
Abstract
Epidemiological studies suggest that an increase of diesel exhaust particles (DEP) in ambient air corresponds to an increase in hospital-recorded myocardial infarctions within 48 h after exposure. Among the many theories to explain this data are endothelial dysfunction and translocation of DEP into vasculature. The mechanisms for such DEP-induced vascular permeability remain unknown. One of the major mechanisms underlying the effects of DEP is suggested to be oxidative stress. Experiments have shown that DEP induce the generation of reactive oxygen species (ROS), such as superoxide anion and H2O2 in the HUVEC tube cells. Transcription factor Nrf2 is translocated to the cell nucleus, where it activates transcription of the antioxidative enzyme HO-1 and sequentially induces the release of vascular permeability factor VEGF-A. Furthermore, a recent study shows that DEP-induced intracellular ROS may cause the release of pro-inflammatory TNF-α and IL-6, which may induce endothelial permeability as well by promoting VEGF-A secretion independently of HO-1 activation. These results demonstrated that the adherens junction molecule, VE-cadherin, becomes redistributed from the membrane at cell–cell borders to the cytoplasm in response to DEP, separating the plasma membranes of adjacent cells. DEP were occasionally found in endothelial cell cytoplasm and in tube lumen. In addition, the induced ROS is cytotoxic to the endothelial tube-like HUVEC. Acute DEP exposure stimulates ATP depletion, followed by depolarization of their actin cytoskeleton, which sequentially inhibits PI3K/Akt activity and induces endothelial apoptosis. Nevertheless, high-dose DEP augments tube cell apoptosis up to 70 % but disrupts the p53 negative regulator Mdm2. In summary, exposure to DEP affects parameters influencing vasculature permeability and viability, i.e., oxidative stress and its upregulated antioxidative and pro-inflammatory responses, which sequentially induce vascular permeability factor, VEGF-A release and disrupt cell–cell junction integrity. While exposure to a low dose of DEP actin triggers cytoskeleton depolarization, reduces PI3K/Akt activity, and induces a p53/Mdm2 feedback loop, a high dose causes apoptosis by depleting Mdm2. Addition of ROS scavenger N-acetyl cysteine suppresses DEP-induced oxidative stress efficiently and reduces subsequent damages by increasing endogenous glutathione.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A very high proportion of the particles in diesel exhaust is PM2.5 (particulate matter ≤2.5 μm), which have been reported as a carcinogen and associated with adverse disorders [1–4]. The cardiopulmonary system is affected most by many substances in diesel exhaust particles (DEP), including carbon black, hydrocarbons, aldehydes, quinones, benzo[a]pyrenes, polycyclic aromatic hydrocarbons (PAHs), and heavy metals, which levels parallel the incidence of allergies, asthma, rhinitis, cardiovascular disorders, as well as mutagenesis and carcinogenesis [5–11].
Vasculature Effects
Epidemiology indicates that long-term exposure to ambient PM2.5 adversely affects the health of those exposed [12]. PM2.5 from diesel vehicles is produced from several processes: (a) It is directly emitted from the tailpipes of on-road vehicles, (b) it is re-entrained from fugitive dust, and (c) it is reacted to form PM2.5 with precursor emissions chemicals such as sulfur dioxide, nitrogen oxides, volatile organic compounds, and ammonia [13]. Items a and b are typically known as primary emissions of PM2.5, for which exposure both indoors and outdoors has been shown to cause acute and chronic health effects [14]. Item c occurs due to chemical reaction in the atmosphere [13]. Some gas phase carbonyl compounds (aldehydes and ketones) are also known to have adverse effects on human health; for example, long-term exposure to high formaldehyde concentrations is known to increase the risk for asthma and cancer [15, 16]. Chronic bronchitis, chronic obstructive pulmonary disease (COPD), asthma, heart failure, and even lung cancer can result, due to the carcinogenic or mutagenic components in inhaled air [11]. Furthermore, for people living in areas where air pollution levels are high, long-term exposure correlates with higher levels of atherosclerosis [17]. Chronic exposure to polluted air indirectly places a tremendous burden on the health care system and is a significant cause of morbidity and mortality [18]. Short-term effects observed within 48 h after exposure include acute eye and nose irritation, neurophysiological symptoms, respiratory symptoms, headache, and fatigue. Ambient particulates have also been correlated with serious cardiovascular events, such as myocardial infarctions or strokes [3, 19]. Short-term elevations of DEP in the air at a concentration as low as 10 μg/m3 increase mortality by 1 % [4]. Acute exposure to a concentration of 50 μg/m3 of DEP causes an average of 1.2 deaths per day in a population of 1 million [4].
Heart and vascular consequences are frequently observed after exposure to pollution [18]. There is a higher incidence of ischemic heart disease in smokers who are chronically exposed to diesel emissions [20]. Men who had a previous myocardial infarction and who were exposed to diesel exhaust during moderate exercise showed an increase in ischemic and thrombotic effects [21]. Furthermore, upon exposure to high levels of traffic air for times as short as 1 h, there is an increased risk of coronary vasoconstriction and altered myocardial energetics [22]. Air pollutants reduce heart rate variability, cause ventricular arrhythmia, and increase left-ventricular end-diastolic pressure in animal models [23, 24]. At levels encountered in an urban environment, inhalation of diesel exhaust impaired two important and complementary aspects of vascular function in humans: the regulation of vascular tone, and endogenous fibrinolysis by increasing fibrinogen and plasminogen activator inhibitor-1 [21, 25]. Blood is not immune from the effects of air pollution. Two components in polluted air, CO and NO2, reduced the prothrombin time (PT) for clotting blood for 1218 healthy people from Lombardy Region, Italy [26]. In addition, air pollutants may significantly increase fibrinogen, factor VIII, and platelet hyperactivity.
Oxidative Stress
Oxidative stress can be defined as the imbalance between cellular oxidant species production and antioxidant capability [27]. Reactive oxygen species (ROS) can be generated under normal cellular condition or can be elicited in response to exposure to environmental stress. Despite the major composition of ROS produced in cell exposed to DEP remaining unelucidated, these free radical species are very transient and cytotoxic [18, 28–30].
