
Abstract
Primary lateral sclerosis (PLS) is classically considered a ‘pure’ upper motor neuron disorder. Motor cortex atrophy and pyramidal tract degeneration are thought to be pathognomonic of PLS, but extra-motor cerebral changes are poorly characterized. In a prospective neuroimaging study, forty PLS patients were systematically evaluated with a standardised imaging, genetic and clinical protocol. Patients were screened for ALS and HSP associated mutations, as well as C9orf72 hexanucleotide repeats. Clinical assessment included composite reflex scores, spasticity scales, functional rating scales, and screening for cognitive and behavioural deficits. The neuroimaging protocol evaluated cortical atrophy patterns, subcortical grey matter changes and white matter alterations in whole-brain and region-of-interest analyses. PLS patients tested negative for known ALS- and HSP-associated mutations and C9orf72 repeat expansions. Voxel-wise analyses revealed anterior cingulate, dorsolateral prefrontal, insular, opercular, orbitofrontal and bilateral mesial temporal grey matter changes and white matter alterations in the fornix, brainstem, temporal lobes, and cerebellum. Significant thalamus, caudate, hippocampus, putamen and accumbens nucleus volume reductions were also identified. Extra-motor clinical manifestations were dominated by verbal fluency deficits, language deficits, apathy and pseudobulbar affect. Our clinical and radiological evaluation confirms considerable extra-motor changes in a population-based cohort of PLS patients. Our data suggest that PLS should no longer be considered a neurodegenerative disorder selectively affecting the pyramidal system. PLS is associated with widespread extra-motor changes and manifestations which should be carefully considered in the multidisciplinary management of this low-incidence condition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
ALS amyotrophic lateral sclerosis.
ALSFRS-r revised amyotrophic lateral sclerosis functional rating scale.
ANCOVA analysis of covariance.
ASS-m modified Ashworth spasticity scale score.
C9orf72 chromosome 9 open reading frame 72.
CNS-LS Center for neurological study-lability scale.
DTI Diffusion Tensor Imaging.
ECAS Edinburgh cognitive and behavioral screen.
ELQ emotional lability questionnaire.
EMM estimated marginal mean.
ELQ Emotional Lability Questionnaire.
FLAIR Fluid-attenuated inversion recovery.
FOV field of view.
FrSBe frontal systems behaviour scale.
FWE familywise error.
GM grey matter.
HADS hospital anxiety and depression scale.
HC healthy control.
HSP hereditary spastic paraplegia.
IR-SPGR inversion recovery prepared spoiled gradient recalled echo.
IR-TSE inversion recovery turbo spin echo sequence.
LMN lower motor neuron.
Lt Left.
MND Motor neuron disease.
MNI152 Montreal Neurological Institute 152 standard space.
MRC Medical Research Council Scale for Muscle Strength.
PBA Pseudobulbar affect.
PCC Pathological crying and laughing.
PLS primary lateral sclerosis.
PUMNS Penn Upper Motor Neuron Score.
ROI region of interest.
Rt Right.
SD standard deviation.
SE-EPI spin-echo echo planar imaging.
SENSE Sensitivity Encoding.
SPIR spectral presaturation with inversion recovery.
T1W T1-weighted imaging.
TE Echo time.
TFCE threshold-free cluster enhancement.
TI Inversion time.
TIV total intracranial volume.
TR repetition time.
UMN Upper motor neuron.
Introduction
Primary lateral sclerosis (PLS) is a low-incidence motor neuron disease characterized by slowly progressive upper motor neuron (UMN) dysfunction (Statland et al. 2015). As clinical signs are dominated by striking upper motor neuron signs, and PLS has a much lower incidence than ALS, there is a prevailing notion that primarily lateral sclerosis solely affects the pyramidal system. The term ‘PLS-plus’ has been sporadically used to describe cases with extra-motor features (Gordon et al. 2006), but unlike in ALS (Burke, Elamin, et al. 2016; Elamin et al. 2013), no systematic studies have been performed to specifically characterize extra-motor disease burden in PLS. While extra-motor manifestations have been described in small case series, the prevalence and severity of these features are not well established. Despite the scarcity of neuropathological studies in PLS, extra-motor pathology has been intermittently described (Gazulla et al. 2019; Kobayashi et al., 2010). Sporadic reports of cognitive and behavioral impairment (Caselli et al. 1995; G. M. Grace et al. 2011; Le Forestier et al. 2001; Piquard et al. 2006) have been recently confirmed by larger case series (Agarwal et al. 2018; de Vries et al. 2019; de Vries et al. 2017). Motor cortex, corticospinal tract and corpus callosum degeneration have been consistently identified by computational imaging studies (Butman and Floeter 2007; Claassen et al. 2010; Finegan et al. 2019a; Muller et al. 2018; Pringle et al. 1992; van der Graaff et al. 2010), but reports of extra-motor involvement are surprisingly inconsistent (Chipika et al. 2020a). Perfusion abnormalities (Murphy et al. 2008), functional connectivity changes (Agosta et al. 2014) and diffusivity alterations (Canu et al. 2013) have been noted in PLS patients with cognitive impairment, but the majority of these studies suffer from sample size limitations. Brainstem (Bede et al. 2019), cerebellar (Finegan et al. 2019a; Tu et al. 2019), thalamic (Chipika et al. 2020b), and basal ganglia (Finegan et al. 2019b) degeneration have also been noted, but seldom linked to clinical manifestations (Floeter et al. 2014). The implications of cognitive and behavioral impairment are well established in other motor neuron diseases and associated with increased caregiver burden, limited compliance with assistive devices, increased fall risk, faster functional decline, shorter survival and a decreased likelihood of participation in clinical trials (Burke et al. 2017; Burke, Pinto-Grau, et al. 2016; Elamin et al. 2015; Elamin et al. 2011; Olney et al. 2005). As survival is much longer in PLS than in ALS (Finegan et al. 2019c), possible extra-motor manifestations are likely to have an enduring impact on multidisciplinary care, rehabilitation efforts, engagement with occupational and physiotherapy, ambulation safety and care preferences. The objective of this study is therefore the systematic evaluation of extra-motor disease burden in PLS using comprehensive cortical, subcortical and white matter analyses in a relatively large cohort of clinically and genetically well-characterized PLS patients. Based on the available literature, our hypothesis is that the multifaceted radiological analysis of quantitative imaging data reveals extra-motor changes in PLS.
