
Abstract
Both increase in cardiac arrhythmia incidence and decrease in heart rate variability (HRV) have been described following human and experimental animal exposures to air pollutants. However, the potential causal relationship between these two factors remains unclear. Incidence of ventricular arrhythmia and HRV were evaluated during and after a 3 h period of Diesel engine exhaust exposure in ten healthy and ten chronic ischemic heart failure (CHF, 3 months after coronary ligation) Wistar rats using implantable ECG telemetry. Air pollutants were delivered to specifically designed whole body individual exposure chambers at particulate matter concentrations similar to those measured inside cabins of cars inserted in congested urban traffic. Recordings were obtained from unrestraint and unsedated vigil rats. Immediate decrease in RMSSD was observed in both healthy (6.64 ± 2.62 vs. 4.89 ± 1.67 ms, P < 0.05) and CHF rats (8.01 ± 0.89 vs. 6.6 ± 1.37 ms, P < 0.05) following exposure. An immediate 200–500% increase in ventricular premature beats was observed in CHF rats only. Whereas HRV progressively returned to baseline values within 2.5 h after exposure start, the proarrhythmic effect persisted as late as 5 h after exposure termination in CHF rats. Persistence of ventricular proarrhythmic effects after HRV normalization suggests that HRV reduction is not the mechanism of cardiac arrhythmias in this model. Our methodological approach, closely reflecting the real clinical situations, appeared to be a unique tool to provide further insight into the pathophysiological mechanisms of traffic related airborne pollution health impact.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Several epidemiological studies have shown a positive association between daily mortality and short-term increases in air pollution especially of particulate matter (PM) (Pope et al. 2004a, b; Samet et al. 2000). On a global scale, the World Health Organization has estimated that 800,000 deaths occur per year due to PM exposure. This increased mortality is mainly of cardiovascular origin. Impact of air pollution on cardiovascular disease thus represents a serious public health problem. Epidemiological studies also pointed out that elderly people and patients with pre-existing cardiorespiratory disease might be at much higher susceptibility than healthy population (Goldberg et al. 2001; von Klot et al. 2005). Accordingly, in several models of rats exposed to air PM, a higher toxic impact was observed in diseased animals (Campen et al. 2002; Wellenius et al. 2002). As in human studies, both increase in cardiac arrhythmia and decrease in heart rate variability (HRV) have been described following exposure to air pollutants. However, the potential causal relationship between these two factors remains unclear. In these experimental studies, air PM was mainly constituted of residual oil fly ash (ROFA). Although being of high interest, these studies with ROFA, or concentrated US ambient air particles, may not be representative of road traffic emissions with a high contribution of Diesel PM and nitrogen oxides are the main source of pollutant at least for European countries. In addition, studies dealing with PM have usually been performed in the absence of the associated gas phase, and/or in sedated or restraint animals which may limit the interpretation of data due to potential interference of these experimental conditions with cardiac control pathways. Recent epidemiological findings (Samoli et al. 2006) also point to a significant role of NO2 as a trigger of acute cardiac insults thus questioning the potential impact of gas phase components on cardiac responses. In order to overcome these methodological problems, we designed an experimental setup allowing to expose unrestraint vigil rats to a continuous flow of continuously sampled and diluted Diesel engine exhausts, with preserved physicochemical properties as a model for “in traffic” pollutant exposure. These diluted emissions were delivered to specifically designed whole body exposure chambers. Importantly, polluted air emissions were delivered at PM concentrations similar to those measured inside cars inserted in urban traffic (Gouriou et al. 2004) in order to mimic the real life situation. The aim of the study was to assess the impact of urban traffic air pollution on arrhythmia incidence and on heart rate variability recorded by ECG telemetry during and after diluted Diesel engine exhaust inhalation exposure in both healthy and chronic ischemic heart failure (CHF) rats to further describe the possible higher susceptibility of chronic heart failure over healthy rodents to airborne pollution.