Exposure to DEP induces the generation of free radicals that lead to a state of cellular oxidative stress [31]. This has been shown to cause significant damage in both cell cultures and animal models [32–36]. In vitro studies demonstrate that DEP upregulate antioxidant enzymes in various types of cells, including bronchial and pulmonary epithelial cells [37, 38], macrophages, lymphocytes [39], and endothelial cells [40]. DEP induce the generation of H2O2 [41], a powerful oxidizer which can be converted into hydroxyl radicals (.OH). In organisms, hydrogen peroxide is naturally produced as a by-product of oxygen metabolism; therefore, enzymes such as catalase catalyze conversion of hydrogen peroxide to water and oxygen. In fact, catalase is the most abundant enzyme in the human body. DEP-induced ROS leads to NO production which associated with human pulmonary artery endothelial cell damage [40]. The major cause of cell damage may be excess NO production that contributes to reduction of NO bioavailability [42]. Endothelial nitric oxide synthase plays a key role in modulating NO production and cardiovascular homeostasis. Furthermore, the metabolites of arachidonic acid, so-called epoxyeicosatrienoic acids (EETs), enhance eNOS phosphorylation and upregulate eNOS protein expression [43]. eNOS not only mediates NO production but also is involved in the release of prostacyclin, which regulates vasodilatation [44]. On the other hand, DEP paralyze eNOS and cause dysfunction in coronary arterioles [21, 44]. Therefore, exposure to DEP would result in higher risk of heart attack, coronary artery disease, or hypertension in those with existing cardiovascular disease.
DEP have a carbonaceous core onto which the toxic components of exhaust are absorbed. These chemicals contain two main families of organic compounds: polycyclic aromatic hydrocarbons (PAHs) and quinines, which can be oxygenated to quinone derivatives that produce ROS in the cells via redox cycling. PAHs desorbed from DEP bind the cytosolic aryl hydrocarbon receptor and induce phase I metabolization enzymes cytochrome P450 1A1 (CYP1A1) and cytochrome P450 1A2 (CYP1A2) in the lung and heart [45–47]. This mechanism produces electrophilic and reactive metabolites such as 1-nitropyrene (1-NP), 1,3-dinitropyrene (1,3-DNP), and 1,8-dinitropyrene (1,8-DNP). Such oxidative stress can induce DNA damage [48]. Furthermore, in the lung, DEP-induced chemical derivatization of quinones causes free radicals and diminishes the antioxidant capacity of redox cycling via the enzymes CYP reductase and NADPH oxidase. Quinones are suspected to be responsible for the production of superoxide anion (O2 −) and hydroxyl radicals [10, 49]. This can occur as follows: Redox cycling quinones undergo a one-electron reduction to form semi-quinones [50], and then semi-quinones are recycled to the original quinones with the formation of O2 −. The detoxification of quinones occurs by a two-electron reduction initiated by the phase II reaction with NADPH-quinone oxidoreductase-1 (NQO-1). Quinones are electrophiles that are able to participate in ROS damage by inducing covalent modification of proteins and DNA strands. Thus, the modification of DEP organics results in DNA adducts and DNA strand breakages, and can result in cell death [36, 51, 52].
DEP exposure has been shown to generate an ROS response that can overwhelm antioxidative proteins [40]. To maintain redox cycling equilibrium for cell survival, the cells release antioxidants such as glutathione S-transferase (GST), superoxide dismutase (SOD), NADPH-quinone oxidoreductase-1 (NQO-1), and heme oxygenase-1 (HO-1). These help neutralize the potent injuries ROS can cause. For example, in response to a 24-h free radical stimulation, endothelial cells upregulate heme oxygenase-1 (HO-1) [53]. This is accomplished by cytoplasmic nuclear factor erythroid 2-related factor 2 (Nrf2) translocating from the cytoplasm to the nucleus, where it binds to the antioxidant response element (ARE) that resides in the promoter regions of antioxidant genes. This upregulates HO-1 mRNA levels via Nrf2/ARE enhancement of transcription [54–56].
DEP also induce lipid peroxidation, as well as massive protein oxidation and mitochondria superoxide production [57, 58]. ROS are involved in a variety of cellular processes, ranging from cell proliferation and carcinogenesis to cell death [27]. Previous studies showed that excessive production of ROS causes irreversible damage to lipids, DNA, and proteins, thus provoking cell death through several modes, including autophagy and apoptosis [59]. Furthermore, recent results have suggested that DEP function by changing the levels of its effector, H2O2, which triggers Nrf2 translocation from the cytoplasm to the nucleus [56, 60]. Downstream heme oxygenase (HO)-1 is then upregulated to facilitate antioxidative stress response in the endothelium [61–64]. HO-1 also functions to induce vascular permeability and contributes to the secretion of vascular endothelial growth factor A (VEGF-A) [65]. VEGF-A, also called vascular permeability factor (VPF), has been shown to induce vascular permeability [66, 67]. Upon exposure of in vitro capillary tube cells to DEP, the VE-cadherin/VEGF receptor 2 (VEGF-R2) complex on the cell membrane dissociates [56]. Partial internalization of VE-cadherin and discontinuity of the cell–cell border are also induced following these junctional alternation [56, 68]. Moreover, these events cause endothelial junctions to become disrupted and may explain how VEGF-A initiates vascular permeability following inhalation of DEP.
Pro-inflammatory
Many reports have suggested that DEP initiate an inflammatory response that ultimately causes injury. In vitro studies have demonstrated that PM2.5 upregulates the secretion of pro-inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) in macrophages, as well as epithelial and endothelial cells [69–72]. Additionally, cultured bronchial epithelial cells exposed to DEP also released interleukin-8 (IL-8) and granulocyte macrophage colony stimulating factor (GM-CSF) in a time- and dose-dependent manner. Both of these are known to be involved in allergic diseases [38, 45].
