenic agents and played a role in the beneficial effects of calcitriol on atherogenesis.
There is growing evidence to suggest that vitamin D status is associated with the development and progression of HF [52]. A recently published study demonstrated that low 25(OH)D levels are common in HF and are independently related to an increased risk for all-cause mortality and HF rehospitalization [53]. Further, a low 25(OH)D concentration was associated with higher plasma rennin activity and CRP levels, which suggests that activated rennin–angiotensin and inflammation play a role in the association between 25(OH)D levels and HF prognosis. Inflammation plays an important role in the pathogenesis of HF. Abnormal inflammatory response may result in increased levels of inflammation, tissue destruction, cardiovascular remodeling, vascular calcification, and loss of function, which may contribute to the development and progression of HF.
The Role of Cytokines in Ischemia-Induced Inflammation
The cytokines engaged in the ischemic myocardium are produced by myocardiocytes, liver cells, activated macrophages, and lymphocytes, as well as adipose tissue. Inflammatory cytokines, such as TNF-α, IL-6, and IL-8, are among the promoters of the humoral post-MI healing process [54–58]. Upregulation and production of these cytokines represent an intrinsic or an innate stress response against myocardial injury.
Under normal conditions, the human heart does not express cytokines; however, during an ischemic event, they may be up to 50 times in the culprit ischemic region and up to 15 times in the adjacent “nonischemic” zones [59]. In the early post-MI phase, a certain degree of cytokine production is physiological because, in this phase, cytokines play an important cytoprotective role by reducing cell apoptosis [60]. The robust upregulation may return to baseline levels if the infarction is small. However, if the infarction is large or if host inflammatory response is exuberant, there can be either sustained cytokine upregulation or a second wave of cytokine upregulation, corresponding to the chronic remodeling phase. The second wave can also extend to involve the non-infarct remote zone, mediating the important remodeling process in the entire myocardium. Cytokines can self-amplify through a positive feedback loop targeting the nuclear factor-κB. In extensive MIs, cytokines may persist at very high levels and remain detectable also in the normal adjacent myocardium. This phenomenon produces an unfavorable myocardial remodeling, eventually worsening clinical outcomes [61].
Cytokine accumulation directly interferes with the myocardial contractility, vascular endothelial (VE) function, and recruitment of other inflammatory cells. In the setting of injury, the reduction in contractility mediated by TNF and IL-6 may be an adaptive response to decrease myocardial energy demand. TNF-α and IL-6 can attenuate myocyte contractility directly through the immediate reduction of systolic cytosolic [Ca2+] via alterations in sarcoplasmic reticulum function, reversible by the removal of cytokine exposure. Experimental studies indicate that inflammatory cytokines (TNF-α and IL-6) are associated with HF, including progressive LV dysfunction, pulmonary edema, LV remodeling, fetal gene expression, myocyte hypertrophy, and myocyte apoptosis [62]. Alteration of endothelial permeability is an important feature during inflammatory conditions and is associated with leukocyte transendothelial migration and accumulation within the tissues. A number of proinflammatory cytokines, such as TNF-α and IFN-γ, have been shown to alter the distribution of adhesion receptors involved in cell–cell adhesion, namely, VE–cadherin–catenin complexes, and prevent the formation of F-actin stress fibers. This results in restructuring of the intercellular junction, leading to loss of endothelial permeability and favoring leukocyte transmigration [63].
TNF-α production in the acute post-MI phase is triggered mainly by ischemia, as well as by other factors, such as mechanical stress deformation of damaged myocytes, reactive oxygen species, and autoregulating self-amplification [64, 65]. Kapadia et al. have shown that direct hemodynamic stretch can trigger myocardial production of TNF-α de novo within 30 min [66]. Upregulation of TNF-α in the infarct myocardium can upregulate the levels of TNF-α in the neighboring normal myocardium, leading to amplified cytokine effects. TNF-α stimulates the expression of proinflammatory cytokines, chemokines, and adhesion molecules by leukocytes and endothelial cells and regulates extracellular matrix metabolism by reducing collagen synthesis and by enhancing matrix metalloproteinase (MMP) activity in cardiac fibroblasts [67]. Schleithoff et al. [27] demonstrated that daily vitamin D supplementation led to lower TNF-α and higher IL-10 levels in HF patients. Recent clinical and experimental studies have suggested that, among inflammatory cytokines, increased release of TNF-α can contribute to the progression of LV remodeling and dysfunction. In adult rats surgically implanted with an osmotic minipump infusing TNF-α for 5 days in order to reach plasma levels comparable to those reported in clinical HF, a decrease in LV fractional shortening occurred and LV end diastolic diameter increased by over 25 % when compared with time-matched controls [68]. In a transgenic mouse model, myocardial TNF-α overexpression caused increased systolic and diastolic LV volumes and a reduced collagen cross-linking in the myocardium [69]. Since the activation of TNF-α receptors increases the production of MMPs, one mechanism by which TNF-α can influence LV remodeling appears to be the induction of myocardial MMPs and subsequent degradation of extracellular matrix components. In patients undergoing postischemic LV remodeling in vitro, TNF-α production immediately after the ischemic event was statistically increased in comparison with patients who did not develop LV remodeling. Furthermore, 6 months after MI, in the same group of patients, TNF-α production was persistently higher than that in the patients not undergoing LV remodeling [70].