Methods
Participants
Forty PLS patients were recruited from a population-based register, diagnosed based on the new consensus diagnostic criteria (Turner et al. 2020). MRI data from one hundred age-matched healthy controls were used for the interpretation of PLS imaging data. Healthy controls were unrelated to the participating PLS patients and had no established neurological or psychiatric diagnoses. The study was approved by the Ethics Committee of Beaumont Hospital, Dublin, Ireland and all participants provided informed consent prior to inclusion.
Clinical evaluation
Age, gender, education and handedness were recorded for each participant. Clinical data were collected at the time of MR imaging and included the total ALSFRS-r (Cedarbaum et al. 1999), ALSFRS sub-scores, symptom duration, Penn Upper Motor Neuron Score (PUMNS), modified Ashworth spasticity scale scores (Bohannon and Smith 1987), Edinburgh Cognitive and Behavioural ALS Screen (ECAS)(Abrahams et al. 2014), the Hospital Anxiety and Depression Scale (HADS) (Zigmond et al. 1983), the Frontal Systems Behavior Scale (J. Grace et al. 2002), the Emotional Lability Questionnaire (ELQ) and the Center for Neurological Study- Lability Scale (CNS-LS) (Moore et al. 1997; Newsom-Davis et al. 1999). The revised ALS functional rating scale (ALSFRS-r) provides a composite score of motor disability in four core domains; bulbar region, upper-limb/fine motor, lower-limb/gross motor and respiratory function (Cedarbaum et al. 1999). The Penn Upper Motor Neuron Score (PUMNS) appraises pathologically increased reflexes in the bulbar region and limbs (Bohannon and Smith 1987; Quinn et al. 2020). The Edinburgh Cognitive and Behavioural Screen (ECAS) evaluates cognitive performance across language, verbal fluency, executive, memory and visuospatial domains (Abrahams et al. 2014). It has been extensively validated in the Irish population and normative data are available (Pinto-Grau et al. 2017). The Frontal Systems Behavior Scale (FrSBe) questionnaire was completed by both 30 participating PLS patients and their caregivers. The FrSBe consists of 46 items across three subscales; apathy, disinhibition and executive dysfunction (Carvalho et al. 2013). Both ‘before’ and ‘after’ scores are awarded to identify disease-related behavioral change. Patients and family members were instructed to complete the questionnaire separately without conferring. Raw scores were converted to T-scores according to the published age, gender and education specific cut-offs. A T-score ≥ 65 on total score is indicative of behavioral impairment.
Magnetic resonance imaging
A 3D Inversion Recovery prepared Spoiled Gradient Recalled echo (IR-SPGR) pulse sequence was utilised to acquire T1-weighted images on a 3 Tesla Philips Achieva system with an 8-channel receive-only head coil. The following parameters were used for the IR-SPGR pulse sequence; TR/TE = 8.5/3.9 ms, TI =1060 ms, field-of-view (FOV): 256 × 256 × 160 mm, spatial resolution: 1 mm3, flip angle = 8°, SENSE factor = 1.5, acquisition time: 7 min 30 s. A spin-echo echo planar imaging (SE-EPI) sequence was used to acquire 32-direction DTI images; FOV = 245 × 245 × 150 mm, spatial resolution = 2.5 mm3, 60 slices were acquired with no interslice gap, TR/TE = 7639 / 59 ms, SENSE factor = 2.5, b-values = 0, 1100 s/mm2. FLAIR images were also acquired for each participant to assess for vascular white matter lesion load and images were individually reviewed. FLAIR images were acquired in axial orientation using an Inversion Recovery Turbo Spin Echo (IR-TSE) sequence: FOV = 230 × 183 × 150 mm, spatial resolution = 0.65 × 0.87 × 4 mm, 30 slices with 1 mm gap, TR/TE = 11,000 / 125 ms, TI = 2800 ms, 120° refocusing pulse, with flow compensation and motion smoothing and a saturation slab covering the neck region. Imaging data were first explored in standard ‘whole-brain’ voxelwise analyses. Subsequently, region-of-interest (ROI) analyses were undertaken to assess a range of imaging metrics in clinically relevant anatomical regions.
