
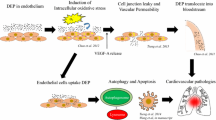
Abstract
Previous studies suggest a direct correlation between exposure to diesel exhaust particles (DEP) and the onset of vascular permeability, presumably through the disruption of the adherens junctions. This would lead to deleterious effects on vasculature, such as acute myocardial infarction and atherosclerosis. Although the mechanism remains unclear, we demonstrate DEP-induced mitochondrial reactive oxygen species generation, which may be a central cause of the above vascular disorders. In vitro capillary-like HUVEC tube cells are used in this study and show that acute DEP exposure stimulates ATP depletion, followed by depolarization of their actin cytoskeleton, which sequentially inhibits PI3K/Akt activity and induces endothelial apoptosis. These events are accompanied by induction of p53/Mdm2 feedback regulation at 10 µg/mL DEP and produce 20 % cell apoptosis. Nevertheless, 100 µg/mL DEP augments tube cell apoptosis up to 70 % but disrupts the p53 negative regulator Mdm2. Addition of N-acetylcysteine provides substantial protection against the cytotoxic effects of DEP. In summary, exposure to a low dose of DEP actin triggers cytoskeleton depolarization, reduces PI3K/Akt activity, and induces a p53/Mdm2 feedback loop, and a high dose causes apoptosis by depleting Mdm2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diesel exhaust particles (DEP) are solid and liquid, are predominantly produced from vehicle exhaust, and contain a high percentage of potential toxins [1–3]. These substances include sulfur compounds, heavy metals, and hydrocarbons, such as aldehydes, quinones, benzo[a]pyrenes. Polycyclic aromatic hydrocarbons (PAHs) have shown mutagenic and carcinogenic properties [4, 5]. Epidemiological studies have suggested that exposure to high-dosage DEP might cause acute cardiovascular symptomatic flares within even 48 h of exposure [6–8]. The underlying mechanism for the acute vasculature failure remains unclear. However, DEP-induced reactive oxygen species (ROS) and pro-inflammatory cytokines might be considered as central in the development of these health issues [9–11]. Chao et al. [12] and Tseng et al. [11] have reported that ROS induces the release of vascular permeability factor/VEGFA (Vascular endothelial growth factor A), which is able to stimulate the permeability of the cell–cell barrier adherens junctions. Then, the particle might slip between the cells via a redistributed VE-cadherin network and travel in the circulation system [13].
Oxidative stress is defined as the imbalance between production of cellular oxidant species and antioxidant capability [14]. ROS can be produced under normal cellular condition or can emerge in response to exposure to environmental stress. Although the major sources of ROS generated in cells exposed to DEP remain unclear, these free radical species are very transient and cytotoxic [15–17]. Because of their high chemical reactivity, they lead to lipid peroxidation and massive protein oxidation and degradation [18, 19]. ROS are involved in a variety of cellular processes, ranging from cell proliferation and carcinogenesis to cell death [14]. Previous studies showed that excessive production of ROS causes irreversible damage to lipids, DNA, and proteins, thus, provoking cell death through several modes, including autophagy and apoptosis [20]. Apoptosis in cells has also been implicated when mitochondrial functions are attenuated or intracellular ATP is depleted [21, 22]. It would link to derangement of actin cytoskeleton [23, 24] and dephosphorylation of cell survival Akt signaling [25], which potentially contribute to induction of myocardial ischemia and infarction [26, 27].
As argued in the literature, DEP mediate the pro-inflammatory cytokines and ROS generation in the 3D capillary-like tube cells and then increase vasculature permeability [11, 12]. Even though it was shown that DEP-induced oxidative stress promoted alterations in endothelial tube barrier junctions, the result cannot definitively differentiate the mechanism. The present study seeks to show that short-term DEP exposure initiates mitochondrial oxidative stress, leading to decrease in ATP. ATP reduction reorganizes actin cytoskeleton and initiates survival PI3K/Akt signaling termination, which might cause endothelial apoptosis with p53 up-regulation. Although low-dose DEP induce Mdm2 to be up-regulated and the Mdm2 feedback inhibits p53 activity, high-dose DEP has been observed to cause apoptosis by depletion of Mdm2. The above implies that anti-apoptotic mechanisms of cellular protection which is effective at low doses of DEP.
Materials and Methods
Cell Culture
Human umbilical vein endothelial cells (HUVEC) were cultured in medium 199 (Sigma-Aldrich) with endothelial cell growth supplement (Millipore), heparin (Sigma-Aldrich), and 10 % FBS (Gibco). HUVEC at passage 5–15 were used to assemble tubes on 10 mg/mL Matrigel, LDEV-free (BD Biosciences). Cells were grown in a 5 % CO2 atmosphere at 37 °C.
Reagents
DEP were collected from a Japanese automobile diesel engine by Dr. Masaru Sagai (Aomori University of Health and Welfare, Aomori, Japan) [28]. The DEP were dispersed by the method described previously [13]. PI3K/Akt pathway inhibitors Wortmannin and LY294002 were purchased from Enzo Life Sciences (Farmingdale, NY, USA). NAC, pifithrin-α (PFT-α, p53 suppressor), SJ-172550 (MDMX inhibitor), and nutlin-3a (MDM2 inhibitor) were purchased from Sigma-Aldrich. Concentration of NAC 10 mM was applied in all experiments.