IL-6 is a proinflammatory cytokine that affects many immune system cells, including B cells, T cells, vascular smooth muscle cells, and endothelial cells. This IL has been shown to enhance fatty lesion development in mice. IL-6 levels appear to be predictive of future coronary artery disease (CAD) and are elevated in patients with unstable angina (UA) compared with those with stable angina [71]. Patients with persistently elevated IL-6 levels demonstrate a worse in-hospital outcome following admission with UA. Raised levels of IL-6 is often correlated to CRP levels, consistent with IL-6 being the main stimulant for the hepatic production of CRP.
IL-6 synthesis is rapidly induced in mononuclear cells and cardiomyocytes of the ischemic myocardium [72]. IL-6 may mediate a biological interplay between various immune responses and exert autocrine, paracrine, and endocrine responses to link clinical risk [73]. IL-6 serves as an endogenous pyrogen, an upregulator of inflammatory reaction and a stimulator of acute-phase reactants. IL-6 is capable of modulating the phenotypic characteristics and gene expression of many cell types involved in infarct healing. IL-6 null mice demonstrated significantly delayed cutaneous wound healing, suggesting a significant role for IL-6 in tissue repair [74].
IL-8 is also an inflammatory cytokine secreted from activated macrophages and by endothelial cells in response to inflammatory stimuli. The prospective EPIC-Norfolk population study provided evidence that elevated plasma levels of IL-8 were associated with an increased risk of CAD in apparently healthy individuals [75]. IL-8 production is activated by the complement cascade, which, together with platelet activated factor produced by the endothelial cells, stimulates endovascular adhesion of neutrophils, thus increasing vascular and tissue inflammation. A significant amount of myocardial injury induced by coronary artery occlusion followed by reperfusion is considered to be neutrophil-dependent [76]. CRP is known to enhance IL-8 production at 8 to 24 h of incubation. It is also conceivable that IL-8 may promote neutrophil–endothelial cell adhesion by increasing the expression of endothelial CAM [77]. The induction of monocyte procoagulant activity with either IL-6 or IL-8 has been proposed as a possible link between the inflammation and thrombosis in patients with CAD.
Our study has shown that, 5 days following an AMI, vitamin D-treated patients had lower serum IL-6 and IL-8, whereas in the control group, there was an increase in circulating levels of these cytokines. These differences were statistically significant for IL-6, but not for IL-8. TNF-α levels decreased slightly after 5 days in both groups with no significant difference.
Systemically circulating cytokines, such as TNF-α and IL-6, stimulate the liver to produce inflammatory markers, such as CRP. These proteins are “acute-phase” reactants, meaning that they change in characteristic ways during the early phase of injury and infarction. High levels of CRP have been associated with a higher risk of cardiovascular events in older patients without CVD, in particular HF events. CRP binds to the damaged myocardial cells, stimulates the complement cascade, and may finally increase the infarcted zone size, worsening the overall post-MI outcomes [78]. CRP is considered not only as an inflammatory marker, but also as a direct inflammatory promoter with proatherogenic and prothrombotic properties [79, 80]. CRP has also been reported to increase IL-8 levels [81, 82]. It has also been recently demonstrated that CRP causes the expression of the adhesion molecules ICAM-1 and VCAM-1 by endothelial cells [83]. CRP levels after an AMI can predict LV remodeling and HF development [84]. In our study, we demonstrated elevated CRP levels following an acute coronary event. The molecule levels were higher 5 days after the event and elevation was significantly attenuated under vitamin D treatment.
Vitamin D as an Effector of Circulating Adhesion Molecule Levels
CAM are surface proteins involved in modulating intercellular communication among a wide variety of different cell types. Several major families of adhesion molecule receptors have been identified and characterized; these include the integrins, cadherins, selectins, membrane-associated proteoglycans, and immunoglobulin superfamily members. CAM mediate the margination, adhesion, and transendothelial migration of circulating mononuclear cells from the bloodstream to the extravascular compartment [85]. Soluble circulating forms of adhesion molecules can be measured from peripheral blood samples, and their levels reflect the amount of membrane-bound adhesion molecules and the degree of local endothelial activation [86].