Grey matter analyses
Voxel-based morphometry was carried out in FMRIB’s FSL environment (Douaud et al. 2007; Good et al. 2001) using standard pre-processing steps; skull-removal, motion-corrections and tissue-type segmentation. Grey-matter partial volume data were aligned to the MNI152 standard space, a study-specific GM template was created to which the grey matter images from each subject were co-registered. To evaluate cortical grey matter changes in PLS compared to healthy controls, permutation based non-parametric inference was utilized using the threshold-free cluster enhancement (TFCE) method. The design matrix included study group membership and the following demeaned covariates; age, gender, years of education and total intracranial volumes.
White matter analyses
Pre-processing of raw diffusion data included eddy current corrections and skull removal (Schuster, Elamin, et al. 2016). Subsequently, a tensor model was fitted to the diffusion data to create maps of fractional anisotropy (FA), axial diffusivity (AD), mean diffusivity (MD) and radial diffusivity (RD). The tract-based statistics (TBSS) pipeline of the FSL image analysis suite was utilized for non-linear registration and skeletonization of diffusion images (Smith et al. 2006). FA, AD, MD and RD images were merged into a 4D file and a mean FA mask was created. Permutation-based non-parametric statistics was used to characterize the voxelwise diffusivity profile of PLS patients in contrast to healthy controls. Covariates included age, gender and years of education.
Subcortical great matter segmentation
The subcortical segmentation and registration tool FIRST of the FMRIB’s Software Library was utilized to segment subcortical grey matter structures (Patenaude et al. 2011). Volumes of subcortical grey matter structures were estimated as described previously (Bede, Finegan, et al. 2018; Bede, Omer, et al. 2018). Total intracranial volume was included as an additional covariate for the comparisons of subcortical grey matter volumes.
Region of interest analyses
The standard pipelines of the FreeSurfer image analysis suite (B. Fischl 2012) was used for cortical thickness measurements including the removal of non-brain tissue, segmentation of the subcortical white matter and deep grey matter structures, intensity normalization, tessellation of the grey matter-white matter boundary, and automated topology correction (Fischl and Dale 2000). The labels of the Desikan-Killiany atlas were utilized to retrieve average cortical thickness values (Schuster, Hardiman, et al. 2016). White matter integrity metrics were retrieved from skeletonized FA, AD, MD and RD maps using atlas-defined labels for the corpus callosum, corticospinal tracts, frontal lobe, cerebellum, forceps major, occipital lobe, parietal lobe, thalamus, temporal lobe and forceps minor (Schuster, Elamin, et al. 2016; Wakana et al. 2007). Values for bilateral structures were averaged.
Genetic testing
Twenty-eight of the 40 PLS patients underwent whole genome sequencing and were screened for ALS and HSP-associated mutations. Thirty-three PLS were screened for C9orf72 GGGGCC repeat expansions using repeat-primed polymerase chain reaction.
Statistical analyses
Demographic variables for PLS and healthy control groups were compared with independent samples t-tests for continuous variables and Chi-square and Fisher’s exact test for categorical variables. All assumptions were verified. Statistical analysis of clinical data was performed using IBM SPSS Statistics Version 26. Voxel-wise imaging data were analyzed using non-parametric permutation-based statistics with FMRIB’s Randomise. Region-of-interest imaging statics were performed on raw data retrieved from individual scans. Regional cortical thickness, subcortical grey matter volumes and white matter metrics were interpreted using IBM SPSS v. 26. Assumptions of normality were examined using the Kolmogorov-Smirnov test. Skewness and kurtosis were assessed separately for each study group. Since all variables followed a normal distribution, parametric statistics were applied. Group differences in imaging metrics retrieved from ROIs were examined using multivariate analysis of covariance (MANCOVA) with age, gender and education as covariates.
Results
Clinical characteristics
The demographic of all participants and the clinical characteristics PLS patients are presented in Table 1. Thirty-nine of the 40 patients reported symptom onset in the lower limbs. Pathologically increased reflexes were identified in all body regions, but UMN burden was particularly high in the lower limbs with a mean UMN score of 9.8 (max = 14). Spasticity was also higher in the lower limbs than upper limbs with a mean ASS-m of 3.3 and 2.6 respectively. Pseudobulbar affect was identified in 17 patients (42%) based on established thresholds for CNS-LS and ELQ. Nine patients (22.5%) showed evidence of cognitive impairment based on the total ECAS score. Language (28%) and verbal fluency (22.5%) deficits were most common, followed by memory impairment (12.5%). Abnormal performance in executive (7.5%) and visuospatial domains (2.5%) were uncommon based on our screening assessment with ECAS. Self and family-reported behavioral change was evident in one third of patients. Self-reported and caregiver-reported behavioral changes were remarkably concordant across all subscales. Apathy was the most commonly identified behavioral change.