It is important to realize that ROS and pro-inflammatory responses go hand in hand. For example, in the bloodstream, TNF-α has pro-oxidative properties and stimulates generation of ROS in the cardiac muscle of patients with heart failure [73]. In such patients, TNF-α enhances platelet superoxide anion (O2 −) production [74]. Also, in airway epithelial cells, the components of DEP adsorbed on particles elicit inflammation through CYP reductase and NADPH oxidase [75]. These activate cytokine secretion as well as an oxidative stress response.
Inhalation of DEP for 24 h upregulates TNF-α and leads to accumulation of large amounts of TNF-α in human plasma [76]. This changes the expression of adhesion molecules on endothelial cells, facilitating the transmigration of neutrophils and thereby leading to changes in vascular permeability [77]. Furthermore, there is a positive correlation between vascular permeability and adherens junction integrity [78]. Nwariaku et al. [79] found that TNF-α-induced tyrosine phosphorylation of VE-cadherin, which permits regulation of microvascular permeability, increases the formation of intercellular gap formation. IL-6 has also been shown to be directly involved in increasing monolayer endothelial permeability [80]. Maruo et al. [81] suggested that IL-6 induces increased endothelial permeability by rearranging VE-cadherin and altering the shape of endothelial cells. This implies that the endothelial cell–cell barrier may also be altered.
Vascular Permeability
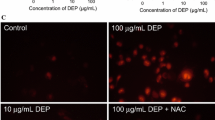
Exposure to DEP is associated with adverse pulmonary and cardiovascular health effect due to its composition and particle size. DEP contribute to fine PM (PM2.5) and ultrafine PM (diameters of 0.1 μm, i.e., 100 nm, or smaller, or PM0.1) [82], both of which are capable of entering the alveolar region. Reports have suggested that inhaled ultrafine titanium dioxide particles were found on the luminal side of airways and alveoli, in lung tissues and cells, and within capillaries. The ultrafine particles translocated into the bloodstream after inhalation by a volunteer [83, 84]. Particle uptake in vitro into cells did not occur by any of the expected endocytic processes, but rather by diffusion or adhesive interactions. Study demonstrated that the role of particles translocated into circulation might be mediated by endothelial cell–cell adherens junctions [82]. VE-cadherin is an endothelial-specific cadherin of adherens junctions that regulates not only vascular permeability, but also leukocyte transmigration [85]. Disruption of VE-cadherin endothelial barrier integrity has also been shown to alter vascular permeability [86, 87]. The pulmonary endothelium acts as a semipermeable barrier, and the integrity of the barrier is necessary for efficient pulmonary function [88]. Furthermore, DEP induce oxidative stress in differential endothelial cells [56]. Cells respond by transporting Nrf2 to the nucleus to facilitate transcription of genes to defend against ROS. HO-1 is a defense enzyme induced by Nrf2, which consequently secretes vascular permeability factor VEGF-A to contribute to vasculature being permeable. Recently, our unpublished results also reported that DEP-induced intracellular ROS is able to release the pro-inflammatory cytokines TNF-α and IL-6, which might also contribute to VEGF-A secretion and disrupt cell–cell borders to increase vasculature permeability. Interestingly, addition of ROS scavenger N-acetyl cysteine (NAC) suppresses DEP-induced ROS efficiently and reduces subsequent damages by increasing endogenous glutathione [89].
Apoptosis
Epidemiological studies have suggested that exposure to high concentration DEP might cause acute cardiovascular symptomatic flares within even 48 h of exposure [58, 90]. The DEP-induced symptoms include myocardial infarction and atherosclerosis. Studies indicated that induction of apoptosis in endothelial cells, smooth muscle cells, and immune cells has been involved in the formation of atheromatous plaques [91]. Apoptosis is a type of cell death characterized by cell shrinkage, membrane blebbing, and chromatin condensation [92]. Apoptosis in cells has also been implicated when mitochondrial functions are attenuated or intracellular ATP is depleted [93]. It would link to derangement of actin cytoskeleton [94, 95] and dephosphorylation of cell survival Akt signaling [96], which potentially contribute to induction of myocardial ischemia and infarction [97, 98].
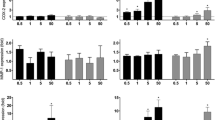
Atherosclerosis can be regarded as an inflammatory disorder which arises from accumulation of chemokines, cytokines, growth factors, and lipoproteins; it gave birth to vascular pathology [99]. On the other hand, the apoptosis of endothelium is also triggered in atherosclerosis. Denudation of the endothelial monolayer in aortic segment is associated with endothelium apoptosis, which initiates the migration of smooth muscle cells to the denuded segment. The smooth muscle cells proliferate and increase the intimal mass of the denuded segment, but the subsequent reendothelialization of the denuded segment replaces the smooth muscle cells and intimal mass which initiate the death of smooth muscle cells into the intima, further damaging the vasculature and propagating plaque development [100]. Atherosclerosis may also depend on increased coagulation of apoptotic endothelial cells. Apoptotic cells are procoagulant of the circulation system, and the activated platelets are aggregated in areas rich in apoptotic cells. Our recent studies showed that DEP induce mitochondrial superoxide anion generation, which leads to ATP depletion followed by depolarization of actin cytoskeleton and prohibits PI3K/Akt activity and contributes to endothelial apoptosis [58]. The performance is accompanied by induction of the p53/Mdm2 feedback regulation at 10 µg/mL DEP and produces 20 % cell apoptosis. Nevertheless, a high dose of DEP (100 µg/mL) augments tube cell apoptosis up to 70 %, but dysfunction of p53 negative regulator, Mdm2.