Materials and methods
Animals
Adult male Wistar rats weighing 200–225 g (Charles River, France) were maintained and studied in accordance with national guidelines for care and use of animals. They were housed (12-h light/dark cycle) in plastic cages and had free access to tap water and standard rat diet chow (UAR, France). All experiments are conducted according to the Helsinki protocol rules.
Surgical protocols
Myocardial infarction
Ischemic myocardial infarction was performed according to the method of Pfeffer et al. (1985) and modified by our laboratory (Mulder et al. 1997). This model allowed to reproduce experimentally hemodynamic and biological conditions of chronic heart failure (Mulder et al. 2004). Briefly, 10-week-old male Wistar rats were anesthetized with 80 mg/kg ketamine and 5 mg/kg xylasine, and ventilated 60 cycles/min with a 1 ml/100 g body weight tidal volume. After a left thoracotomy and pericardectomy, ligation of left descending coronary artery was performed 2 mm below its origin (6–0 Prolene). Myocardial ischemia was confirmed visually by cyanosis. About 10 min after ligation, thorax was closed and pneumothorax evacuated. Rats were allowed to wake up and recover in individual cages.
Sham rats underwent thoracotomy and pericardectomy and no ligation constitute the healthy rat group.
Telemetry sensor implantation
Two months after cardiac surgery, telemetry sensor implantation was performed under general anesthesia with 80 mg/kg of ketamine and 5 mg/kg of xylasine. Physiotel TA-F40 telemetry sensors (Transoma Medical, USA) were implanted intra-peritoneally. Lead wires were conducted subcutaneously to place current sensitive extremities in a pseudo lead II position, which allowed to record the best ECG signals for further quantification. Rats were allowed to recover for at least 8 days post-implantation before experimentation.
Pollution exposure
Experiments were started 3 months after left coronary ligation. Previous studies from our laboratory have shown that at this stage rats are in chronic heart failure, with reduced cardiac output and left ventricular shortening, cardiac hypertrophy and infarct zone fibrosis (Mulder et al. 2004). To investigate the cardiac impact of Diesel engine emissions, rats were exposed for 3 h to either clean room air or for constant flow of continuously sampled and diluted emissions from a monocylinder Diesel engine using Euro 4 ELF 85A reference gasoline. A two step dilution procedure was used to prepare reproducible and constant dilution ratio (1:50) leading to PM concentration of 0.5 mg/m3, (AVL 415 opacimeter) PM levels which were routinely measured by our group inside cars inserted in the urban traffic flow or in road tunnels (Gouriou et al. 2004). Other regulated emissions levels measured in inhalation cages (Horiba analyzer) were 7.7 ppm for non-methane hydrocarbons, 1.1 ppm for NO2 and 4.3 ppm for carbon monoxide.
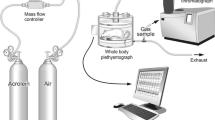
The dedicated inhalation chamber design has been calculated using 2D fluid dynamics computational software in order to deliver homogeneous emission concentration throughout the whole rat housing volume. Attention was paid to avoid transfer of potentially stressful noise from engine exhaust line to the exposure chamber (Pictures 1, 2).

Flow velocities in the chamber design. Calculations were made using Fluent 3D software. Aerosol inlet from the left handside. Clear recirculation zones are seen close to the inlet (left handside) while in the right handside where rats are to be housed, constant velocities are found suggesting homogeneous aerosol distribution and concentrations throughout the housing volume. Authors are indebted to Dr. A. Konstandopoulos (CERTH-CPERI Thessaloniki Grece) for calculating these data using Fluent 3D software

Continuous flow through whole body inhalation cages. Rats were individually housed for individual telemetry purpose. Cages were made of PVC and polycarbonate to be suitable for telemetry signal recordings. Receprion antenna are placed under the rat housing volume for most efficient telemetry signal recording
Particulate matter size distributions were measured in the exposure chambers under experimental conditions using scanning mobility particle sizer (SMPS) which operates for particle sizes ranging from 10 to 650 nm. Figure 1 shows typical spectrum demonstrating that rats were exposed to a typical submicron combustion PM aerosol with a mean mobility diameter of 85 nm. Particulate matter mass concentration was calculated from both SMPS and AVL 415 opacimeter which exhibited highly correlated measurements.