Determination of Mitochondrial Activity
Mitochondrial activity was detected using a commercial MTS assay purchased from Promega (Madison, WI), measuring mitochondrial succinate dehydrogenase activity via conversion of MTS and phenazine methosulfate to formazan. After treatment, cells were rinsed three times with PBS; then, a mixture of 10 μL water-soluble kit reagent plus 190 μL fresh medium was added to each well for a 1-h incubation at 37 C in the dark. Supernatants (100 μL/well) were collected, and the absorbance of the generated formazan was measured at 490 nm with a microreader.
Measurement of Mitochondrial ROS Generation
Mitochondrial superoxide formation was detected by incubating cells in the dark with 5 μM MitoSOX red dye (Gibco) (excitation/emission at λ = 510 nm/580 nm) for 30 min. After treatment, mitochondrial ROS formation was assessed according to the manufacturer’s instructions. Changes in mitochondrial oxidant production were measured using a microreader. The values were normalized and presented as the percentage of the negative control. Images were observed on an Olympus IX51 inverted fluorescence microscope.
Measurement of ATP Depletion
After DEP exposure, the remaining live tube cells were treated with ApoSENSOR Cell Viability kit reagent (Biovision, Milpitas, CA, USA) to differentiate apoptosis and living cells. The samples were treated with 200 µL of nuclear releasing reagent and shaken for 5 min at room temperature. Two microliters of ATP Monitoring Enzyme was added into the cell lysate. The assay method utilized the enzyme luciferase to catalyze the formation of luminescence from ATP and luciferin in the cells, and the light was measured using a microreader.
Immunofluorescence Microscopy
After treatments with DEP, nonspecific reactivity was blocked by the addition of 2 % normal goat serum with 0.02 % NaN3 and 0.05 % Triton X-100 in PBS. The capillary tube cells were incubated with primary rabbit polyclonal anti-p53 (Abcam) and mouse monoclonal anti-Mdm2 (Abnova) antibodies at a 1:100 dilution. Slides were transferred to goat anti-rabbit secondary antibody labeled with Alexa Fluor 488 (Invitrogen) or Alexa Fluor 594 (Invitrogen) at a 1:200 dilution and were covered with Prolong Gold (Invitrogen) anti-fade mounting media with DAPI and stored at 4° C overnight. Images were observed on a Leica TCS SP5 spectral confocal microscope. For actin cytoskeleton detection, the cells were probed with phalloidin-Alexa 594 (Invitrogen). Images were observed on an Olympus IX51 inverted fluorescence microscope. The extent of tube formation function was quantified by measuring the tube length in the actin-stained images with software ImageJ (National Institutes of Health).
Western Blot
For extraction of the total cell, the protein was collected and the concentration was measured using the BCA method; then, the samples were denatured and loaded 40 μg/well onto SDS–polyacrylamide gels for electrophoresis. Proteins separated electrophorectically on SDS gels were transferred to nitrocellulose membranes. Nonspecific reactivity of the membranes was blocked, and primary anti-PI3K (1:500, Abcam), anti-Akt (1:1,000, Cell Signaling Technology), anti-phospho-Akt (1:1,000, Cell Signaling Technology), anti-p53 (1:1,000, Abcam), anti-p21 (1:1,000, Cell Signaling Technology), anti-Mdm2 (phosphor S166) (1:500, Abnova), anti-Bcl-XL (1:1,000, Abcam), and anti-GAPDH (1:5,000, Sigma-Aldrich) were probed. Secondary goat anti-mouse IgG or anti-rabbit IgG conjugated with HRP were used, and the protein products were visualized on X-ray film using chemiluminescent reagent containing luminol.
Measurement of Caspase 3 Activity
Caspase activity was assessed using the Caspase-Glo 3/7 assay kit (Promega). After treatment, the plates were removed from the incubator and allowed to equilibrate to room temperature for approximately 30 min; then, the Caspase-Glo 3/7 assay reagent (200 μL/well) was applied according to the manufacturer’s instructions. Relative luminance unit (RLU) emitted by the product was measured using a microreader.
Quantification of Apoptosis
Apoptosis was determined using a fluorometric TdT-mediated dUTP nick end labeling assay kit (TUNEL, Promega) according to the manufacturer’s instructions. The nuclei were stained with DAPI. The tube cells were assessed by incorporation of fluorescein-12-dUTP at the 3′-OH ends of the DNA, visualized by immunofluorescence microscopy (Olympus IX51 Inverted Microscope). Magnification was 100×. The percentage of apoptotic cells was calculated by dividing the number of cells stained with dUTP fluorescein label by the number of cells stained with DAPI. As a positive control for apoptosis, cells were exposed to UV light (5 mJ/min) for 10 min.