Autophagy
There is growing interest in the role of autophagic flux in maintaining normal vessel wall biology and a growing suspicion that autophagic dysregulation may be a normal mechanism through which vascular abnormalities and associated pathologies develop [101]. Autophagy is considered a protective process which proceeds cell survival by recycling organelles and long-lived proteins during nutrient deprivation, hypoxia, and infection [102]. Nevertheless, autophagy may be a type of cell death under certain circumstances [103]. Lysosomal coordination regulates the late state of autophagic flux [104]. Some nanoscale materials were identified with contribution of lysosomal dysfunction, including multi-wall carbon nanotube (MWCNT) [105], glass wool [106], titanium dioxide (TiO2) [107], polystyrene [108], and zinc oxide [109]. Nanoparticles are usually sequestered within the lysosomal compartment; therefore, the nanoparticles inhibit lysosomal enzyme activity and cause biopersistence; the above legions contribute to autophagy dysfunction and cell death. Accordingly, DEP are nanoscale-like particles; our unpublished research indicates that DEP are uptaken and accumulated in the endothelial cells within 2 h exposure and induce autophagy, while p62 was simultaneously accumulated in the cytoplasm for 8 h, suggesting that autophagosome is not able to digest DEP and lead to autophagy-independent endothelium apoptosis. On the other hand, the upregulated expression of antioxidative enzymes was observed at various time points as well, suggesting that these undigested DEP cause oxidative stress in the endothelial cytoplasm and sequentially lead to endothelial apoptosis.
Conclusion
The mechanisms for DEP-induced endothelial dysfunction that possibly result in atherosclerosis remain unknown. One of the major mechanisms underlying the effects of DEP is suggested to be oxidative stress. As shown in Fig. 1, investigations have suggested that DEP induces the generation of oxidative stress in the HUVEC tube cells. Transcription factor Nrf2 is translocated to the cell nucleus and activates transcription of the antioxidative enzyme HO-1 and sequentially induces the release of vascular permeability factor VEGF-A. Additionally, DEP-induced intracellular ROS may cause the release of pro-inflammatory TNF-α and IL-6, which may induce endothelial permeability as well by promoting VEGF-A secretion independently of HO-1 activation. These effects cause the adherens junction molecule, VE-cadherin, to become redistributed from the membrane at cell–cell borders to the cytoplasm in response to DEP, separating the plasma membranes of adjacent cells. DEP translocate occasionally in the endothelial cell cytoplasm and in the tube lumen. Furthermore, acute DEP exposure stimulates ATP depletion, followed by depolarization of their actin cytoskeleton, which sequentially inhibits PI3K/Akt activity and induces endothelial apoptosis. Nevertheless, while exposure to a low dose of DEP actin triggers cytoskeleton depolarization, reduces PI3K/Akt activity, and induces a p53/Mdm2 feedback loop, a high dose causes apoptosis by depleting Mdm2. N-acetyl cysteine suppresses DEP-induced oxidative stress efficiently and reduces subsequent damages by increasing endogenous glutathione. Although autophagy is considered a protective process which proceeds cell survival by recycling organelles and long-lived proteins during nutrient deprivation, hypoxia, and infection, DEP are sequestered within the lysosomal compartment and inhibit lysosomal enzyme activity and contribute to autophagy dysfunction and apoptosis. It might be the mechanism that these undigested DEP cause oxidative stress in the endothelial cytoplasm and sequentially lead to endothelial apoptosis.

Mechanism of DEP contributes to cardiovascular pathologies. Exposure of endothelial cells to DEP results in two potential pathways to cardiovascular disorders. First, the DEP-induced intracellular ROS production causes cell–cell junction leakage with VEGF-A release and vascular permeability. Then, DEP translocate into bloodstream and result in vasculature illness. Second, the DEP-induced ROS trigger endothelial phagocytosis of the particles. The ingested DEP will be embedded by lysosome and sequentially fused with autophagosome, which results in autophagy. However, the DEP is not able to be digested, and these autolysosome-embedded DEP accumulate in the cytosol, leading to higher oxidative stress and the eventual causation of apoptosis
PM2.5-DEP threaten our daily life. It is important to elucidate how DEP affect our bodies. The continued expansion of the field of respiratory and cardiovascular toxicology requires a thorough understanding of the mechanism of DEP for appropriate safety assessment and identification of exposure biomarkers. With increasing research of respiratory toxicology, the comprehensive mechanism of several respiratory toxicants has begun to emerge. Researchers should be conscious that air pollution like DEP can have deleterious effect on inflammatory, apoptosis, and autophagy pathways, giving rise to respiratory toxicity and lung pathology. Overall, expanding knowledge of the implications and biological significance of DEP-induced reactive oxidative stress, inflammatory response, apoptosis, and autophagy pathways has tremendous potential to aid in our understanding of respiratory toxicology and design of suitable pharmaceutical therapy and chemoprevention.
References
IARC. (2012). Diesel engine exhaust carcinogenic. Central European Journal of Public Health, 20, 120–138.
Ghio, A. J., Sobus, J. R., Pleil, J. D., & Madden, M. C. (2012). Controlled human exposures to diesel exhaust. Swiss Medical Weekly, 142, w13597.
Brook, R. D. (2008). Cardiovascular effects of air pollution. Clinical Science (London), 115, 175–187.
Pope, C. A, I. I. I., & Dockery, D. W. (2006). Health effects of fine particulate air pollution: Lines that connect. Journal of the Air & Waste Management Association (1995), 56, 709–742.
Sydbom, A., Blomberg, A., Parnia, S., Stenfors, N., Sandstrom, T., & Dahlen, S. E. (2001). Health effects of diesel exhaust emissions. European Respiratory Journal, 17, 733–746.
Krivoshto, I. N., Richards, J. R., Albertson, T. E., & Derlet, R. W. (2008). The toxicity of diesel exhaust: Implications for primary care. The Journal of the American Board of Family Medicine, 21, 55–62.
Mamessier, E., Nieves, A., Vervloet, D., & Magnan, A. (2006). Diesel exhaust particles enhance T-cell activation in severe asthmatics. Allergy, 61, 581–588.
Ishinishi, N., Kuwabara, N., Nagase, S., Suzuki, T., Ishiwata, S., & Kohno, T. (1986). Long-term inhalation studies on effects of exhaust from heavy and light duty diesel engines on F344 rats. Developments in Toxicology and Environmental Science, 13, 329–348.
McClellan, R. O. (1987). Health effects of exposure to diesel exhaust particles. Annual Review of Pharmacology and Toxicology, 27, 279–300.