The first contact between leukocytes and the vessel wall is established by members of the selectin family of adhesion molecules, among them E-selectin which is expressed exclusively by endothelial cells. E-selectin is found on endothelial cells stimulated by inflammatory cytokines, such as TNF-α, IL-1, or platelet factor 4. Plasma E-selectin levels are elevated in patients with ACS [87] and in patients after undergoing coronary angioplasty [88], and E-selectin is also detected on human atherosclerosis-prone endothelial cells and on the surface of fibrous and lipid-containing human plaques [89]. After selectin-mediated slowing down of the leukocytes, their adhesion to the vascular wall involves interaction between the leukocyte integrins and members of the immunoglobulin superfamily, ICAM-1 and VCAM-1 [90–92].
VCAM-1 (or CD106) and ICAM-1 (or CD54) are two members of the immunoglobulin gene superfamily that are critical in the recruitment and infiltration of inflammatory cells to sites of injury. VCAM-1 binds circulating monocytes and lymphocytes expressing the integrins α4β1 and α4β7, whereas ICAM-1 is the counterreceptor for several leukocyte β2 integrins (e.g., lymphocyte function-associated antigen [CD11a/CD18] and Mac-1 [CD11b/CD18]). The interaction of ICAM-1 with leukocyte integrins also plays an important role in leukocyte trafficking and the initiation of antigen-specific immune responses. ICAM-1 can be expressed by a variety of cells, mostly endothelial cells and leukocytes, whereas VCAM-1 expression is restricted only to endothelial cells. VCAM-1 is expressed on endothelial cells in response to inflammatory cytokines, and like ICAM-1, it interacts with integrins on the surface of leukocytes. Both ICAM-1 and VCAM-1 promote firm adhesion and subsequent arrest of leukocytes on the surface of endothelial cells [92].
The importance of adhesion molecules in the inflammatory process has been confirmed by several in vitro and in vivo studies. Specifically, monoclonal antibodies and antisense oligonucleotides to adhesion molecules and their ligands, as well as the absence of CAM expression in knockout mice, prevent tissue injury associated with both acute and chronic inflammation [93]. Plasma concentrations of CAM may be higher in patients with atherosclerosis, and the predisposition of the old vessels to develop atherosclerotic lesions may be related to an age-dependent increase of the expression of CAM on the surface of VE cells [94].
Levels of soluble adhesion molecules have been shown to correlate to various cardiovascular risk factors, such as smoking, hypertension, and dyslipidemia [95, 96]. The expression of VCAM-1 is induced by arterial endothelial cells in response to the accumulation of cholesterol within the intima of aortas [97]. VCAM-1 is also rapidly induced in proatherosclerotic conditions in animal models and humans [96]. ICAM-1 is expressed in ischemic myocardial segments as early as 1 h after reperfusion, with marked elevations after longer time intervals.
Since the initial discovery that cytokines induce VCAM-1 expression on endothelial cells, many cytokines, including IL-1 and TNF-α, have been implicated in the induction of an array of adhesion molecules and chemokines in the vascular wall. Patients with ACS show significantly higher levels of both VCAM-1 and ICAM-1, compared with those with stable angina pectoris (AP) and healthy controls. Circulating VCAM-1 molecules have been found to be good predictors of future cardiovascular events in patient with ACS, regardless of troponin levels [98]. ICAM-1 may facilitate both emigration of neutrophils in reperfused myocardium and their adherence-dependent cytotoxic behavior. In contrast to VCAM-1, circulating ICAM-1 levels are not considered to relate to future cardiovascular risk in AMI patients. The ARMYDA-CAM study, examining CAM levels in stable AP patients undergoing elective PCI after 1 week of statin treatment (atorvastatin, 40 mg/day vs. placebo) [99], showed an attenuation of postprocedural increase of ICAM-1 and E-selectin levels. The authors concluded that reduction of the endothelial inflammatory response might explain some of the statin protective effects. In a canine model, selectin blockade as an adjunct to thrombolysis significantly reduced infarct size and myocardial neutrophil infiltration well beyond the thrombolytic effect alone [100].
In our study, VCAM-1 levels were highly detectable close to the cardiac event. The levels continued to increase in untreated patients, compared to the patients who received vitamin D. ICAM-1 levels followed a similar pattern, although the differences did not reach statistical significance. The high circulating CAM levels represented a continuous inflammatory process after the coronary event, and vitamin D supplementation effectively reduced the inflammatory burden. E-selectin levels were high shortly after an acute coronary event. After 5 days, E-selectin levels declined in both patient groups. Vitamin D treatment was associated with a higher rate of decline, although the changes were not statistically significant.