Statistics
For statistical analysis, each experiment was performed in triplicate and repeated three times. The results were expressed as mean ± SD for three independent experiments, and the differences between the groups were analyzed using Student t tests with GraphPad statistics software.
Results and Discussion
DEP Reduced Mitochondrial Activity and Induced Mitochondrial O2 − Generation
Studies have demonstrated that DEP can cause mitochondrial toxicity and apoptosis [22, 29]. MTS is a tetrazolium salt that is reduced to purple water-soluble formazan, mainly by mitochondrial succinate dehydrogenase. The color intensity of the formazan dye is correlated to the enzyme activity of mitochondria and number of viable cells [30]. In addition, the DEP doses used in our experiments were designed according to previous in vivo and in vitro studies [12, 28, 31, 32]. As shown in Fig. 1a, the tube cells exposed to 1 µg/mL DEP reduce mitochondrial activity by 6.5 %. DEP at 10 µg/mL decreases the activity to 77 % of control. A still higher dose of DEP (100 µg/mL) presented a 35 % activity ratio to the tube cells’ mitochondria. The activity values decreased in a dose-dependent manner, and adding NAC entirely prevented mitochondria dysfunction at 1 and 10 µg/mL DEP. NAC provided protection up to 75 % against the deleterious effects caused by 100 µg/mL DEP in mitochondria, suggesting that intracellular ROS might be the factor in determining cell survival. On the other hand, to examine mitochondria-specific ROS generation in response to DEP, tube cells were treated with DEP ± NAC, and superoxide generation in live cells was measured with MitoSOX Red, a mitochondria-specific fluorogenic dye that produces red fluorescence upon being oxidized by superoxide. DEP treatment for 24 h brought a significant rise in mitochondrial superoxide production in HUVEC tubes, from 122 ± 7.8 to 332 ± 16.7 % relative to the negative control (set to 100 %; Fig. 1b). Co-treating DEP with NAC resulted in a trend toward a decrease in endogenous levels of mitochondrial oxidants. Live cell imaging of MitoSOX Red oxidation using immunofluorescent microscopy showed that, relative to the negative control, DEP treatments at 10 and 100 µg/mL resulted in an increase in mitochondrial oxidants as imaged at 24 h (Fig. 1c). Compared with this result, DEP plus NAC treatment resulted in a remarkable reduction in mitochondrial oxidants. The MitoSOX experiments not only confirm the results generated from Chao et al. [12] but suggest that tube cell viability is directly correlated to the oxidative stress generated by DEP exposure.

DEP reduced mitochondrial activity and induced mitochondrial ROS production a HUVEC tube cultures were treated with DEP (1, 10, 100 μg/mL) ± 10 mM NAC for 24 h, as assessed by MTS assays. The values were normalized and shown as the percentage change relative to the control. b Measurement of fluorescence intensity of oxidized MitoSOX Red in a plate reader assay. Data are presented as the percentage change relative to the untreated control. c Immunofluorescent live imaging of MitoSOX Red. Representative images of control untreated cells and cells after 10 μg/mL DEP and 100 μg/mL DEP ± NAC treatments are shown. The 594-nm excitation wavelength was detected for imaging the MitoSOX Red fluorescence signal. Scale bar = 20 μm. Magnification = ×200. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the control, mean ± SD, n = 3
The Effects of Depletion of Intracellular ATP in Response to DEP-induced ROS
ATP degrades rapidly in dead cells and declines in injured cells. Previous studies have indicated that excessive production of ROS provokes mitochondrial dysfunction by depletion of intracellular ATP [20–22]. Therefore, the amount of ATP present in the cells treated with DEP is proportional to the mitochondrial activity and number of viable cells in culture. As shown in Fig. 2a, tube cells treated without DEP retained high ATP concentrations because of the normal mitochondrial function. Addition of DEP decreased ATP, causing the cells to cease basic metabolic functions and therefore die. Nevertheless, no significant differences were observed in ATP level between untreated and 1 μg/mL DEP-treated cells over the course of 24 h, which shows 26.88 and 25.73 μM, respectively. At 5, 10, and 50 μg/mL, the cells retained 80 (20.9 μM), 60 (17.1 μM), and 35 % (9.5 μM) of their original ATP, respectively. At 100 μg/mL DEP, the level of ATP was only 25 % (5.9 μM). However, NAC mitigated the mitochondrial damage induced by DEP. Even at 50 and 100 μg/mL of DEP, NAC was able to restore ATP up to 22 and 17 μM, respectively. Furthermore, since intracellular ATP concentration was reduced, actin cytoskeleton was depolymerized simultaneously after exposure to DEP (Fig. 2b). However, immunoblot results shown in Chao et al. [13] revealed an unaffected actin cytoskeleton in the protein level. The extent of tube formation function was quantified by measuring the tube length in Fig. 2b. As shown in Fig. 2c, untreated control and 10 μg/mL-treated samples showed the average tube length are 70 and 65 μm, respectively. At 100 μg/mL DEP, the length of tube formation significantly shrunk to 11 μm, while addition of NAC restored tube length back to 60 μm. Moreover, Chao et al. [13] has indicated that total cellular actin amount is unaffected in response to DEP exposure. Thus, these results together suggest that DEP might cause ATP depletion and actin cytoskeleton rearrangement, but it would not lead to decrease in actin expression. Meanwhile, derangement of actin cytoskeleton might initiate activation of apoptosis [23].