Size distribution curve of particulate matter. Typical size distribution curve of particulate matter recorded in exposure chamber: showing log normal distribution centered on a mean mobility diameter of 85 nm. Measurement performed using a scanning mobility particle sizer
Electrocardiographic data acquisition and analysis
Electrocardiogram signals transmitted from Physiotel sensors (Transoma Medical) were collected through RC1 antenna and stored in a PC using ART gold 2.0 acquisition system (Transoma Medical) after digitalization (1,000 Hz). Stable ECG recordings were of high quality in both healthy and CHF rats and measures were consistent for successive 5 min step analysis.
Digital ECG tracings were analyzed using EMKA ECG-Auto 1.5.12.0 software which allowed P-QRS-T shape recognition and HRV analysis. Ventricular extrasystoles (VPB) were recognized as abnormal shape premature beats with increased QRS duration. An automated procedure of arrhythmia beat invalidation (the arrhythmia beat and the following normal beat) was performed allowing extraction of the normal sinus beat interval from the raw time series (Fig. 2a, b). Less than 15% of total beats were invalidated as arrhythmia beats during this “cleaning process”. Then, beat-to-beat analysis allowed construction of temporal series of normal RR intervals (NN intervals). The variability of these NN intervals was evaluated using RMSSD (the square root of the mean squared differences of successive NN intervals) according to the recommendations of the taskforce of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. We have chosen to analyze long period of time in uncontrolled conditions (unconstraint and non-sedated rats), SDNN or HRV triangular index were not used in our study since they appeared to be highly dependent on rat physical activity (mobility or resting phases) more than on actual cardiac autonomic control (data not shown).

Poincaré representation of RR intervals. a Poincaré representation from raw RR times series. b Poincaré representation from cleaned up RR time series
Study protocol
Rats were individually placed in the inhalation chambers 1 h before starting the experiment. ECG recording started at t 0 and the exhaust exposure at t 30 minutes for a 3 h period. Cage volume was 28 l and diluted aerosol flowrate was 10 l/min thus ensuring ca. 20 cage volume renewal per hour (Pictures 1, 2).
The number of VPBs, as well as RMSSD was calculated for each 30 min period during normal air (baseline) exhaust and post-exhaust exposure periods according to taskforce recommendations (1996). An early (from t 210 to t 300 min) and a late (from t 480 to t 540 min) post-exposure periods were analyzed. The number of VPBs observed during the 30 min baseline period prior to exposure was considered as 100% for each rat and referred as its own baseline. From our experience, analysis steps of 30 min were required to minimize the impact of animal behavior on HRV parameters, especially on SDNN and HRV triangular index which appear to be very sensitive to physical activity.
Statistical analysis
The effect of PM exposure on ventricular arrhythmia frequency was assessed by comparing the number of VPBs recorded during the baseline with that of the recorded one during exposure and post-exposure periods in each animal. The RMSSD was compared for the same periods. The Mann–Whitney non-parametric test was used for statistical analysis. Results were considered significant when the P value was less than 0.05.
Results
ECG changes in sham rats
Neither mean RR duration (191.3 ± 12.3 vs. 197.8 ± 17.62, P = ns, Fig. 3) nor QT interval duration (56 ± 5.35 vs. 52.43 ± 1.35, P = ns) did significantly change during the course of exposure compared to the pre-exposure period.
No arrhythmia occurrence was evidenced during engine emission exposure in healthy rats.

Mean RR interval duration. Mean RR interval duration at baseline (Air) and during Diesel exhaust exposure (Exhaust), in both normal and CHF rats
However, a significant reduction in RMSSD (6.64 ± 2.62 vs. 4.89 ± 1.67 ms, P < 0.05, Fig. 4) was observed, which was graphically visible on Poincaré plots as a thinning distribution upon exposure (Fig. 2). Poincaré plot is a first return application on RR time series. Thinning of the distribution corresponds to a reduction of beat-to-beat variation, which is directly correlated to RMSSD reduction.