Cortical Grey matter profiles
Voxel-wise analyses revealed widespread multifocal extra-motor atrophy including the bilateral anterior cingulate, dorsolateral prefrontal cortex, insula, operculum, orbitofrontal and anterior mesial temporal regions. (Fig. 1). Region-of-interest cortical thickness analyses also demonstrated cortical thinning in multiple frontotemporal regions. (Table 2a). Significant atrophy was identified in multiple frontal lobe regions including orbitofrontal, caudal middle frontal, cingulate and insular areas. Considerable fusiform gyrus, temporal pole, transverse temporal, and superior temporal changes were also identified. Within the parietal lobe, precuneus and supramarginal cortical thinning was observed.

Motor and extra-motor grey matter pathology in PLS at p < 0.01 FWE TFCE.
Subcortical grey matter profiles
The volumetric comparisons of subcortical grey regions are shown in Table 2b. The thalamus was the most significantly affected subcortical grey matter structure, followed by the caudate and hippocampus. The putamen and accumbens were also affected, but no significant globus pallidus or amygdala atrophy was observed.
White matter profiles
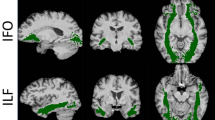
Significant, symmetrical white matter alterations were identified in PLS group, primarily involving the corticospinal tracts and the corpus callosum. Cerebellar, fornix, brainstem and temporal lobe alterations were also captured. Fig. 2. ROI analyses also demonstrated widespread diffusion abnormalities in the PLS group (Table 3). Across all diffusion metrics, the most significant changes were observed in the corticospinal tracts and in the body of the corpus callosum. Thalamic white matter involvement was also detected across all diffusivity measures. While no white matter region-of-interest was entirely preserved, parietal and occipital lobe involvement was relatively spared.

Tract-based diffusivity alterations in PLS at p < 0.0125 FWE TFCE. Regions of reduced fractional anisotropy (FA), increased axial diffusivity (AD), mean diffusivity (MD) and radial diffusivity (RD) are presented
Discussion
The study confirms widespread extra-motor pathology in PLS which is consistent with the growing literature of cognitive and behavioral changes in this condition. Our imaging analyses provide evidence of widespread frontotemporal cortical changes, subcortical grey matter degeneration and extra-motor white matter pathology.
Our imaging data confirm the motor cortex (Butman and Floeter 2007; Finegan 2019a; Schuster et al. 2013), corpus callosum (Muller et al. 2012; van der Graaff et al. 2011) and corticospinal tract (Salameh et al. 2013; Suh et al. 2006; Tzarouchi et al. 2011) findings of previous imaging papers in PLS (Clark et al. 2018; Müller et al. 2018; Turner et al. 2007). However, the novelty of our study is the description of widespread, multifocal and symmetric frontotemporal degeneration is a cohort of established PLS patients. PLS is not classically regarded as part of the ALS-FTD spectrum, yet, our cohort of C9orf72 negative PLS patients exhibit patterns of grey matter atrophy reminiscent of FTD (Bede, Omer, et al. 2018; Omer et al. 2017) involving insular, orbitofrontal, superior temporal regions as well as primary language regions; the pars opercularis and pars triangularis of the inferior frontal lobe. Considerable anterior and posterior cingulate pathology is also evident on both whole-brain morphometry and region-of-interest cortical thickness analyses. Despite the anatomical concordance of morphometric and ROI approaches, one of the benefits of the standard VBM method is its ability to highlight the strikingly symmetric patterns of grey matter degeneration at a given statistical threshold. (Fig. 1.) ALS studies often identify some degree of frontotemporal pathology (Christidi et al., 2019; Christidi et al. 2017; Omer et al. 2017), but they are remarkably inconsistent with regard to extra-motor pathology depending on the phenotypes and genotypes included (Bede, Querin et al. 2018; Christidi, Karavasilis, Rentzos, et al. 2018; Nasseroleslami et al. 2019). ALS is a relatively heterogeneous condition, especially with regard to frontotemporal pathology (Burke, Pinto-Grau, et al. 2016; Elamin et al. 2017). Our radiological and clinical findings suggest that heterogeneity in PLS may be much less prominent than previously suggested and patients exhibit relatively uniform frontotemporal changes (Zhai et al. 2003). The relative homogeneity of PLS patients is also evidenced by their invariably long survival, symmetric lower limb onset, limited respiratory involvement and fairly stereotypical disability profiles.