The effects of depleting intracellular ATP in response to DEP-induced ROS. a Measurement of ATP amount in the tube cells treated with DEP using the ATP depletion method. The values were normalized to the standard curve and presented as the concentration of intracellular ATP. b DEP-induced ROS caused actin cytoskeleton depolarization. Images showed the distribution of actin cytoskeleton in response to DEP (10 and 100 μg/mL) exposure for 24 h. Shown is a representative image from three independent experiments. Scale bar = 50 μm. Magnification is ×100. c Experiments were performed as in B, and the tube length was measured with using ImageJ. d Effects of DEP on PI3K/Akt expression. e Quantification showed that PI3K and pAkt were decreased in a dose-dependent manner, while Akt was unaffected by DEP. *p < 0.05 and **p < 0.01 compared with the control, mean ± SD, n = 3
Furthermore, ATP depletion might also decrease the level of phosphorylated Akt. As shown in Fig. 2d, the levels of PI3K and pAkt were significantly decreased by DEP treatment in a dose-dependent manner. Quantification (Fig. 2e) shows that PI3K was unchanged at 1 μg/mL DEP, but it significantly decreased to 0.8 and 0.6 of the control at 10 and 100 μg/mL, respectively. Again, pAkt was unaffected at 1 μg/mL DEP. However, at the higher concentrations of DEP (10 and 100 μg/mL), it declined to 0.3 and 0.18 of the control (p < 0.01), respectively. Furthermore, for the comparison of DEP exposure, the cells were treated with PI3K/Akt inhibitors Wortmannin (1 μM) and LY294002 (20 μM) for 24 h. NAC prevented 100 μg/mL DEP from causing PI3K inhibition and restored phosphorylation of Akt. These results suggest that DEP-induced ROS might diminish tube cell survivability by down-regulating PI3K/Akt activity. Although studies revealed that VEGFR2 functions with Akt to activate extracellular signal-regulated kinase (ERK1/2) as protection against caspase 3/7 expression and subsequent cell death [33], the MEK/ERK1/2 pathway was not detectable in our system (data not shown).
DEP Caused HUVEC Tube Cell Apoptosis
Chao et al. [12] indicated that exposure to DEP would cause capillary tube cell death. To characterize further mechanisms resulting in DEP-induced cell death, apoptotic cells were identified by TUNEL and Caspase-Glo-3/7 methods analysis. As shown in Fig. 3a, the cells treated with DEP manifested apoptotic cells (green spots) more frequently than did the untreated control. While addition of NAC, the DEP-caused apoptotic fraction was reduced. Figure 3b shows the quantification of apoptotic tube cells assessed from Fig. 3a by counting the number of green spots, dividing by the number of nuclei in the same field, and normalizing the quotient to per 100 cells. In the untreated control, 4 % of the cells underwent apoptosis. At 1 and 10 μg/mL DEP, 7 and 15 % apoptotic tube cells appeared, respectively. At 100 μg/mL, about 70 % of the tube cells were induced into apoptosis. Oppositely, DEP + NAC-treated samples showed <10 % of the cells in apoptosis. In contrast, the caspase 3 activity of tube cells exposed 24 h earlier to 1–100 µg/mL of DEP increased in a dose-dependent manner, to as much as five times that of the control (Fig. 3c). Additional NAC inhibited caspase 3 activation. These values suggest that DEP were able to cause cell death of the endothelial tube cells by inducing apoptosis.