Kumagai, Y., Arimoto, T., Shinyashiki, M., Shimojo, N., Nakai, Y., Yoshikawa, T., & Sagai, M. (1997). Generation of reactive oxygen species during interaction of diesel exhaust particle components with NADPH-cytochrome P450 reductase and involvement of the bioactivation in the DNA damage. Free Radical Biology and Medicine, 22, 479–487.
Wichmann, H. E. (2007). Diesel exhaust particles. Inhalation Toxicology, 19(Suppl 1), 241–244.
Sun, Y., Bochmann, F., Nold, A., & Mattenklott, M. (2014). Diesel exhaust exposure and the risk of lung cancer—A review of the epidemiological evidence. International Journal of Environmental Research and Public Health, 11, 1312–1340.
Hodan, W.M., Barnard, W. R. (2004). Evaluating the contribution of PM2.5 precursor gases and re-entrained road emissions to mobile source PM2.5 particulate matter emissions. MACTEC Federal Programs, Research Triangle Park, NC.
Monn, C., Fuchs, A., Hogger, D., Junker, M., Kogelschatz, D., Roth, N., & Wanner, H. U. (1997). Particulate matter less than 10 microns (PM10) and fine particles less than 2.5 microns (PM2.5): Relationships between indoor, outdoor and personal concentrations. Science of the Total Environment, 208, 15–21.
Nordman, H., Keskinen, H., & Tuppurainen, M. (1985). Formaldehyde asthma—rare or overlooked? Journal of Allergy and Clinical Immunology, 75, 91–99.
Kerns, W. D., Pavkov, K. L., Donofrio, D. J., Gralla, E. J., & Swenberg, J. A. (1983). Carcinogenicity of formaldehyde in rats and mice after long-term inhalation exposure. Cancer Research, 43, 4382–4392.
Suwa, T., Hogg, J. C., Quinlan, K. B., Ohgami, A., Vincent, R., & van Eeden, S. F. (2002). Particulate air pollution induces progression of atherosclerosis. Journal of the American College of Cardiology, 39, 935–942.
Miller, M. R., Shaw, C. A., & Langrish, J. P. (2012). From particles to patients: Oxidative stress and the cardiovascular effects of air pollution. Future Cardiology, 8, 577–602.
Pope, C. A, 3rd. (2007). Mortality effects of longer term exposures to fine particulate air pollution: Review of recent epidemiological evidence. Inhalation Toxicology, 19(Suppl 1), 33–38.
Finkelstein, M. M., Verma, D. K., Sahai, D., & Stefov, E. (2004). Ischemic heart disease mortality among heavy equipment operators. American Journal of Industrial Medicine, 46, 16–22.
Mills, N. L., Tornqvist, H., Gonzalez, M. C., Vink, E., Robinson, S. D., Soderberg, S., et al. (2007). Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. The New England Journal of Medicine, 357, 1075–1082.
Peters, A., von Klot, S., Heier, M., Trentinaglia, I., Hormann, A., Wichmann, H. E., & Lowel, H. (2004). Exposure to traffic and the onset of myocardial infarction. The New England Journal of Medicine, 351, 1721–1730.
Anselme, F., Loriot, S., Henry, J. P., Dionnet, F., Napoleoni, J. G., Thuillez, C., & Morin, J. P. (2007). Inhalation of diluted diesel engine emission impacts heart rate variability and arrhythmia occurrence in a rat model of chronic ischemic heart failure. Archives of Toxicology, 81, 299–307.
Wold, L. E., Simkhovich, B. Z., Kleinman, M. T., Nordlie, M. A., Dow, J. S., Sioutas, C., & Kloner, R. A. (2006). In vivo and in vitro models to test the hypothesis of particle-induced effects on cardiac function and arrhythmias. Cardiovascular Toxicology, 6, 69–78.
Mills, N. L., Tornqvist, H., Robinson, S. D., Gonzalez, M., Darnley, K., MacNee, W., et al. (2005). Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation, 112, 3930–3936.
Baccarelli, A., Zanobetti, A., Martinelli, I., Grillo, P., Hou, L., Giacomini, S., et al. (2007). Effects of exposure to air pollution on blood coagulation. Journal of Thrombosis and Haemostasis, 5, 252–260.
Mates, J. M., Segura, J. A., Alonso, F. J., & Marquez, J. (2008). Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis, and carcinogenesis. Archives of Toxicology, 82, 273–299.
Garza, K. M., Soto, K. F., & Murr, L. E. (2008). Cytotoxicity and reactive oxygen species generation from aggregated carbon and carbonaceous nanoparticulate materials. International Journal of Nanomedicine, 3, 83–94.
Matsunaga, T., Arakaki, M., Kamiya, T., Endo, S., El-Kabbani, O., & Hara, A. (2009). Involvement of an aldo-keto reductase (AKR1C3) in redox cycling of 9,10-phenanthrenequinone leading to apoptosis in human endothelial cells. Chemico-Biological Interactions, 181, 52–60.
Matsuo, M., Shimada, T., Uenishi, R., Sasaki, N., & Sagai, M. (2003). Diesel exhaust particle-induced cell death of cultured normal human bronchial epithelial cells. Biological and Pharmaceutical Bulletin, 26, 438–447.
Hawley, B., L’Orange, C., Olsen, D. B., Marchese, A. J., & Volckens, J. (2014). Oxidative stress and aromatic hydrocarbon response of human bronchial epithelial cells exposed to petro- or biodiesel exhaust treated with a diesel particulate filter. Toxicological Sciences, 141, 505–514.
Hiura, T. S., Kaszubowski, M. P., Li, N., & Nel, A. E. (1999). Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. The Journal of Immunology, 163, 5582–5591.
Hiura, T. S., Li, N., Kaplan, R., Horwitz, M., Seagrave, J. C., & Nel, A. E. (2000). The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. The Journal of Immunology, 165, 2703–2711.
Li, N., Kim, S., Wang, M., Froines, J., Sioutas, C., & Nel, A. (2002). Use of a stratified oxidative stress model to study the biological effects of ambient concentrated and diesel exhaust particulate matter. Inhalation Toxicology, 14, 459–486.