Heart rate variability assessment. Mean RMSSD (in ms) at baseline (Air) and during Diesel exhaust exposure (Exhaust), in both normal and CHF rats. *Statistical significance P < 0.05
ECG changes in CHF rats
As in healthy rats, neither mean heart rate (198.3 ± 10.35 vs. 192.0 ± 11.61 ms, P = ns, Fig. 3) nor QT segment duration (77.1 ± 2.95 vs. 76.7 ± 1.95 ms, P = ns) did significantly change during the course of exposure compared to the pre-exposure period.
Of note, QT interval duration was significantly prolonged in CHF as compared to healthy rats both at baseline (77.1 ± 2.95 vs. 56 ± 5.35 ms, P < 0.001) and during exposure (76.7 ± 1.95 vs. 52.43 ± 1.35 ms, P < 0.001).
Conversely, a marked impact of engine emissions exposure was observed on VPB incidence (Fig. 5), which increased by a factor of 2–5. The increase of VPB frequency was already significant as early as 30 min after the onset of exposure and lasted for at least 4–5 h after exposure termination.

Arrhythmia assessment in CHF rats. Percentage of increase in VPBs over time as compared to baseline value in CHF rats (VPBs % control period). Gray columns represent the Diesel exhaust exposure period
All CHF rats showed increased frequencies of isolated VPBs. However, rats exhibiting the highest baseline numbers of VPBs tend to develop more complex arrhythmia as VPB couplets and triplets, ventricular bigeminy, non-sustained ventricular tachycardia episodes which were recognized on Poincaré plot as more complex figures. Over the entire period of Diesel engine emissions exposure, RMSSD (Fig. 4) was significantly reduced compared to baseline (8.01 ± 0.89 vs. 6.6 ± 1.37 ms, P < 0.05).
When RR intervals and HRV parameter were calculated every 30 min, we observed a marked and immediate decrease of RMSSD with a return to baseline values within 2 h (Fig. 6a, b). Of interest, RMSSD values were similar to those measured at baseline, 4 h after engine emission exposure termination.

Time-course evolution of RR interval duration and RMSSD. a Evolution of the mean RR interval duration over time in CHF rats. Gray columns represent the Diesel exhaust exposure period. Note that the mean RR interval duration was significantly reduced during the first 30 min of exposure only (*P < 0.05). b Evolution of the mean RMSSD over time in CHF rats. Gray columns represent the Diesel exhaust exposure period. Note that the mean RMSSD was significantly reduced during the first 2 h of exposure († P < 0.001; *P < 0.05) and then returned within the baseline values
HRV impacts in Sham and CHF rats
There was no statistical difference in HRV reduction amplitude nor kinetics between exposed sham and CHF rats.
Discussion
In this experimental study, we have observed that inhalation of ambient air polluted with diluted Diesel engine emissions at concentrations similar to those observed in congested urban traffic induced immediate autonomic dysfunction in both healthy and chronic heart failure rats. In addition, a significant immediate and persistent pro-arrhythmic effect was observed in diseased rats only. We have observed here for the first time that modification of HRV was dissociated in time from the proarrhythmic effect induced by Diesel exhaust exposure.
Methodological breakthrough
Because of an easier technical feasibility, many of the experimental studies evaluating the potential proarrhythmic effect of ambient air pollution were performed on sedated (Wellenius et al. 2002, 2004) or anesthetized animals (Nemmar et al. 2002a, b, 2003)). Although experiments using intrapulmonary instillation provided insight into basic mechanisms, the ability to extrapolate findings to humans is limited owing to the route of administration of usually high quantity particulate matter. Extrapolation from experiments using sedated animals is also difficult because the status of sedation does not correspond to the real life and the drugs used for sedation are known, even at low doses, to interact with the autonomic nervous system control and may impact HRV control. In our study, we used a whole body individual inhalation chamber and an ECG telemetry system in order to avoid these confounding factors. This method allowed us to reproducibly expose unconstraint rats to ambient Diesel combustion aerosol at concentrations similar to those measured in urban traffic, and to analyze the dynamics of arrhythmia events as well as heart rate variability on long-term ECG recordings.