VEGF is a vascular permeability factor which is typically considered to be an endothelial-specific growth factor with both angiogenic and antiapoptotic activities [101]. VEGF levels are elevated during a thrombotic event, such as an ST elevation MI. In vitro, vitamin D has been shown to induce VEGF expression and release in vascular smooth muscle cells. VEGF can be induced by IL-6 and TNF-α, which were inhibited by vitamin D treatment. In this study, VEGF levels were elevated following an ACS and continued rising for at least 5 days. There were no significant differences between the groups regarding VEGF levels.
This investigation had several limitations. Plasma levels of cytokines and adhesion molecules showed high variability, indicating substantial interindividual variation. The study was limited by the small number of participants, short duration of treatment, and short follow-up time. We do not know if the trends we found in our study conferred any clinical significance on the patients’ outcome, and finally, this study also did not assess the possibility of a dose effect for vitamin D treatment.
Conclusion
Myocardial ischemia and especially MI lead to a systemic inflammatory response, as well as to a local–regional inflammatory response. The purpose of this inflammation is to promote a complex physiological myocardial healing process. The acute inflammation itself may paradoxically have deleterious effects on myocardial cells, especially in the case of an inflammatory response. For many years, inflammation has been a pharmacological therapeutic target; however, treatment with specific anti-inflammatory agents and cytokine inhibitors has been unsuccessful in clinical practice [102]. Even if interventions targeting the inflammatory response do not reduce cardiomyocyte death, modulation of the reparative process in order to optimize the mechanical and functional properties of the infarcted heart remains an interesting direction. Experimental evidence suggests that inhibition of chemokine signaling, modulation of extracellular matrix remodeling, and enhancement of the endogenous protective mechanisms that contribute to the resolution and containment of the inflammatory response may be promising therapeutic strategies.
Understanding the inflammatory response to ischemic myocardial injury and the role of cytokines after MI may allow us to promote improved healing and cardiac remodeling after MI. Low levels of 25(OH)D are known to be associated with increased atherosclerosis and higher risk of MI [31]. The results of this study demonstrated that vitamin D can attenuate the inflammatory process following AMI.
As vitamin D deficiency is widespread in many populations and is in theory amenable to potential interventions, clarification of its role in myocardial ischemia and infarction may be of great public health relevance. The possible clinical benefits of vitamin D for patients after an ischemic event warrant future studies.
References
Behar S, Battler A, Porath A, Leor J, Grossman E, Hasin Y, Mittelman M, Feigenberg Z, Rahima-Maoz C, Green M, Caspi A, Rabinowitz B, Garty M (2003) A prospective national survey of management and clinical outcome of acute myocardial infarction in Israel, 2000. Isr Med Assoc J 5(4):249–254
Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D'Agostino RB, Levy D, Kannel WB, Vasan RS (2008) Long-term trends in the incidence of heart failure after myocardial infarction. Circulation 118(20):2057–2062
Jennings RB, Murry CE, Steenbergen C Jr, Reimer KA (1990) Development of cell injury in sustained acute ischemia. Circulation 82(3 Suppl):II2–II12
Frangogiannis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, Michael LH, Ballantyne CM, Smith CW, Entman ML (1998) Cytokines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol 30(12):2567–2576
Sitia S, Atzeni F, Sarzi-Puttini P, Di Bello V, Tomasoni L, Delfino L, Antonini-Canterin F, Di Salvo G, De Gennaro CV, La Carrubba S, Carerj S, Turiel M (2009) Cardiovascular involvement in systemic autoimmune diseases. Autoimmun Rev 8(4):281–286
Amital H, Szekanecz Z, Szucs G, Danko K, Nagy E, Csepany T, Kiss E, Rovensky J, Tuchynova A, Kozakova D, Doria A, Corocher N, Agmon-Levin N, Barak V, Orbach H, Zandman-Goddard G, Shoenfeld Y (2010) Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis 69(6):1155–1157