DEP-induced ROS caused endothelial tube cell apoptosis. a Apoptosis detection was performed using the TUNEL assay, which was to analyze the accumulation of fragmental DNA labeled with fluorescent dUTP by TdT. The assays were visualized with immunofluorescence microscopy ×100 magnifications (Olympus IX51 Inverted Microscope) with emission at 495–529 nm. Nuclei were stained with DAPI, shown in the left panel. Green spots in the right panel presented apoptosis in each tube cell. Negative control was the tube cells treated without DEP (0 μg/mL). The cells exposed to UV (5 mJ/min) were defined as the positive control. Shown is a representative image from three independent experiments. b Semi-quantitation of TUNEL assay after DEP exposure. Values were divided by the amount of nuclei stain in the assessed region and expressed as the number of apoptosis events per 100 cells. *p < 0.05 and ***p < 0.001 compared with the control, means ± SD, n = 3. c CASPASE 3 activity measurement in the tube cells treated with DEP ± NAC, using a Caspase-Glo-3/7 assay kit. The values were normalized to the untreated control and shown as the percentage change relative to the control. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the negative control. § and † represent p < 0.01 and p < 0.001, respectively, and compared DEP-treated samples with DEP + NAC samples
DEP Mediated the Activation of p53/Mdm2 in Endothelial Tube Cells
Previous studies showed that DEP is able to induce tumor genesis by causing ROS production and DNA damage in an in vitro model [34, 35]. Upon DNA damage, p53 accumulates immediately through a posttranscriptional process that leads to growth arrest or apoptosis [36, 37]. However, p53 activity is feedback-inhibited by the oncoprotein Mdm2, which is transcribed by p53 and translocates into nuclei regulated by PI3K [38, 39]. Our findings (Fig. 4a) showed that both p53 and Mdm2 have a dose-dependent increase at 1 and 10 μg/mL DEP. However, at high dose of DEP, Mdm2 entirely disappeared. As shown in Fig. 4b, quantification analysis shows a similar protein expression level for both p53 (2.7-fold of control) and Mdm2 (2.6-fold of control) at 1 μg/mL DEP. Although the TUNEL assay showed that DEP induced merely 7 % tube cell apoptosis for this concentration of DEP, at 10 μg/mL DEP 15 % apoptosis was apparent (Fig. 3a). In addition, for the higher dose of DEP, there was no significant difference between p53 (5.5-fold of control) and Mdm2 (5.95-fold of control) protein. Over 70 % cell apoptosis was observed at 100 μg/mL DEP, when p53 was increased 8.5-fold, but Mdm2 merely showed 0.6-fold when compared to the controls. Thus, we propose that at low concentration, DEP might induce p53, which increases Mdm2 and in turn down-regulates p53 activity. However, 100 μg/mL DEP might cause severe DNA damage or destroy normal mitochondrial functions, consequently increasing p53 and decreasing Mdm2 levels. Furthermore, Bcl-XL, a pro-survival protein by preventing the release of mitochondrial cytochrome c, which would lead to caspase activation, was detected to represent mitochondrial function with using Western blot as well. And the expression level obviously declined to 0.7 and 0.2-fold of control at 10 and 100 μg/mL DEP, respectively. It matches the results that DEP cause a decrease in mitochondrial activity and an increase in caspase 3 activation results as shown in Figs. 1a and 3c.

DEP-mediated activation of p53/Mdm2 in endothelial tube cells. a The level of p53, Mdm2, p21, and Bcl-XL were determined by Western blot. b Quantification showed a dose-dependent increase in p53 and Mdm2 at 1 and 10 μg/mL as the relative change of the control. DEP continuously induced p53 at 100 μg/mL, which was accompanied by the disappearance of Mdm2. p21 was unaffected by DEP, and Bcl-XL decreased in a DEP dose-dependent manner. c Images show the distribution of Mdm2 and p53 in response to DEP (10 and 100 μg/mL), DEP (100 μg/mL) + NAC, or PFT-α (p53 inhibitor) exposure for 24 h. Tube cells were analyzed by Leica spectral confocal microscopy using anti-Mdm2 and anti-p53 as primary antibodies. Mdm2 and p53 subcellular localizations were visualized probing with secondary antibodies labeled with Alexa Fluor 488 (Green) and Texas-Red (Red), respectively. The left-side images are representative of an untreated control. Nuclei were stained with DAPI. Shown is a representative image from three independent experiments. Scale bar = 10 μm. Magnification is ×630. d The tube cells were treated with DEP ± Mdm2 inhibitor (nutlin-3a) or MDMX inhibitor (SJ172550), and their cell viability was determined with using MTS assay. *p < 0.05, **p < 0.01, and **p < 0.001 compared with the control, § and † represent p < 0.05 and p < 0.01, respectively, compared to DEP-treated samples, mean ± SD, n = 3. Statistical analysis was by student t test (Color figure online)
Alternatively, DEP might activate deubiquitinating enzymes (USP2a) that degrade various proteins including Mdm2, while at the same time stabilizing p53 activity [40]. USP2a might promote Mdm2 interaction with the nuclear corepressor KAP1 to contribute to p53 inactivation [41]. Further studies will be required to distinguish these various possibilities. However, p21, a known regulator of cell cycle, was unaffected by DEP exposure. Because tubes have a differentiated morphology more similar to in vivo capillaries, these in vitro cultures might respond to DEP in a more biologically relevant manner than do monolayer cultures. Previous studies suggested that cell viability was blocked when PI3K/Akt and MEK/ERK1/2 pathways were inhibited simultaneously [33], and, furthermore, ultrafine particles induced ROS, which leads to activation of endothelial MEK through induced phosphorylation of p38 and ERK1/2 [42]. However, MEK/ERK1/2 cannot be observed in our model (data not shown). These results might suggest that differentiated endothelial cells might lose their proliferative ability and stop going through the MEK/ERK1/2 pathway.