Heart rate variability
Previous investigations have shown that cardiopulmonary compromised rats exhibited greater changes in ECG parameters upon PM exposure compared to normal animals. Campen et al. (2003) reported that exposure to Diesel exhaust 6 h/day for seven consecutive days led to an increased heart rate in spontaneously hypertensive rats as compared to controls (290 vs. 265 bpm). These findings were also observed by Chang et al. (2003) using repeated exposures to concentrated particulate matter (PM 2.5) in spontaneous hypertensive rats. In a rat model of acute myocardial infarction, Wellenius et al. (2004) have reported a significant decrease in heart rate variability during exposure to residual oil fly ash particles. In contrary to our observations, this phenomenon was not observed in their Sham operated animals.
In our study, HRV assessed by RMSSD measurement was significantly reduced in both CHF and healthy rats when measured over the entire period of Diesel exhaust exposure. This discrepancy might be explained by the difference in the methodological approach and the use of sedation by benzodiazepine in Wellenius study. As mentioned by the authors, because of its direct myocardial action (reduction in contractility and oxygen consumption), benzodiazepine could have masked the cardiac effects of particulate matter exposure, especially in potentially less sensitive animals like healthy rats.
Since the decrease in RMSSD occurred immediately after Diesel exhaust exposure, our observation supports the hypothesis of an activation of pulmonary neural reflexes secondary to a direct aerosol interactions with lung receptors.
Induction of ventricular arrhythmia by diluted diesel engine exhaust exposure in CHF rats
Although air pollutants have been shown to induce cardiac arrhythmias in few animal models (Wellenius et al. 2002; Nadziezko et al. 2004), we demonstrated here that urban traffic air pollution induced ventricular proarrhythmic effect in unrestraint CHF animals. A significant increase in VPBs was documented immediately following Diesel engine combustion aerosol exposure in CHF rats. This level of increase in VPBs remained stable over the time of exposure and persisted up until at least 5 h after the end of exposure. Conversely, no such cardiac impact was observed in healthy rats. Nevertheless, Watkinson et al. (1998) reported that instillation of residual oil fly ash caused dose-related increases in the incidence and duration of serious cardiac arrhythmic events in normal rats. This suggests that the absence of proarrhythmic effect in our healthy rats was likely due to either a difference of particulate matter source and chemical composition, and/or most probably the rather low concentration of air pollutants we used in our study compared to the instilled doses. Our experimental study thus underlines the notion of levels of sensitivity of the studied population to air pollution and to some extent mimics the clinical or epidemiological situation.
Mechanisms of ventricular arrhythmia provocation
In contrast to prior studies, our methodological approach allowed us to analyze the dynamics of both HRV modification and arrhythmia occurrence over several hours during and after Diesel exhaust exposure. Although decrease in HRV and induction of cardiac arrhythmia have been documented during air pollutants exposure, the relationship between these two phenomenon has never been elucidated. In our study, a rapid and pronounced reduction in HRV was observed within 30 min after the beginning of exposure, followed by a progressive and linear normalization and a return to the baseline values at 2.5 h. Persistence of ventricular proarrhythmic effects after HRV parameter normalization suggests that HRV reduction is not the mechanism of cardiac arrhythmias in this model. HRV reduction could possibly promote cardiac arrhythmia at the initial phase of Diesel combustion aerosol exposure. However, in our study, the increase in VPBs was not superior during the early phase of exposure as compared to the remaining recording period. Based on these results, HRV modification is likely to be a marker of pollution exposure rather than a mechanism of cardiac arrhythmia provocation. In addition, since HRV modification was documented without ventricular proarrhythmic effect during exposure to Diesel engine combustion aerosol in normal animal, this marker of air pollutant exposure appeared to be more sensitive than arrhythmia development. Myocardial ischemia has also been proposed as one of the potential mechanism of cardiac arrhythmia following air pollutants exposure. In our experiment, no