Arnson Y, Amital H, Shoenfeld Y (2007) Vitamin D and autoimmunity: new aetiological and therapeutic considerations. Ann Rheum Dis 66(9):1137–1142
Arnson Y, Amital H, Agmon-Levin N, Alon D, Sanchez-Castanon M, Lopez-Hoyos M, Matucci-Cerinic M, Szucs G, Shapira Y, Szekanecz Z, Shoenfeld Y (2011) Serum 25-OH vitamin D concentrations are linked with various clinical aspects in patients with systemic sclerosis: a retrospective cohort study and review of the literature. Autoimmun Rev 10(8):490–494
Arnson Y, Amital H (2011) Is vitamin D a new therapeutic agent in autoinflammatory and pain syndromes? Isr Med Assoc J 13(4):234–235
Agmon-Levin N, Blank M, Zandman-Goddard G, Orbach H, Meroni PL, Tincani A, Doria A, Cervera R, Miesbach W, Stojanovich L, Barak V, Porat-Katz BS, Amital H, Shoenfeld Y (2011) Vitamin D: an instrumental factor in the anti-phospholipid syndrome by inhibition of tissue factor expression. Ann Rheum Dis 70(1):145–150
Agmon-Levin N, Kivity S, Tzioufas AG, Lopez-Hoyos M, Rozman B, Efes I, Shapira Y, Shamis A, Amital H, Youinou P, Shoenfeld Y (2012) Low levels of vitamin-D are associated with neuropathy and lymphoma among patients with Sjogren's syndrome. J Autoimmun 39:234–239
Holick MF (2007) Vitamin D, deficiency. N Engl J Med 357(3):266–281
Orbach H, Zandman-Goddard G, Amital H, Barak V, Szekanecz Z, Szucs G, Danko K, Nagy E, Csepany T, Carvalho JF, Doria A, Shoenfeld Y (2007) Novel biomarkers in autoimmune diseases: prolactin, ferritin, vitamin D, and TPA levels in autoimmune diseases. Ann N Y Acad Sci 1109:385–400
Shoenfeld N, Amital H, Shoenfeld Y (2009) The effect of melanism and vitamin D synthesis on the incidence of autoimmune disease. Nat Clin Pract Rheumatol 5(2):99–105
Twig G, Shina A, Amital H, Shoenfeld Y (2012) Pathogenesis of infertility and recurrent pregnancy loss in thyroid autoimmunity. J Autoimmun 38(2–3):J275–J281
Antico A, Tozzoli R, Giavarina D, Tonutti E, Bizzaro N (2012) Hypovitaminosis d as predisposing factor for atrophic type a gastritis: a case–control study and review of the literature on the interaction of Vitamin D with the immune system. Clin Rev Allergy Immunol 42(3):355–364
Cutolo M, Pizzorni C, Sulli A (2011) Vitamin D endocrine system involvement in autoimmune rheumatic diseases. Autoimmun Rev 11(2):84–87
Dobnig H, Pilz S, Scharnagl H, Renner W, Seelhorst U, Wellnitz B, Kinkeldei J, Boehm BO, Weihrauch G, Maerz W (2008) Independent association of low serum 25-hydroxyvitamin d and 1,25-dihydroxyvitamin d levels with all-cause and cardiovascular mortality. Arch Intern Med 168(12):1340–1349
Gonzalez-Molero I, Rojo-Martinez G, Morcillo S, Gutierrez-Repiso C, Rubio-Martin E, Almaraz MC, Olveira G, Soriguer F (2012) Vitamin D and incidence of diabetes: a prospective cohort study. Clin Nutr 31(4):571–573
Holick MF (2012) The D-lightful vitamin D for child health. JPEN J Parenter Enter Nutr 36(1 Suppl):9S–19S
Lerner A, Shapira Y, Agmon-Levin N, Pacht A, Ben-Ami SD, Lopez-Hoyos M, Sanchez-Castanon M, Shoenfeld Y (2012) The clinical significance of 25OH-vitamin D status in celiac disease. Clin Rev Allergy Immunol 42(3):322–330
Lund B, Badskjaer J, Lund B, Soerensen OH (1978) Vitamin D and ischaemic heart disease. Horm Metab Res 10(6):553–556
Mutlu A, Mutlu GY, Ozsu E, Cizmecioglu FM, Hatun S (2011) Vitamin D deficiency in children and adolescents with type 1 diabetes. J Clin Res Pediatr Endocrinol 3(4):179–183
Peelen E, Knippenberg S, Muris AH, Thewissen M, Smolders J, Tervaert JW, Hupperts R, Damoiseaux J (2011) Effects of vitamin D on the peripheral adaptive immune system: a review. Autoimmun Rev 10(12):733–743
Pelajo CF, Lopez-Benitez JM, Miller LC (2010) Vitamin D and autoimmune rheumatologic disorders. Autoimmun Rev 9(7):507–510
Sadovnick AD (2012) Genetic background of multiple sclerosis. Autoimmun Rev 11(3):163–166
Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R (2006) Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 83(4):754–759
Scragg R, Jackson R, Holdaway IM, Lim T, Beaglehole R (1990) Myocardial infarction is inversely associated with plasma 25-hydroxyvitamin D3 levels: a community-based study. Int J Epidemiol 19(3):559–563
Sellner J, Kraus J, Awad A, Milo R, Hemmer B, Stuve O (2011) The increasing incidence and prevalence of female multiple sclerosis—a critical analysis of potential environmental factors. Autoimmun Rev 10(8):495–502
Vik B, Try K, Thelle DS, Forde OH (1979) Tromso Heart Study: vitamin D metabolism and myocardial infarction. Br Med J 2(6183):176