Sagai, M., Saito, H., Ichinose, T., Kodama, M., & Mori, Y. (1993). Biological effects of diesel exhaust particles. I. In vitro production of superoxide and in vivo toxicity in mouse. Free Radical Biology and Medicine, 14, 37–47.
Moller, P., Jensen, D. M., Christophersen, D. V., Kermanizadeh, A., Jacobsen, N. R., Hemmingsen, J. G., et al. (2015). Measurement of oxidative damage to DNA in nanomaterial exposed cells and animals. Environmental and Molecular Mutagenesis, 56, 97–110.
Sugimoto, R., Kumagai, Y., Nakai, Y., & Ishii, T. (2005). 9,10-Phenanthraquinone in diesel exhaust particles downregulates Cu, Zn-SOD and HO-1 in human pulmonary epithelial cells: Intracellular iron scavenger 1,10-phenanthroline affords protection against apoptosis. Free Radical Biology and Medicine, 38, 388–395.
Takizawa, H., Ohtoshi, T., Kawasaki, S., Abe, S., Sugawara, I., Nakahara, K., et al. (2000). Diesel exhaust particles activate human bronchial epithelial cells to express inflammatory mediators in the airways: A review. Respirology (Carlton Vic), 5, 197–203.
Al-Humadi, N. H., Siegel, P. D., Lewis, D. M., Barger, M. W., Ma, J. Y., Weissman, D. N., & Ma, J. K. (2002). Alteration of intracellular cysteine and glutathione levels in alveolar macrophages and lymphocytes by diesel exhaust particle exposure. Environmental Health Perspectives, 110, 349–353.
Bai, Y., Suzuki, A. K., & Sagai, M. (2001). The cytotoxic effects of diesel exhaust particles on human pulmonary artery endothelial cells in vitro: Role of active oxygen species. Free Radical Biology and Medicine, 30, 555–562.
Park, S., Nam, H., Chung, N., Park, J. D., & Lim, Y. (2006). The role of iron in reactive oxygen species generation from diesel exhaust particles. Toxicology in Vitro, 20, 851–857.
Langrish, J. P., Unosson, J., Bosson, J., Barath, S., Muala, A., Blackwell, S., et al. (2013). Altered nitric oxide bioavailability contributes to diesel exhaust inhalation-induced cardiovascular dysfunction in man. Journal of the American Heart Association, 2, e004309.
Jiang, J. G., Chen, R. J., Xiao, B., Yang, S., Wang, J. N., Wang, Y., et al. (2007). Regulation of endothelial nitric-oxide synthase activity through phosphorylation in response to epoxyeicosatrienoic acids. Prostaglandins & Other Lipid Mediators, 82, 162–174.
Cherng, T. W., Paffett, M. L., Jackson-Weaver, O., Campen, M. J., Walker, B. R., & Kanagy, N. L. (2011). Mechanisms of diesel-induced endothelial nitric oxide synthase dysfunction in coronary arterioles. Environmental Health Perspectives, 119, 98–103.
Bonvallot, V., Baeza-Squiban, A., Baulig, A., Brulant, S., Boland, S., Muzeau, F., et al. (2001). Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. American Journal of Respiratory Cell and Molecular Biology, 25, 515–521.
Rengasamy, A., Barger, M. W., Kane, E., Ma, J. K., Castranova, V., & Ma, J. Y. (2003). Diesel exhaust particle-induced alterations of pulmonary phase I and phase II enzymes of rats. Journal of Toxicology and Environmental Health, 66, 153–167.
Bradley, J. M., Cryar, K. A., El Hajj, M. C., El Hajj, E. C., & Gardner, J. D. (2013). Exposure to diesel exhaust particulates induces cardiac dysfunction and remodeling. Journal of Applied Physiology (1985), 115, 1099–1106.
Landvik, N. E., Gorria, M., Arlt, V. M., Asare, N., Solhaug, A., Lagadic-Gossmann, D., & Holme, J. A. (2007). Effects of nitrated-polycyclic aromatic hydrocarbons and diesel exhaust particle extracts on cell signalling related to apoptosis: Possible implications for their mutagenic and carcinogenic effects. Toxicology, 231, 159–174.
Risom, L., Moller, P., & Loft, S. (2005). Oxidative stress-induced DNA damage by particulate air pollution. Mutation Research, 592, 119–137.
Monks, T. J., & Lau, S. S. (1992). Toxicology of quinone-thioethers. Critical Reviews in Toxicology, 22, 243–270.
Valavanidis, A., Vlachogianni, T., Fiotakis, K., & Loridas, S. (2013). Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. International Journal of Environmental Research and Public Health, 10, 3886–3907.
Chuang, H. C., Cheng, Y. L., Lei, Y. C., Chang, H. H., & Cheng, T. J. (2013). Protective effects of pulmonary epithelial lining fluid on oxidative stress and DNA single-strand breaks caused by ultrafine carbon black, ferrous sulphate and organic extract of diesel exhaust particles. Toxicology and Applied Pharmacology, 266, 329–334.
Fredenburgh, L. E., Perrella, M. A., & Mitsialis, S. A. (2007). The role of heme oxygenase-1 in pulmonary disease. American Journal of Respiratory Cell and Molecular Biology, 36, 158–165.
Chen, X. L., Varner, S. E., Rao, A. S., Grey, J. Y., Thomas, S., Cook, C. K., et al. (2003). Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. Journal of Biological Chemistry, 278, 703–711.
Hsieh, C. Y., Hsiao, H. Y., Wu, W. Y., Liu, C. A., Tsai, Y. C., Chao, Y. J., et al. (2009). Regulation of shear-induced nuclear translocation of the Nrf2 transcription factor in endothelial cells. Journal of Biomedical Science, 16, 12.
Chao, M. W., Po, I. P., Laumbach, R. J., Koslosky, J., Cooper, K., & Gordon, M. K. (2012). DEP induction of ROS in capillary-like endothelial tubes leads to VEGF-A expression. Toxicology, 297, 34–46.
Hengstler, J. G., & Bolt, H. M. (2008). Oxidative stress: From modification of cell-cycle related events, secondary messenger function, dysregulation of small GTPases, protein kinases and phosphatases to redox-sensitive cancer models. Archives of Toxicology, 82, 271–272.