Our voxelwise and ROI white matter analyses not only confirm the pathognomonic degeneration of the pyramidal tracts, but capture extra-motor white matter changes across multiple diffusivity metrics. Tract-based spatial statistics readily capture the degeneration of the body of the corpus callosum, but the ROI analyses also demonstrate the forceps minor (anterior forceps) and forceps major (posterior forceps) are also significantly affected. The tract-based analysis of AD, MD and RD captured considerable fornix pathology which has not been previously reported in PLS. The fornix is a key output tract of the hippocampus and its degeneration has been linked to memory deficits in ALS (Christidi et al. 2019). The observation that both the hippocampus and its output bundle are affected illustrates the concomitant degeneration of interconnected brain regions (Bak and Chandran 2012). Tract-based spatial statistics also identified diffusivity changes within the thalamus, which is typically by either evaluated by volumetric (Finegan et al. 2020c) or vertex-wise (Finegan et al. 2019d) approaches relying on T1-wieghted raw data. In this study we found both thalamic diffusivity alterations – reduced FA, increased AD, MD, RD - as well as volume reductions on structural analyses. Both imaging and post mortem studies suggest that thalamic changes are driven by the focal degeneration of specific nuclei as opposed to global atrophy (Chipika et al. 2020b; Chipika et al. 2020c). One the benefits of combining whole-brain and ROI analyses is the ability to rank the most affected and least affected brain regions. Similarly to ALS (Bede et al. 2016), PLS also exhibits a strikingly selective pattern of frontotemporal vulnerability with the relative sparing of postcentral and lateral occipital brain regions. Despite the considerable symptom duration profile of this cohort, parietal and occipital white matter involvement is limited in this patient group. The white matter changes identified in the brainstem (Fig. 2.) are not surprising in light of the UMN burden and pseudobulbar manifestations observed in the cohort and are consistent with dedicated brainstem studies (Bede et al. 2020; Bede et al. 2019). Cerebellar grey matter atrophy has been previously described (Finegan et al. 2019a) based on imaging data, but it is seldom assessed specifically clinically. The clinical relevance of cerebellar pathology may be underestimated in PLS where a significant proportion of patients use walking aids for safe ambulation and may be at a relatively high risk of falls. Cerebellar pathology was also detected on tract-based and ROI white matter analyses. While the clinical detection of subtle cerebellar signs may be confounded by prominent pyramidal signs the target evaluation of cerebellar function may be judicious. Cerebellar degeneration has also been implicated in the aetiology of pseudobulbar affect through impaired gate-control mechanisms (Bede and Finegan 2018; Christidi, Karavasilis, Ferentinos, et al. 2018; Finegan et al. 2019b; Floeter et al. 2014).
While direct correlations between imaging metrics and neuropsychological performance are often considered contentious (Verstraete et al. 2015), the clinical profile of the patients shows remarkable concordance with our imaging findings. The considerable paracentral gyrus cortical thinning is in line with the lower limb predominant motor disability of our cohort as the medial segment of the motor cortex provides the somatotopic representation of the lower limbs in the motor homunculus. The language deficits observed on our screening tests are also concordant with the degenerative changes of the pars opercularis and pars triangularis. The high prevalence of apathy observed in our cohort may be anatomically linked to the considerable volume reductions identified in the accumbens nucleus, which, under physiological conditions, mediates motivation and contributes to reward processing (Ikemoto et al. 1999; Machts et al. 2015). Linking impairments in specific cognitive domains to single cortical or subcortical regions however is simplistic, as the majority of cognitive functions are mediated by multi-synaptic corticobasal networks (Bonelli and Cummings 2007; O’Callaghan et al. 2013). A high prevalence of language and verbal fluency deficits was identified while memory and visuospatial deficits were relatively uncommon. The identified verbal fluency deficits cannot be attributed to spastic dysarthria as the ECAS corrects for motor disability through the ‘verbal fluency index’. Although the ECAS assessment can be undertaken in either spoken or written format, all patients in this cohort were able to complete it verbally. In our experience, anarthria is not a common clinical feature of PLS despite the presence of some degree of spastic dysarthria (Yunusova et al. 2019). Unlike in ALS, where executive dysfunction is the commonest cognitive deficit (Christidi, Karavasilis, Rentzos, et al. 2018), impaired executive function was not readily identified on ECAS in our cohort of PLS patients. By contrast, patients and family consistently reported executive dysfunction on FrSBe.