Giovannucci E, Liu Y, Hollis BW, Rimm EB (2008) 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 168(11):1174–1180
Autier P, Gandini S (2007) Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med 167(16):1730–1737
Gannage-Yared MH, Azoury M, Mansour I, Baddoura R, Halaby G, Naaman R (2003) Effects of a short-term calcium and vitamin D treatment on serum cytokines, bone markers, insulin and lipid concentrations in healthy post-menopausal women. J Endocrinol Investig 26(8):748–753
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD (2012) Third universal definition of myocardial infarction. Circulation 126(16):2020–2035
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion—from mechanism to translation. Nat Med 17(11):1391–1401
Sitia S, Atzeni F, Sarzi-Puttini P, Di Bello V, Tomasoni L, Delfino L, Antonini-Canterin F, Di Salvo G, De Gennaro Colonna V, La Carrubba S, Carerj S, Turiel M (2009) Cardiovascular involvement in systemic autoimmune diseases. Autoimmun Rev 8(4):281–286
Frangogiannis NG (2006) The mechanistic basis of infarct healing. Antioxid Redox Signal 8(11–12):1907–1939
Nathan C (2002) Points of control in inflammation. Nature 420(6917):846–852
Atzeni F, Turiel M, Caporali R, Cavagna L, Tomasoni L, Sitia S, Sarzi-Puttini P (2010) The effect of pharmacological therapy on the cardiovascular system of patients with systemic rheumatic diseases. Autoimmun Rev 9(12):835–839
de Carvalho JF, Viana VS, Neto EF, Santos RD, Bonfa E (2011) Anti-lipoprotein lipase antibodies in patients with hypertriglyceridemia without associated autoimmune disease. Isr Med Assoc J 13(6):350–353
Liao YH, Fu M (2001) Autoimmunity in the pathogenesis of cardiomyopathy. J Autoimmun 16(1):1–2
Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW (1998) The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res 13(3):325–349
Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC (1983) 1,25-Dihydroxyvitamin D3 receptors in human leukocytes. Science 221(4616):1181–1183
Overbergh L, Decallonne B, Valckx D, Verstuyf A, Depovere J, Laureys J, Rutgeerts O, Saint-Arnaud R, Bouillon R, Mathieu C (2000) Identification and immune regulation of 25-hydroxyvitamin D-1-alpha-hydroxylase in murine macrophages. Clin Exp Immunol 120(1):139–146
Takeuchi A, Reddy GS, Kobayashi T, Okano T, Park J, Sharma S (1998) Nuclear factor of activated T cells (NFAT) as a molecular target for 1alpha,25-dihydroxyvitamin D3-mediated effects. J Immunol 160(1):209–218
Gouni-Berthold I, Krone W, Berthold HK (2009) Vitamin D and cardiovascular disease. Curr Vasc Pharmacol 7(3):414–422
Mattila C, Knekt P, Mannisto S, Rissanen H, Laaksonen MA, Montonen J, Reunanen A (2007) Serum 25-hydroxyvitamin D concentration and subsequent risk of type 2 diabetes. Diabetes Care 30(10):2569–2570
Forman JP, Giovannucci E, Holmes MD, Bischoff-Ferrari HA, Tworoger SS, Willett WC, Curhan GC (2007) Plasma 25-hydroxyvitamin D levels and risk of incident hypertension. Hypertension 49(5):1063–1069
Lee JH, O'Keefe JH, Bell D, Hensrud DD, Holick MF (2008) Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor? J Am Coll Cardiol 52(24):1949–1956
Trivedi DP, Doll R, Khaw KT (2003) Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ 326(7387):469
Takeda M, Yamashita T, Sasaki N, Nakajima K, Kita T, Shinohara M, Ishida T, Hirata K (2010) Oral administration of an active form of vitamin D3 (calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler Thromb Vasc Biol 30(12):2495–2503
Meems LM, van der Harst P, van Gilst WH, de Boer RA (2011) Vitamin D biology in heart failure: molecular mechanisms and systematic review. Curr Drug Targets 12(1):29–41
Liu LC, Voors AA, van Veldhuisen DJ, van der Veer E, Belonje AM, Szymanski MK, Sillje HH, van Gilst WH, Jaarsma T, de Boer RA (2011) Vitamin D status and outcomes in heart failure patients. Eur J Heart Fail 13(6):619–625
Daskalopoulos EP, Janssen BJ, Blankesteijn WM (2012) Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal 18(1):35–49
Liehn EA, Postea O, Curaj A, Marx N (2011) Repair after myocardial infarction, between fantasy and reality: the role of chemokines. J Am Coll Cardiol 58(23):2357–2362
Liu SQ, Tefft BJ, Zhang D, Roberts D, Schuster DJ, Wu A (2011) Cardioprotective mechanisms activated in response to myocardial ischemia. Mol Cell Biomech 8(4):319–338