significant ST segment modification was observed which does not support myocardial ischemia as the cause of arrhythmia. Although QT interval was significantly prolonged in CHF rats, no further QT interval prolongation was documented during air pollution exposure. These results excluded modification of ventricular depolarization as a mechanism of arrhythmia. The rapid initiation of proarrhythmic effect following air pollution exposure supports a potential direct myocardial effect via agents (i.e., gases, ultrafine particles, soluble constituents of PM) that may cross the pulmonary epithelium into the circulation (Nemmar et al. 2002a, b). The persistence of increase VPBs late after exposure termination may be related to indirect effects of air pollutants mediated through pulmonary oxidative stress (Kelly 2003) and inflammatory responses (Salvi et al. 1999). Preliminary data from our laboratory to be published elsewhere revealed that increased plasma levels of TNFalpha and increased activities of renal and hepatic anti-oxidant enzymes were observed in exposed animals thus confirming the occurrence of systemic inflammation and oxidant stress in our experimental conditions. Indeed, Diesel emissions have been shown to induce oxidant stress in vitro on organotypic cultures of lung tissue, which was attributed to gas phase components and pro-inflammatory reaction which was attributed to particulate matter (Bion et al. 2002). These impacts seem to occur in vivo after inhalation exposure and may further imbalance the pertubated redox status in CHF rats.
Clinical implications
The present results corroborated the epidemiological study of Mann et al. (2002), in which people with ischemic heart disease and accompanying CHF and/or cardiac arrhythmia constituted a sensitive subgroup for hospitalization in relation to the effects of air pollutants associated with combustion engine emissions. These are also in accordance with the increased number of implantable cardiac defibrillator (ICD) interventions that have been observed in 100 ICD patients few days after an increase in NO2 concentration (Peters et al. 2000). In this study, increased arrhythmias were associated with NO2, CO, black carbon and fine particle mass, in sicker patients (with ten or more ICD interventions). From our data, where CHF rats exhibit similar clinical and functional symptoms as human undergoing myocardial infarction, we could extrapolate that in patients with CHF and pre-existing cardiac arrhythmia, exposure to combustion engine emissions would likely increase the risk of fatal ventricular arrhythmia. Besides the claimed impact of particulate matter, an increase in coefficient of haze and NO2 concentration has been found to be associated with an increase in daily mortality among CHF patients (Goldberg et al. 2003; von Klot et al. 2006), this hypothesis seems to be further supported by the recent epidemiological findings (Samoli et al. 2006) which however should be validated by further clinical studies. These observations point to a multipollutant potential impact and to the necessity of a more global approach of airborne pollutant health related issues.
In several epidemiological studies (Devlin et al. 2003; Pope et al. 2004b) an association between an increase in various types of air pollutants and a reduction in HRV has been documented, especially in the elderly population. Modifications in HRV appeared to persist for up to 24 h after exposure. However, this association was not found in all studies (Sullivan et al. 2005). These discrepancies might be related to variability in air pollutant sources involved and to the methodology of HRV measurement which varied from one study to the other. The absence of a strong and constant association between air pollution and HRV modification rather argues against an increased cardiovascular morbidity and mortality due to HRV reduction. Since HRV reduction and increase in ventricular arrhythmia were dissociated in time, the results of our study support this hypothesis.
Conclusion
An immediate and persistent ventricular proarrhythmic effect has been demonstrated during and after inhalation exposure to diluted Diesel engine emissions in CHF rats only. A marked decrease in HRV was observed during the early phase of exposure only, irrespective of the presence of CHF, which did not support HRV modification as the mechanism of ventricular arrhythmia provocation. Our methodological approach, closely reflecting the real clinical situations appeared to be a unique tool to provide further insight into the pathophysiological mechanisms involved in traffic related air pollution health impacts.