Mdm2 is well known to negatively regulate p53-mediated transcription. Mdm2 not only processes p53 response elements but can be activated by p53 as well [43]. Recent mechanistic studies and animal models have demonstrated how functional interactions in these p53 and Mdm2 networks are crucial for maintaining normal tissue homeostasis and for determining responses to oncogenic and therapeutic challenges [44–46]. Furthermore, PI3K/Akt were reported to serve as important anti-apoptotic signaling molecules by phosphorylating Mdm2 at Ser −166 and −186, and the resulting phospho-Mdm2 may localize into nuclei and serve to protect cells from p53-induced apoptosis [47, 48]. Thus, confocal microscopy confirmed the distribution of endogenous p53 and Mdm2 in this model. As shown in Fig. 4c, Mdm2 and p53 were unexpressed in the negative control. Addition of 10 μg/mL DEP caused tube cells to up-regulate Mdm2 (green), which overlapped with p53 (red) in the cytoplasm and nucleus. At 100 μg/mL DEP, the intensity of p53 was higher, but Mdm2 had entirely disappeared. This suggests that Mdm2 might function as a regulator to inhibit p53 at the lower dose of DEP (10 μg/mL) for maintaining normal capillary homeostasis. However, the cells might lose Mdm2 function because DEP (100 μg/mL) causes extreme cytotoxicity. p53 and Mdm2 expressions were attenuated in response to 100 μg/mL DEP plus additional NAC, demonstrating that ROS is central in the signaling mechanism to bring about endothelium death. As shown in Fig. 4d, addition of Mdm2 inhibitors (nutlin-3a and SJ172550) with DEP markedly reduces cell viability from 80–65 and 38 % (10 μg/mL) to 40–20 and 1 % (100 μg/mL). It means Mdm2 undoubtedly has capacity for 15 and 20 % cell survivability maintenance via p53 function inhibition at 10 and 100 μg/mL. Once herein Mdm2 function is blocked, p53 would exert apoptosis immediately [39]. Our findings here suggest that up-regulated Mdm2 may function as a regulator to feedback inhibition of p53 at lower doses of DEP (1 and 10 μg/mL). Meanwhile, at the highest dose of DEP (100 μg/mL), the tube cells may lose Mdm2 because of the extreme cytotoxicity of DEP. On the other hand, Mdm2 has been reported to maintain cell survivability by phosphorylating at Ser −166 and −186 and to inhibit p53 function by translocating from the cytoplasm to the nucleus [39]. Our supplementary fluorescent images show Mdm2 overlapped with p53 in both the nucleus and cytoplasm after 24-h treatment by Wortmannin and LY294002 (Fig. S1).
Conclusion
It is tempting to speculate that vasculature possesses mechanisms to modulate apoptosis when tube cells are exposed to DEP. Our results are consistent with a potential mechanism model of in vitro capillary tubes. Here, we suggest that DEP might cause mitochondrial oxidative stress and dysfunction and depolarize actin cytoskeleton rearrangement by depleting ATP. Furthermore, DEP inhibit PI3K activation and cause endothelium apoptosis with caspase 3 and p53 up-regulation. However, p53 and its regulator, Mdm2, act to form a feedback regulation loop for the maintenance of tube cell survivability at low doses of DEP. In contrast, tube cells augment the induction of apoptosis by inhibition of the negative regulator Mdm2 after exposure to a high dose of DEP. This result may also reflect a situation that if the endothelial cells have abnormal Mdm2, the vasculature system might lose the capacity against the damages generated from DEP. Even exposure to very low dose DEP might cause possible disorders. The endothelial tube model represents a simplistic view of endothelial DEP treatment, since there are no gaseous components of DEP being considered, nor is there any decent correlation with actual in vivo exposure levels. Still, despite these pitfalls, the data that can be obtained may illuminate different ideas and approaches for investigations of humans or animals exposed to DEP. There is a wealth of information on endothelial cell biology that should be used to uncover potential mechanisms of DEP toxicity.
Abbreviations
- DEP:
-
Diesel exhaust particles
- HUVEC:
-
Human umbilical vein endothelial cells
- ROS:
-
Reactive oxygen species
- NAC:
-
N-acetylcysteine
- PI3K:
-
Phosphoinositide 3-kinase
References
Oberdorster, G., & Utell, M. J. (2002). Ultrafine particles in the urban air: To the respiratory tract–and beyond? Environmental Health Perspectives, 110, A440–441.
Nel, A. (2005). Atmosphere. Air pollution-related illness: Effects of particles. Science, 308, 804–806.
Cheng, W. Y., Currier, J., Bromberg, P. A., Silbajoris, R., Simmons, S. O., & Samet, J. M. (2012). Linking oxidative events to inflammatory and adaptive gene expression induced by exposure to an organic particulate matter component. Environmental Health Perspectives, 120, 267–274.
Kumagai, Y., Arimoto, T., Shinyashiki, M., Shimojo, N., Nakai, Y., Yoshikawa, T., & Sagai, M. (1997). Generation of reactive oxygen species during interaction of diesel exhaust particle components with NADPH-cytochrome P450 reductase and involvement of the bioactivation in the DNA damage. Free Radical Biology & Medicine, 22, 479–487.
Wichmann, H. E. (2007). Diesel exhaust particles. Inhalation Toxicology, 19(Suppl 1), 241–244.
Samet, J. M. (2000). Epidemiology and policy: The pump handle meets the new millennium. Epidemiologic Reviews, 22, 145–154.