While the neuropsychological profile of ALS has been extensively studied, disease-specific screening instruments have been developed, large longitudinal studies have been undertaken, cognitive profiles linked to specific genotypes and imaging changes, extra-motor changes in PLS remain woefully underrecognized and they are not routinely tested for. It is clear that the considerable apathy detected in a significant minority of our patients is likely to impact on the quality of life of the patients, their caregivers and affect the management of this cohort. The recognition of language deficits in PLS is also hugely relevant with regard to maintaining interpersonal communication and employment. We must acknowledge the limitations of the cognitive screening tests utilized in this cohort which does not substitute comprehensive neuropsychological testing. Furthermore, the prolonged mean symptom duration in this PLS group must be considered when interpreting the prevalence and pattern of extra-motor features in comparison with previous studies in ALS with much shorter symptom duration. We also concede that a multi-timepoint study design, enrolling from first presentation, would provide crucial longitudinal insights, in particular with respect to the chronology of motor and extra-motor involvement (Chipika et al. 2019). A recent natural history study of PLS revealed ceiling-effects; progressive UMN degeneration in the first 8 years followed by a plateau (Floeter and Wu 2020). A recent analysis of imaging data of patients with a symptom duration of less than 4 years suggests motor cortex pathology without detectable extra-motor atrophy (Finegan et al. 2020a; Finegan et al. 2020c), but large perspective studies are needed to verify if precentral gyrus changes truly precede extra-motor expansion. We also acknowledge that the comparison of a single patient group to healthy controls only permits the description of group-level changes as opposed to the precision categorization of individual MND patients (Bede et al. 2017; Grollemund et al. 2019; Querin et al. 2018; Verde et al. 2020). Despite these limitations, our data provide compelling evidence of considerable frontotemporal pathology in a population-based sample of C9orf72 negative PLS patients.
Conclusions
Primary lateral sclerosis is associated with widespread frontotemporal and subcortical pathology and neuropsychological deficits. Accordingly, it should no longer be regarded as a pure upper motor neuron disorder. Patients should be meticulously screened for cognitive deficits and apathy, and the impact of these deficits on decision making and clinical management should be urgently studied in dedicated prospective research protocols.
References
Abrahams, S., Newton, J., Niven, E., Foley, J., & Bak, T. H. (2014). Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener, 15(1–2), 9–14. https://doi.org/10.3109/21678421.2013.805784.
Agarwal, S., Highton-Williamson, E., Caga, J., Matamala, J. M., Dharmadasa, T., Howells, J., Zoing, M. C., Shibuya, K., Geevasinga, N., Vucic, S., Hodges, J. R., Ahmed, R. M., & Kiernan, M. C. (2018). Primary lateral sclerosis and the amyotrophic lateral sclerosis-frontotemporal dementia spectrum. Journal of Neurology, 265(8), 1819–1828. https://doi.org/10.1007/s00415-018-8917-5.
Agosta, F., Canu, E., Inuggi, A., Chio, A., Riva, N., Silani, V., et al. (2014). Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiology of Aging, 35(4), 916–925.
Bak, T. H., & Chandran, S. (2012). What wires together dies together: Verbs, actions and neurodegeneration in motor neuron disease. Cortex, 48(7), 936–944. https://doi.org/10.1016/j.cortex.2011.07.008.
Bede, P., Chipika, R. H., Finegan, E., Li Hi Shing, S., Chang, K. M., Doherty, M. A., Hengeveld, J. C., Vajda, A., Hutchinson, S., Donaghy, C., McLaughlin, R. L., & Hardiman, O. (2020). Progressive brainstem pathology in motor neuron diseases: Imaging data from amyotrophic lateral sclerosis and primary lateral sclerosis. Data in Brief, 29, 105229. https://doi.org/10.1016/j.dib.2020.105229.
Bede, P., Chipika, R. H., Finegan, E., Li Hi Shing, S., Doherty, M. A., Hengeveld, J. C., Vajda, A., Hutchinson, S., Donaghy, C., McLaughlin, R. L., & Hardiman, O. (2019). Brainstem pathology in amyotrophic lateral sclerosis and primary lateral sclerosis: A longitudinal neuroimaging study. Neuroimage Clin, 24, 102054. https://doi.org/10.1016/j.nicl.2019.102054.
Bede, P., & Finegan, E. (2018). Revisiting the pathoanatomy of pseudobulbar affect: Mechanisms beyond corticobulbar dysfunction. Amyotroph Lateral Scler Frontotemporal Degener, 19(1–2), 4–6. https://doi.org/10.1080/21678421.2017.1392578.
Bede, P., Finegan, E., Chipika, R. H., Li Hi Shing, S., Lambe, J., Meaney, J., & Redmond, J. (2018). Occulomotor neural integrator dysfunction in multiple sclerosis: Insights from neuroimaging. Frontiers in Neurology, 9, 691. https://doi.org/10.3389/fneur.2018.00691.
Bede, P., Iyer, P. M., Finegan, E., Omer, T., & Hardiman, O. (2017). Virtual brain biopsies in amyotrophic lateral sclerosis: Diagnostic classification based on in vivo pathological patterns. Neuroimage Clin, 15, 653–658. https://doi.org/10.1016/j.nicl.2017.06.010.
Bede, P., Iyer, P. M., Schuster, C., Elamin, M., McLaughlin, R. L., Kenna, K., & Hardiman, O. (2016). The selective anatomical vulnerability of ALS: 'disease-defining' and 'disease-defying' brain regions. Amyotroph Lateral Scler Frontotemporal Degener, 17(7–8), 561–570. https://doi.org/10.3109/21678421.2016.1173702.