Abbreviations
- CHF:
-
Chronic heart failure (at least 3 months after left coronary artery ligation)
- ECG:
-
Electrocardiogram
- HRV:
-
Heart rate variability
- RMSSD:
-
The square root of the mean squared differences of successive sinusal beats
- SDNN:
-
Standard deviation of sinusal beat-to-beat intervals
- SMPS:
-
Scanning mobility particle sizer (apparatus measuring particle number and size distribution)
References
Bion A, Fall M, Gouriou F, Le Prieur E, Dionnet F, Morin JP (2002) Biphasic culture of rat lung slices for pharmacotoxicological evaluation of complex atmospheres. Cell Biol Toxicol 18:301–314
Campen MJ, Nolan JP, Schladweiler MC, Kodavanti UP, Costa DL, Watkinson WP (2002) Cardiac and thermoregulatory effects of instilled particulate matter-associated transition metals in healthy and cardiopulmonary-compromised rats. Toxicol Environ Health A 65(20):1615–1631
Campen MJ, McDonald JD, Gigliotti AP, Seilkop SK, Reed MD, Benson JM (2003) Cardiovascular effects of inhaled diesel exhaust in spontaneously hypertensive rats. Cardiovasc Toxicol 3(4):353–361
Chang CC, Hwang JS, Chan CC, Wang PY, Hu TH, Cheng TJ (2004) Effects of concentrated ambient particles on heart rate, blood pressure, and cardiac contractility in spontaneously hypertensive rats. Inhal Toxicol 16(6–7):421–429
Devlin RB, Ghio AJ, Kehrl H, Sanders G, Cascio W (2003) Elderly humans exposed to concentrated air pollution particles have decreased heart rate variability. Eur Respir J Suppl 40:76s–80s
Goldberg MS, Burnett RT, Bailar JC 3rd, Tamblyn R, Ernst P, Flegel K, Brook J, Bonvalot Y, Singh R, Valois MF, Vincent R (2001) Identification of persons with cardiorespiratory conditions who are at risk of dying from the acute effects of ambient air particles. Environ Health Perspect 109(Suppl 4):487–494
Goldberg MS, Burnett RT, Valois MF, Flegel K, Bailar JC 3rd, Brook J, Vincent R, Radon K (2003) Associations between ambient air pollution and daily mortality among persons with congestive heart failure. Environ Res 91(1):8–20
Gouriou F, Morin JP, Weill ME (2004) On road measurements of particle number concentrations and size distributions in urban and tunnel environments. Atmos Environ 38:2831–2840
Kelly FJ (2003) Oxidative stress: its role in air pollution and adverse health effects. Occup Environ Med 60:612–616
Mann JK, Tager IB, Lurmann F, Segal M, Quesenberry CP Jr, Lugg MM, Shan J, Van Den Eeden SK (2002) Air pollution and hospital admissions for ischemic heart disease in persons with congestive heart failure or arrhythmia. Environ Health Perspect 110(12):1247–1252
Mulder P, Richard V, Derumeaux G, Hogie M, Henry JP, Lallemand F, Compagnon P, Mace B, Comoy E, Letac B, Thuillez C (1997) Role of endogenous endothelin in chronic heart failure: effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation 96(6):1976–1982
Mulder P, Barbier S, Chagraoui A, Richard V, Henry JP, Lallemand F, Renet S, Lerebours G, Mahlberg-Gaudin F, Thuillez C (2004) Long-term heart rate reduction induced by the selective I(f) current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation 109(13):1674–1679
Nadziejko C, Fang K, Narciso S, Zhong M, Su WC, Gordon T, Nadas A, Chen LC (2004) Effect of particulate and gaseous pollutants on spontaneous arrhythmias in aged rats. Inhal Toxicol 16(6–7):373–380
Nemmar A, Hoet M, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B (2002a) Passage of inhaled particles into the blood circulation in humans. Circulation 105:411–414
Nemmar A, Hoylaerts MF, Hoet PH, Dinsdale D, Smith T, Xu H, Vermylen J, Nemery B (2002b) Ultrafine particles affect experimental thrombosis in an in vivo hamster model. Am J Respir Crit Care Med 166(7):998–1004