Pope, C. A, I. I. I., & Dockery, D. W. (2006). Health effects of fine particulate air pollution: Lines that connect. Journal of the Air & Waste Management Association(1995), 56, 709–742.
Pope, C. A, I. I. I., Renlund, D. G., Kfoury, A. G., May, H. T., & Horne, B. D. (2008). Relation of heart failure hospitalization to exposure to fine particulate air pollution. The American Journal of Cardiology, 102, 1230–1234.
Kelly, F. J., & Fussell, J. C. (2011). Air pollution and airway disease. Clinical and Experimental Allergy, 41, 1059–1071.
Ristovski, Z. D., Miljevic, B., Surawski, N. C., Morawska, L., Fong, K. M., Goh, F., & Yang, I. A. (2012). Respiratory health effects of diesel particulate matter. Respirology(Carlton Victoria), 17, 201–212.
Tseng, C.Y., Chang, J.F., Wang, J.S., Chen, Y.J., Gordon, M.K., & Chao, M.W. (2014). Contribution of diesel exhaust particles-induced intracellular ROS generated Pro-inflammatory cytokines to capillary-like endothelial tubes permeability. in press.
Chao, M. W., Po, I. P., Laumbach, R. J., Koslosky, J., Cooper, K., & Gordon, M. K. (2012). DEP induction of ROS in capillary-like endothelial tubes leads to VEGF-A expression. Toxicology, 297, 34–46.
Chao, M. W., Kozlosky, J., Po, I. P., Strickland, P. O., Svoboda, K. K., Cooper, K., et al. (2011). Diesel exhaust particle exposure causes redistribution of endothelial tube VE-cadherin. Toxicology, 279, 73–84.
Mates, J. M., Segura, J. A., Alonso, F. J., & Marquez, J. (2008). Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis, and carcinogenesis. Archives of Toxicology, 82, 273–299.
Garza, K. M., Soto, K. F., & Murr, L. E. (2008). Cytotoxicity and reactive oxygen species generation from aggregated carbon and carbonaceous nanoparticulate materials. International Journal of Nanomedicine, 3, 83–94.
Matsunaga, T., Arakaki, M., Kamiya, T., Endo, S., El-Kabbani, O., & Hara, A. (2009). Involvement of an aldo-keto reductase (AKR1C3) in redox cycling of 9,10-phenanthrenequinone leading to apoptosis in human endothelial cells. Chemico-Biological Interactions, 181, 52–60.
Matsuo, M., Shimada, T., Uenishi, R., Sasaki, N., & Sagai, M. (2003). Diesel exhaust particle-induced cell death of cultured normal human bronchial epithelial cells. Biological & Pharmaceutical Bulletin, 26, 438–447.
Hengstler, J. G., & Bolt, H. M. (2008). Oxidative stress: From modification of cell-cycle related events, secondary messenger function, dysregulation of small GTPases, protein kinases and phosphatases to redox-sensitive cancer models. Archives of Toxicology, 82, 271–272.
Mates, J. M., Segura, J. A., Alonso, F. J., & Marquez, J. (2012). Oxidative stress in apoptosis and cancer: An update. Archives of Toxicology, 86, 1649–1665.
Azad, M. B., Chen, Y., & Gibson, S. B. (2009). Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxidants & Redox Signaling, 11, 777–790.
Orrenius, S. (2007). Reactive oxygen species in mitochondria-mediated cell death. Drug Metabolism Reviews, 39, 443–455.
Shen, B., He, P. J., & Shao, C. L. (2013). Norcantharidin induced DU145 cell apoptosis through ROS-mediated mitochondrial dysfunction and energy depletion. PLoS One, 8, e84610.
White, S. R., Williams, P., Wojcik, K. R., Sun, S., Hiemstra, P. S., Rabe, K. F., & Dorscheid, D. R. (2001). Initiation of apoptosis by actin cytoskeletal derangement in human airway epithelial cells. American Journal of Respiratory Cell and Molecular Biology, 24, 282–294.
Rubtsova, S. N., Kondratov, R. V., Kopnin, P. B., Chumakov, P. M., Kopnin, B. P., & Vasiliev, J. M. (1998). Disruption of actin microfilaments by cytochalasin D leads to activation of p53. FEBS Letters, 430, 353–357.
Carmeliet, P., Lampugnani, M. G., Moons, L., Breviario, F., Compernolle, V., Bono, F., et al. (1999). Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell, 98, 147–157.
Kerr, B. A., Ma, L., West, X. Z., Ding, L., Malinin, N. L., Weber, M. E., et al. (2013). Interference with akt signaling protects against myocardial infarction and death by limiting the consequences of oxidative stress. Science Signaling, 6, ra67.
Robertson, S., Thomson, A. L., Carter, R., Stott, H. R., Shaw, C. A., Hadoke, P. W., et al. (2014). Pulmonary diesel particulate increases susceptibility to myocardial ischemia/reperfusion injury via activation of sensory TRPV1 and beta1 adrenoreceptors. Particle and Fibre Toxicology, 11, 12.