Bede, P., Omer, T., Finegan, E., Chipika, R. H., Iyer, P. M., Doherty, M. A., Vajda, A., Pender, N., McLaughlin, R. L., Hutchinson, S., & Hardiman, O. (2018). Connectivity-based characterisation of subcortical grey matter pathology in frontotemporal dementia and ALS: A multimodal neuroimaging study. Brain Imaging and Behavior, 12(6), 1696–1707. https://doi.org/10.1007/s11682-018-9837-9.
Bede, P., Querin, G., & Pradat, P. F. (2018). The changing landscape of motor neuron disease imaging: The transition from descriptive studies to precision clinical tools. Current Opinion in Neurology, 31(4), 431–438. https://doi.org/10.1097/wco.0000000000000569.
Bohannon, R. W., & Smith, M. B. (1987). Interrater reliability of a modified Ashworth scale of muscle spasticity. Physical Therapy, 67(2), 206–207.
Bonelli, R. M., & Cummings, J. L. (2007). Frontal-subcortical circuitry and behavior. Dialogues in Clinical Neuroscience, 9(2), 141–151.
Burke, T., Elamin, M., Bede, P., Pinto-Grau, M., Lonergan, K., Hardiman, O., & Pender, N. (2016). Discordant performance on the 'Reading the mind in the Eyes' test, based on disease onset in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener, 1-6. https://doi.org/10.1080/21678421.2016.1177088.
Burke, T., Pinto-Grau, M., Lonergan, K., Bede, P., O'Sullivan, M., Heverin, M., Vajda, A., McLaughlin, R. L., Pender, N., & Hardiman, O. (2017). A cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: Subphenotypes, staging, cognitive predictors, and survival. Annals of Clinical Translational Neurology, 4(5), 305–317. https://doi.org/10.1002/acn3.407.
Burke, T., Pinto-Grau, M., Lonergan, K., Elamin, M., Bede, P., Costello, E., Hardiman, O., & Pender, N. (2016). Measurement of social cognition in amyotrophic lateral sclerosis: A population based study. PLoS One, 11(8), e0160850. https://doi.org/10.1371/journal.pone.0160850.
Butman, J. A., & Floeter, M. K. (2007). Decreased thickness of primary motor cortex in primary lateral sclerosis. American Journal of Neuroradiology, 28(1), 87–91.
Canu, E., Agosta, F., Galantucci, S., Chio, A., Riva, N., Silani, V., et al. (2013). Extramotor damage is associated with cognition in primary lateral sclerosis patients. PLoS ONE [Electronic Resource], 8(12), e82017.
Carvalho, J. O., Ready, R. E., Malloy, P., & Grace, J. (2013). Confirmatory factor analysis of the frontal systems behavior scale (FrSBe). Assessment, 20(5), 632–641. https://doi.org/10.1177/1073191113492845.
Caselli, R. J., Smith, B. E., & Osborne, D. (1995). Primary lateral sclerosis: A neuropsychological study. Neurology, 45(11), 2005–2009.
Cedarbaum, J. M., Stambler, N., Malta, E., Fuller, C., Hilt, D., Thurmond, B., & Nakanishi, A. (1999). The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study group (phase III). Journal of the Neurological Sciences, 169(1–2), 13–21.
Chipika, R. H., Christidi, F., Finegan, E., Li Hi Shing, S., McKenna, M. C., Chang, K. M., Karavasilis, E., Doherty, M. A., Hengeveld, J. C., Vajda, A., Pender, N., Hutchinson, S., Donaghy, C., McLaughlin, R. L., Hardiman, O., & Bede, P. (2020a). Amygdala pathology in amyotrophic lateral sclerosis and primary lateral sclerosis. Journal of the Neurological Sciences, 117039, 117039. https://doi.org/10.1016/j.jns.2020.117039.
Chipika, R. H., Finegan, E., Li Hi Shing, S., Hardiman, O., & Bede, P. (2019). Tracking a fast-moving disease: Longitudinal markers, monitoring, and clinical trial endpoints in ALS. Frontiers in Neurology, 10, 229. https://doi.org/10.3389/fneur.2019.00229.
Chipika, R. H., Finegan, E., Li Hi Shing, S., McKenna, M. C., Christidi, F., Chang, K. M., Doherty, M. A., Hengeveld, J. C., Vajda, A., Pender, N., Hutchinson, S., Donaghy, C., McLaughlin, R. L., Hardiman, O., & Bede, P. (2020b). "switchboard" malfunction in motor neuron diseases: Selective pathology of thalamic nuclei in amyotrophic lateral sclerosis and primary lateral sclerosis. Neuroimage Clin, 27, 102300. https://doi.org/10.1016/j.nicl.2020.102300.
Chipika, R. H., Siah, W. F., Shing, S. L. H., Finegan, E., McKenna, M. C., Christidi, F., Chang, K. M., Karavasilis, E., Vajda, A., Hengeveld, J. C., Doherty, M. A., Donaghy, C., Hutchinson, S., McLaughlin, R. L., Hardiman, O., & Bede, P. (2020c). MRI data confirm the selective involvement of thalamic and amygdalar nuclei in amyotrophic lateral sclerosis and primary lateral sclerosis. Data in Brief, 106246, 106246. https://doi.org/10.1016/j.dib.2020.106246.