Suffee N, Richard B, Hlawaty H, Oudar O, Charnaux N, Sutton A (2011) Angiogenic properties of the chemokine RANTES/CCL5. Biochem Soc Trans 39(6):1649–1653
van Kimmenade RR, Januzzi JL Jr (2012) Emerging biomarkers in heart failure. Clin Chem 58(1):127–138
Alwi I, Santoso T, Suyono S, Sutrisna B, Kresno SB (2007) The cut-off point of interleukin-6 level in acute coronary syndrome. Acta Med Indones 39(4):174–178
Maggio M, Guralnik JM, Longo DL, Ferrucci L (2006) Interleukin-6 in aging and chronic disease: a magnificent pathway. J Gerontol A Biol Sci Med Sci 61(6):575–584
Puhakka M, Magga J, Hietakorpi S, Penttila I, Uusimaa P, Risteli J, Peuhkurinen K (2003) Interleukin-6 and tumor necrosis factor alpha in relation to myocardial infarct size and collagen formation. J Card Fail 9(4):325–332
Baumgarten G, Knuefermann P, Mann DL (2000) Cytokines as emerging targets in the treatment of heart failure. Trends Cardiovasc Med 10(5):216–223
Edens HA, Parkos CA (2000) Modulation of epithelial and endothelial paracellular permeability by leukocytes. Adv Drug Deliv Rev 41(3):315–328
Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML (1999) Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 99(4):546–551
Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML (1998) Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 98(7):699–710
Kapadia SR, Oral H, Lee J, Nakano M, Taffet GE, Mann DL (1997) Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res 81(2):187–195
Siwik DA, Chang DL, Colucci WS (2000) Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res 86(12):1259–1265
Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL (1998) Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation 97(14):1382–1391
Li YY, Feng YQ, Kadokami T, McTiernan CF, Draviam R, Watkins SC, Feldman AM (2000) Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor alpha can be modulated by anti-tumor necrosis factor alpha therapy. Proc Natl Acad Sci U S A 97(23):12746–12751
Pasqui AL, Di Renzo M, Maffei S, Pastorelli M, Pompella G, Auteri A, Puccetti L (2010) Pro/anti-inflammatory cytokine imbalance in postischemic left ventricular remodeling. Mediat Inflamm 2010:974694
Biasucci LM, Vitelli A, Liuzzo G, Altamura S, Caligiuri G, Monaco C, Rebuzzi AG, Ciliberto G, Maseri A (1996) Elevated levels of interleukin-6 in unstable angina. Circulation 94(5):874–877
Kukielka GL, Smith CW, Manning AM, Youker KA, Michael LH, Entman ML (1995) Induction of interleukin-6 synthesis in the myocardium. Potential role in postreperfusion inflammatory injury. Circulation 92(7):1866–1875
Bermudez EA, Rifai N, Buring J, Manson JE, Ridker PM (2002) Interrelationships among circulating interleukin-6, C-reactive protein, and traditional cardiovascular risk factors in women. Arterioscler Thromb Vasc Biol 22(10):1668–1673
Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, Sugawara T, Luster MI (2000) Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J 14(15):2525–2531
Boekholdt SM, Peters RJ, Hack CE, Day NE, Luben R, Bingham SA, Wareham NJ, Reitsma PH, Khaw KT (2004) IL-8 plasma concentrations and the risk of future coronary artery disease in apparently healthy men and women: the EPIC-Norfolk prospective population study. Arterioscler Thromb Vasc Biol 24(8):1503–1508
Jordan JE, Zhao ZQ, Vinten-Johansen J (1999) The role of neutrophils in myocardial ischemia–reperfusion injury. Cardiovasc Res 43(4):860–878
Kokura S, Wolf RE, Yoshikawa T, Granger DN, Aw TY (2000) Postanoxic T lymphocyte–endothelial cell interactions induce tumor necrosis factor-alpha production and neutrophil adhesion: role of very late antigen-4/vascular cell adhesion molecule-1. Circ Res 86(12):1237–1244
Barrett TD, Hennan JK, Marks RM, Lucchesi BR (2002) C-reactive-protein-associated increase in myocardial infarct size after ischemia/reperfusion. J Pharmacol Exp Ther 303(3):1007–1013
Pasceri V, Willerson JT, Yeh ET (2000) Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation 102(18):2165–2168
Verma S, Li SH, Badiwala MV, Weisel RD, Fedak PW, Li RK, Dhillon B, Mickle DA (2002) Endothelin antagonism and interleukin-6 inhibition attenuate the proatherogenic effects of C-reactive protein. Circulation 105(16):1890–1896
Srinivasan S, Yeh M, Danziger EC, Hatley ME, Riggan AE, Leitinger N, Berliner JA, Hedrick CC (2003) Glucose regulates monocyte adhesion through endothelial production of interleukin-8. Circ Res 92(4):371–377