Tseng, C. Y., Wang, J. S., Chang, Y. J., Chang, J. F., & Chao, M. W. (2015). Exposure to high-dose diesel exhaust particles induces intracellular oxidative stress and causes endothelial apoptosis in cultured in vitro capillary tube cells. Cardiovascular Toxicology, 15, 345–354.
Azad, M. B., Chen, Y., & Gibson, S. B. (2009). Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxidants & Redox Signaling, 11, 777–790.
Li, Y. J., Kawada, T., & Azuma, A. (2013). Nrf2 is a protective factor against oxidative stresses induced by diesel exhaust particle in allergic asthma. Oxidative Medicine and Cellular Longevity, 2013, 323607.
Brouard, S., Berberat, P. O., Tobiasch, E., Seldon, M. P., Bach, F. H., & Soares, M. P. (2002). Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. The Journal of Biological Chemistry, 277, 17950–17961.
Silva, G., Cunha, A., Gregoire, I. P., Seldon, M. P., & Soares, M. P. (2006). The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38 alpha MAPK isoform. The Journal of Immunology, 177, 1894–1903.
Wang, Z., Armando, I., Asico, L. D., Escano, C., Wang, X., Lu, Q., et al. (2007). The elevated blood pressure of human GRK4gamma A142V transgenic mice is not associated with increased ROS production. American Journal of Physiology, 292, H2083–H2092.
Wilson, S. J., & Keenan, A. K. (2003). Role of hemin in the modulation of H2O2-mediated endothelial cell injury. Vascular Pharmacology, 40, 109–118.
Dulak, J., Loboda, A., & Jozkowicz, A. (2008). Effect of heme oxygenase-1 on vascular function and disease. Current Opinion in Lipidology, 19, 505–512.
Nagy, J. A., Vasile, E., Feng, D., Sundberg, C., Brown, L. F., Manseau, E. J., et al. (2002). VEGF-A induces angiogenesis, arteriogenesis, lymphangiogenesis, and vascular malformations. Cold Spring Harbor Symposia on Quantitative Biology, 67, 227–237.
Weis, S. M., & Cheresh, D. A. (2005). Pathophysiological consequences of VEGF-induced vascular permeability. Nature, 437, 497–504.
Vandenbroucke, E., Mehta, D., Minshall, R., & Malik, A. B. (2008). Regulation of endothelial junctional permeability. Annals of the New York Academy of Sciences, 1123, 134–145.
Auger, F., Gendron, M. C., Chamot, C., Marano, F., & Dazy, A. C. (2006). Responses of well-differentiated nasal epithelial cells exposed to particles: Role of the epithelium in airway inflammation. Toxicology and Applied Pharmacology, 215, 285–294.
Jalava, P., Salonen, R. O., Halinen, A. I., Sillanpaa, M., Sandell, E., & Hirvonen, M. R. (2005). Effects of sample preparation on chemistry, cytotoxicity, and inflammatory responses induced by air particulate matter. Inhalation Toxicology, 17, 107–117.
Veranth, J. M., Cutler, N. S., Kaser, E. G., Reilly, C. A., & Yost, G. S. (2008). Effects of cell type and culture media on Interleukin-6 secretion in response to environmental particles. Toxicology in Vitro, 22, 498–509.
Rusznak, C., Mills, P. R., Devalia, J. L., Sapsford, R. J., Davies, R. J., & Lozewicz, S. (2000). Effect of cigarette smoke on the permeability and IL-1beta and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology, 23, 530–536.
Eleuteri, E., Magno, F., Gnemmi, I., Carbone, M., Colombo, M., La Rocca, G., et al. (2009). Role of oxidative and nitrosative stress biomarkers in chronic heart failure. Frontiers in Bioscience, 14, 2230–2237.
De Biase, L., Pignatelli, P., Lenti, L., Tocci, G., Piccioni, F., Riondino, S., et al. (2003). Enhanced TNF alpha and oxidative stress in patients with heart failure: Effect of TNF alpha on platelet O2-production. Thrombosis and Haemostasis, 90, 317–325.
Baulig, A., Garlatti, M., Bonvallot, V., Marchand, A., Barouki, R., Marano, F., & Baeza-Squiban, A. (2003). Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. American Journal of Physiology, 285, L671–L679.
Tornqvist, H., Mills, N. L., Gonzalez, M., Miller, M. R., Robinson, S. D., Megson, I. L., et al. (2007). Persistent endothelial dysfunction in humans after diesel exhaust inhalation. American Journal of Respiratory and Critical Care Medicine, 176, 395–400.
Forchhammer, L., Loft, S., Roursgaard, M., Cao, Y., Riddervold, I. S., Sigsgaard, T., & Moller, P. (2012). Expression of adhesion molecules, monocyte interactions and oxidative stress in human endothelial cells exposed to wood smoke and diesel exhaust particulate matter. Toxicology Letters, 209, 121–128.
Yokota, S., Ohara, N., & Kobayashi, T. (2008). The effects of organic extract of diesel exhaust particles on ischemia/reperfusion-related arrhythmia and on pulmonary inflammation. Journal of Toxicological Sciences, 33, 1–10.
Nwariaku, F. E., Liu, Z., Zhu, X., Turnage, R. H., Sarosi, G. A., & Terada, L. S. (2002). Tyrosine phosphorylation of vascular endothelial cadherin and the regulation of microvascular permeability. Surgery, 132, 180–185.
Cromer, W. E., Zawieja, S. D., Tharakan, B., Childs, E. W., Newell, M. K., & Zawieja, D. C. (2014). The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis, 17, 395–406.
Maruo, N., Morita, I., Shirao, M., & Murota, S. (1992). IL-6 increases endothelial permeability in vitro. Endocrinology, 131, 710–714.
Chao, M. W., Kozlosky, J., Po, I. P., Strickland, P. O., Svoboda, K. K., Cooper, K., et al. (2011). Diesel exhaust particle exposure causes redistribution of endothelial tube VE-cadherin. Toxicology, 279, 73–84.