Nemmar A, Hoet PH, Dinsdale D, Vermylen J, Hoylaerts MF, Nemery B (2003) Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation 107(8):1202–1208
Peters A, Liu E, Verrier RL, Schwartz J, Gold DR, Mittleman M, Baliff J, Oh JA, Allen G, Monahan K, Dockery DW (2000) Air pollution and incidence of cardiac arrhythmia. Epidemiology 11(1):11–17
Pfeffer MA, Pfeffer JM, Steinberg C, Finn P (1985) Survival after an experimental myocardial infarction: beneficial effects of long-term therapy with captopril. Circulation 72(2):406–412
Pope CA 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ (2004a) Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation 109(1):71–77
Pope CA 3rd, Hansen ML, Long RW, Nielsen KR, Eatough NL, Wilson WE, Eatough DJ (2004b) Ambient particulate air pollution, heart rate variability, and blood markers of inflammation in a panel of elderly subjects. Environ Health Perspect 112(3):339–345
Salvi S, Blomberg A, Rudell B, Kelly F, sandstorm T, Holgate ST, Frew A (1999) Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med 159:702–709
Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL (2000) Fine particulate air pollution and mortality in 20 US cities, 1987–1994. N Engl J Med 343(24):1742–1749
Samoli E, Aga E, Touloumi G, Nisiotis K, Forsberg B, Lefranc A, Pekkanen J, Wojtyniak B, Schindler C, Niciu E, Brunstein R, Dodic Fikfak M, Schwartz J, Katsouyanni K (2006) Short-term effects of nitrogen dioxide on mortality: an analysis within the APHEA project. Eur Respir J 27:1129–1137
Sullivan JH, Schreuder AB, Trenga CA, Liu SL, Larson TV, Koenig JQ, Kaufman JD (2005) Association between short term exposure to fine particulate matter and heart rate variability in older subjects with and without heart disease. Thorax 60(6):462–466
Task Force European Society Cardiology (1996) Heart rate Variability: standards of measurements, physiological interpretations and clinical use. Circulation 93:1043–1065
Von Klot S, Peters A, Aalto P, Bellander T, Berglind N, D’Ippoliti D, Elosua R, Hormann A, Kulmala M, Lanki T, Lowel H, Pekkanen J, Picciotto S, Sunyer J, Forastiere F (2005) Health effects of particles on susceptible subpopulations (HEAPSS) study group. Ambient air pollution is associated with increased risk of hospital cardiac readmissions of myocardial infarction survivors in five European cities. Circulation. 112(20):3073–3079. Erratum in: Circulation 2006 Feb 7;113(5):e71
Watkinson WP, Campen MJ, Costa DL (1998) Cardiac arrhythmia induction after exposure to residual oil fly ash particles in a rodent model of pulmonary hypertension. Toxicol Sci 41:209–216
Wellenius GA, Saldiva PH, Batalha JR, Krishna Murthy GG, Coull BA, Verrier RL, Godleski JJ (2002) Electrocardiographic changes during exposure to residual oil fly ash (ROFA) particles in a rat model of myocardial infarction. Toxicol Sci 66(2):327–335
Wellenius GA, Batalha JR, Diaz EA, Lawrence J, Coull BA, Katz T, Verrier RL, Godleski JJ (2004) Cardiac effects of carbon monoxide and ambient particles in a rat model of myocardial infarction. Toxicol Sci 80(2):367–376
Acknowledgments
Financial supports from EC QLK4-CT62002-02357 MAAPHRI program, ADEME, Direction Generale de la Sante and Région Haute Normandie are greatly acknowledged. Dr Konstandopoulos (CERTH Thessaloniki Grece) for fluid dynamics computation and skilled technical assistance from Mrs. Annie Lejeune are greatly acknowledged
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anselme, F., Loriot, S., Henry, JP. et al. Inhalation of diluted diesel engine emission impacts heart rate variability and arrhythmia occurrence in a rat model of chronic ischemic heart failure. Arch Toxicol 81, 299–307 (2007). https://doi.org/10.1007/s00204-006-0147-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-006-0147-4