Pradhan AD, Cook NR, Buring JE, Manson JE, Ridker PM (2003) C-reactive protein is independently associated with fasting insulin in nondiabetic women. Arterioscler Thromb Vasc Biol 23(4):650–655
Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Saito T, Manabe I, Imai Y, Nagai R (2006) C-reactive protein induces VCAM-1 gene expression through NF-kappaB activation in vascular endothelial cells. Atherosclerosis 185(1):39–46
Uehara K, Nomura M, Ozaki Y, Fujinaga H, Ikefuji H, Kimura M, Chikamori K, Nakaya Y, Ito S (2003) High-sensitivity C-reactive protein and left ventricular remodeling in patients with acute myocardial infarction. Hear Vessel 18(2):67–74
Hillis GS, Flapan AD (1998) Cell adhesion molecules in cardiovascular disease: a clinical perspective. Heart 79(5):429–431
Gearing AJ, Hemingway I, Pigott R, Hughes J, Rees AJ, Cashman SJ (1992) Soluble forms of vascular adhesion molecules, E-selectin, ICAM-1, and VCAM-1: pathological significance. Ann N Y Acad Sci 667:324–331
Lu HH, Sheng ZQ, Wang Y, Zhang L (2010) Levels of soluble adhesion molecules in patients with various clinical presentations of coronary atherosclerosis. Chin Med J (Engl) 123(21):3123–3126
Siminiak T, Dye JF, Egdell RM, More R, Wysocki H, Sheridan DJ (1997) The release of soluble adhesion molecules ICAM-1 and E-selectin after acute myocardial infarction and following coronary angioplasty. Int J Cardiol 61(2):113–118
Galkina E, Ley K (2007) Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol 27(11):2292–2301
Entman ML, Smith CW (1994) Postreperfusion inflammation: a model for reaction to injury in cardiovascular disease. Cardiovasc Res 28(9):1301–1311
Luscinskas FW, Gimbrone MA Jr (1996) Endothelial-dependent mechanisms in chronic inflammatory leukocyte recruitment. Annu Rev Med 47:413–421
Muller WA (2002) Leukocyte–endothelial cell interactions in the inflammatory response. Lab Investig 82(5):521–533
Kubes P, Jutila M, Payne D (1995) Therapeutic potential of inhibiting leukocyte rolling in ischemia/reperfusion. J Clin Invest 95(6):2510–2519
Richter V, Rassoul F, Purschwitz K, Hentschel B, Reuter W, Kuntze T (2003) Circulating vascular cell adhesion molecules VCAM-1, ICAM-1, and E-selectin in dependence on aging. Gerontology 49(5):293–300
Blankenberg S, Barbaux S, Tiret L (2003) Adhesion molecules and atherosclerosis. Atherosclerosis 170(2):191–203
O'Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, McCarty J, Ferguson M, Hudkins K, Benjamin CD (1993) Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest 92(2):945–951
Couffinhal T, Duplaa C, Moreau C, Lamaziere JM, Bonnet J (1994) Regulation of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in human vascular smooth muscle cells. Circ Res 74(2):225–234
Postadzhiyan AS, Tzontcheva AV, Kehayov I, Finkov B (2008) Circulating soluble adhesion molecules ICAM-1 and VCAM-1 and their association with clinical outcome, troponin T and C-reactive protein in patients with acute coronary syndromes. Clin Biochem 41(3):126–133
Patti G, Chello M, Pasceri V, Colonna D, Nusca A, Miglionico M, D'Ambrosio A, Covino E, Di Sciascio G (2006) Protection from procedural myocardial injury by atorvastatin is associated with lower levels of adhesion molecules after percutaneous coronary intervention: results from the ARMYDA-CAMs (Atorvastatin for Reduction of MYocardial Damage during Angioplasty-Cell Adhesion Molecules) substudy. J Am Coll Cardiol 48(8):1560–1566
Silver MJ, Sutton JM, Hook S, Lee P, Malycky JL, Phillips ML, Ellis SG, Topol EJ, Nicolini FA (1995) Adjunctive selectin blockade successfully reduces infarct size beyond thrombolysis in the electrolytic canine coronary artery model. Circulation 92(3):492–499
Ferrara N, Henzel WJ (1989) Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 161(2):851–858
Nah DY, Rhee MY (2009) The inflammatory response and cardiac repair after myocardial infarction. Korean Circ J 39(10):393–398
Author information
Authors and Affiliations
Corresponding author
Additional information
Yoav Arnson and Dganit Itzhaky contributed equally to this manuscript.
Rights and permissions
About this article
Cite this article
Arnson, Y., Itzhaky, D., Mosseri, M. et al. Vitamin D Inflammatory Cytokines and Coronary Events: A Comprehensive Review. Clinic Rev Allerg Immunol 45, 236–247 (2013). https://doi.org/10.1007/s12016-013-8356-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-013-8356-0