Higgins, K. J., Jung, H., Kittelson, D. B., Roberts, J. T., & Zachariah, M. R. (2003). Kinetics of diesel nanoparticle oxidation. Environmental Science and Technology, 37, 1949–1954.
Burch, W. M. (2002). Passage of inhaled particles into the blood circulation in humans. Circulation, 106, e141–E142. (author reply e141–142).
Corada, M., Liao, F., Lindgren, M., Lampugnani, M. G., Breviario, F., Frank, R., et al. (2001). Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood, 97, 1679–1684.
Harris, E. S., & Nelson, W. J. (2010). VE-cadherin: At the front, center, and sides of endothelial cell organization and function. Current Opinion in Cell Biology, 22, 651–658.
Gavard, J. (2013). Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adhesion & Migration, 7, 455–461.
Dudek, S. M., & Garcia, J. G. (2001). Cytoskeletal regulation of pulmonary vascular permeability. Journal of Applied Physiology, 91, 1487–1500.
Tseng, C. Y., Chang, J. F., Wang, J. S., Chang, Y. J., & Chao, M. W. (2015). Protective effects of N-acetyl cysteine against diesel exhaust particles-induced intracellular ROS generates pro-inflammatory cytokines to mediate the vascular permeability of capillary-like endothelial tubes. PLoS One, 10, e0131911.
Chow, J. C., Watson, J. G., Mauderly, J. L., Costa, D. L., Wyzga, R. E., Vedal, S., et al. (2006). Health effects of fine particulate air pollution: Lines that connect. Journal of the Air & Waste Management Association (1995), 56, 1368–1380.
Choy, J. C., Granville, D. J., Hunt, D. W., & McManus, B. M. (2001). Endothelial cell apoptosis: Biochemical characteristics and potential implications for atherosclerosis. Journal of Molecular and Cellular Cardiology, 33, 1673–1690.
Kerr, J. F., Wyllie, A. H., & Currie, A. R. (1972). Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer, 26, 239–257.
Shen, B., He, P. J., & Shao, C. L. (2013). Norcantharidin induced DU145 cell apoptosis through ROS-mediated mitochondrial dysfunction and energy depletion. PLoS One, 8, e84610.
Rubtsova, S. N., Kondratov, R. V., Kopnin, P. B., Chumakov, P. M., Kopnin, B. P., & Vasiliev, J. M. (1998). Disruption of actin microfilaments by cytochalasin D leads to activation of p53. FEBS Letters, 430, 353–357.
White, S. R., Williams, P., Wojcik, K. R., Sun, S., Hiemstra, P. S., Rabe, K. F., & Dorscheid, D. R. (2001). Initiation of apoptosis by actin cytoskeletal derangement in human airway epithelial cells. American Journal of Respiratory Cell and Molecular Biology, 24, 282–294.
Carmeliet, P., Lampugnani, M. G., Moons, L., Breviario, F., Compernolle, V., Bono, F., et al. (1999). Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell, 98, 147–157.
Kerr, B. A., Ma, L., West, X. Z., Ding, L., Malinin, N. L., Weber, M. E., et al. (2013). Interference with akt signaling protects against myocardial infarction and death by limiting the consequences of oxidative stress. Science Signaling, 6, ra67.
Robertson, S., Thomson, A. L., Carter, R., Stott, H. R., Shaw, C. A., Hadoke, P. W., et al. (2014). Pulmonary diesel particulate increases susceptibility to myocardial ischemia/reperfusion injury via activation of sensory TRPV1 and beta1 adrenoreceptors. Particle and Fibre Toxicology, 11, 12.
Liuzzo, G. (2001). Atherosclerosis: an inflammatory disease. Rays, 26, 221–230.
Bochaton-Piallat, M. L., Gabbiani, F., Redard, M., Desmouliere, A., & Gabbiani, G. (1995). Apoptosis participates in cellularity regulation during rat aortic intimal thickening. The American Journal of Pathology, 146, 1059–1064.
Nussenzweig, S. C., Verma, S., & Finkel, T. (2015). The role of autophagy in vascular biology. Circulation Research, 116, 480–488.
Benbrook, D. M., & Long, A. (2012). Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Experimental Oncology, 34, 286–297.
Gustafsson, A. B., & Gottlieb, R. A. (2008). Recycle or die: The role of autophagy in cardioprotection. Journal of Molecular and Cellular Cardiology, 44, 654–661.
Yogalingam, G., & Pendergast, A. M. (2008). Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. The Journal of Biological Chemistry, 283, 35941–35953.
Tsukahara, T., Matsuda, Y., Usui, Y., & Haniu, H. (2013). Highly purified, multi-wall carbon nanotubes induce light-chain 3B expression in human lung cells. Biochemical and Biophysical Research Communications, 440, 348–353.
Koehler, A., Marx, U., Broeg, K., Bahns, S., & Bressling, J. (2008). Effects of nanoparticles in Mytilus edulis gills and hepatopancreas—A new threat to marine life? Marine Environmental Research, 66, 12–14.
Halamoda Kenzaoui, B., Chapuis Bernasconi, C., Guney-Ayra, S., & Juillerat-Jeanneret, L. (2012). Induction of oxidative stress, lysosome activation and autophagy by nanoparticles in human brain-derived endothelial cells. The Biochemical Journal, 441, 813–821.
Kobayashi, S., Kojidani, T., Osakada, H., Yamamoto, A., Yoshimori, T., Hiraoka, Y., & Haraguchi, T. (2010). Artificial induction of autophagy around polystyrene beads in nonphagocytic cells. Autophagy, 6, 36–45.
Roy, R., Singh, S. K., Chauhan, L. K., Das, M., Tripathi, A., & Dwivedi, P. D. (2014). Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicology Letters, 227, 29–40.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tseng, CY., Wang, JS. & Chao, MW. Causation by Diesel Exhaust Particles of Endothelial Dysfunctions in Cytotoxicity, Pro-inflammation, Permeability, and Apoptosis Induced by ROS Generation. Cardiovasc Toxicol 17, 384–392 (2017). https://doi.org/10.1007/s12012-016-9364-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-016-9364-0