Sagai, M., Saito, H., Ichinose, T., Kodama, M., & Mori, Y. (1993). Biological effects of diesel exhaust particles. I. In vitro production of superoxide and in vivo toxicity in mouse. Free Radical Biology & Medicine, 14, 37–47.
Hiura, T. S., Li, N., Kaplan, R., Horwitz, M., Seagrave, J. C., & Nel, A. E. (2000). The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. The Journal of Immunology, 165, 2703–2711.
Wang, H. Z., Chang, C. H., Lin, C. P., & Tsai, M. C. (1996). Using MTT viability assay to test the cytotoxicity of antibiotics and steroid to cultured porcine corneal endothelial cells. Journal of Ocular Pharmacology and Therapeutics, 12, 35–43.
Niwa, Y., Hiura, Y., Murayama, T., Yokode, M., & Iwai, N. (2007). Nano-sized carbon black exposure exacerbates atherosclerosis in LDL-receptor knockout mice. Circulation Journal, 71, 1157–1161.
Yokota, S., Ohara, N., & Kobayashi, T. (2008). The effects of organic extract of diesel exhaust particles on ischemia/reperfusion-related arrhythmia and on pulmonary inflammation. The Journal of Toxicological Sciences, 33, 1–10.
Gomes, E., Papa, L., Hao, T., & Rockwell, P. (2007). The VEGFR2 and PKA pathways converge at MEK/ERK1/2 to promote survival in serum deprived neuronal cells. Molecular and Cellular Biochemistry, 305, 179–190.
Risom, L., Moller, P., & Loft, S. (2005). Oxidative stress-induced DNA damage by particulate air pollution. Mutation Research, 592, 119–137.
Murata, M., Ohnishi, S., Seike, K., Fukuhara, K., Miyata, N., & Kawanishi, S. (2004). Oxidative DNA damage induced by carcinogenic dinitropyrenes in the presence of P450 reductase. Chemical Research in Toxicology, 17, 1750–1756.
Levine, A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331.
Vousden, K. H., & Prives, C. (2009). Blinded by the light: The growing complexity of p53. Cell, 137, 413–431.
Freedman, D. A., Wu, L., & Levine, A. J. (1999). Functions of the MDM2 oncoprotein. Cellular and Molecular Life Sciences, 55, 96–107.
Mayo, L. D., & Donner, D. B. (2001). A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proceedings of the National Academy of Sciences, 98, 11598–11603.
Stevenson, L. F., Sparks, A., Allende-Vega, N., Xirodimas, D. P., Lane, D. P., & Saville, M. K. (2007). The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO Journal, 26, 976–986.
Wang, C., Ivanov, A., Chen, L., Fredericks, W. J., Seto, E., Rauscher, F. J, 3rd, & Chen, J. (2005). MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO Journal, 24, 3279–3290.
Mo, Y., Wan, R., Chien, S., Tollerud, D. J., & Zhang, Q. (2009). Activation of endothelial cells after exposure to ambient ultrafine particles: The role of NADPH oxidase. Toxicology and Applied Pharmacology, 236, 183–193.
Wu, X., Bayle, J. H., Olson, D., & Levine, A. J. (1993). The p53-mdm-2 autoregulatory feedback loop. Genes & Development, 7, 1126–1132.
Wade, M., Li, Y. C., & Wahl, G. M. (2013). MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nature Reviews, 13, 83–96.
Huang, L., Yan, Z., Liao, X., Li, Y., Yang, J., Wang, Z. G., et al. (2011). The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proceedings of the National Academy of Sciences, 108, 12001–12006.
Zhuang, C., Miao, Z., Zhu, L., Zhang, Y., Guo, Z., Yao, J., et al. (2011). Synthesis and biological evaluation of thio-benzodiazepines as novel small molecule inhibitors of the p53-MDM2 protein-protein interaction. European Journal of Medicinal Chemistry, 46, 5654–5661.
Ogawara, Y., Kishishita, S., Obata, T., Isazawa, Y., Suzuki, T., Tanaka, K., et al. (2002). Akt enhances Mdm2-mediated ubiquitination and degradation of p53. Journal of Biological Chemistry, 277, 21843–21850.
Manning, B. D., & Cantley, L. C. (2007). AKT/PKB signaling: Navigating downstream. Cell, 129, 1261–1274.
Acknowledgments
This work was supported by Grant from Ministry of Science and Technology (NSC-102-2320-B-033-001-MY3) in Taiwan. We thank Dr. Marion K. Gordon from Joint Program of Toxicology, EOHSI center, Rutgers University, for providing the DEP, which was collected from Dr. Masaru Sagai in Japan.
Conflict of interest
The authors have declared that no conflict of interests exist.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tseng, CY., Wang, JS., Chang, YJ. et al. Exposure to High-Dose Diesel Exhaust Particles Induces Intracellular Oxidative Stress and Causes Endothelial Apoptosis in Cultured In Vitro Capillary Tube Cells. Cardiovasc Toxicol 15, 345–354 (2015). https://doi.org/10.1007/s12012-014-9302-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-014-9302-y