Christidi, F., Karavasilis, E., Ferentinos, P., Xirou, S., Velonakis, G., Rentzos, M., Zouvelou, V., Zalonis, I., Efstathopoulos, E., Kelekis, N., & Evdokimidis, I. (2018). Investigating the neuroanatomical substrate of pathological laughing and crying in amyotrophic lateral sclerosis with multimodal neuroimaging techniques. Amyotroph Lateral Scler Frontotemporal Degener, 19(1–2), 12–20. https://doi.org/10.1080/21678421.2017.1386689.
Christidi, F., Karavasilis, E., Rentzos, M., Kelekis, N., Evdokimidis, I., & Bede, P. (2018). Clinical and radiological markers of extra-motor deficits in amyotrophic lateral sclerosis. Frontiers in Neurology, 9, 1005. https://doi.org/10.3389/fneur.2018.01005.
Christidi, F., Karavasilis, E., Rentzos, M., Velonakis, G., Zouvelou, V., Xirou, S., Argyropoulos, G., Papatriantafyllou, I., Pantolewn, V., Ferentinos, P., Kelekis, N., Seimenis, I., Evdokimidis, I., & Bede, P. (2019). Hippocampal pathology in amyotrophic lateral sclerosis: Selective vulnerability of subfields and their associated projections. Neurobiology of Aging, 84, 178–188. https://doi.org/10.1016/j.neurobiolaging.2019.07.019.
Christidi, F., Karavasilis, E., Zalonis, I., Ferentinos, P., Giavri, Z., Wilde, E. A., Xirou, S., Rentzos, M., Zouvelou, V., Velonakis, G., Toulas, P., Efstathopoulos, E., Poulou, L., Argyropoulos, G., Athanasakos, A., Zambelis, T., Levin, H. S., Karandreas, N., Kelekis, N., & Evdokimidis, I. (2017). Memory-related white matter tract integrity in amyotrophic lateral sclerosis: An advanced neuroimaging and neuropsychological study. Neurobiology of Aging, 49, 69–78. https://doi.org/10.1016/j.neurobiolaging.2016.09.014.
Claassen, D. O., Josephs, K. A., & Peller, P. J. (2010). The stripe of primary lateral sclerosis: Focal primary motor cortex hypometabolism seen on fluorodeoxyglucose F18 positron emission tomography. Archives of Neurology, 67(1), 122–125.
Clark, M. G., Smallwood Shoukry, R., Huang, C. J., Danielian, L. E., Bageac, D., & Floeter, M. K. (2018). Loss of functional connectivity is an early imaging marker in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener, 19(7–8), 562–569. https://doi.org/10.1080/21678421.2018.1517180.
de Vries, B. S., Rustemeijer, L. M. M., Bakker, L. A., Schröder, C. D., Veldink, J. H., van den Berg, L. H., Nijboer, T. C. W., & van Es, M. A. (2019). Cognitive and behavioural changes in PLS and PMA:Challenging the concept of restricted phenotypes. Journal of Neurology, Neurosurgery, and Psychiatry, 90(2), 141–147. https://doi.org/10.1136/jnnp-2018-318788.
de Vries, B. S., Rustemeijer, L. M. M., van der Kooi, A. J., Raaphorst, J., Schröder, C. D., Nijboer, T. C. W., Hendrikse, J., Veldink, J. H., van den Berg, L. H., & van Es, M. A. (2017). A case series of PLS patients with frontotemporal dementia and overview of the literature. Amyotroph Lateral Scler Frontotemporal Degener, 18(7–8), 534–548. https://doi.org/10.1080/21678421.2017.1354996.
Douaud, G., Smith, S., Jenkinson, M., Behrens, T., Johansen-Berg, H., Vickers, J., James, S., Voets, N., Watkins, K., Matthews, P. M., & James, A. (2007). Anatomically related grey and white matter abnormalities in adolescent-onset schizophrenia. Brain, 130(Pt 9), 2375–2386.
Elamin, M., Bede, P., Byrne, S., Jordan, N., Gallagher, L., Wynne, B., O'Brien, C., Phukan, J., Lynch, C., Pender, N., & Hardiman, O. (2013). Cognitive changes predict functional decline in ALS: A population-based longitudinal study. Neurology, 80(17), 1590–1597. https://doi.org/10.1212/WNL.0b013e31828f18ac.
Elamin, M., Bede, P., Montuschi, A., Pender, N., Chio, A., & Hardiman, O. (2015). Predicting prognosis in amyotrophic lateral sclerosis: A simple algorithm. Journal of Neurology, 262(6), 1447–1454. https://doi.org/10.1007/s00415-015-7731-6.
Elamin, M., Phukan, J., Bede, P., Jordan, N., Byrne, S., Pender, N., & Hardiman, O. (2011). Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology, 76(14), 1263–1269.
Elamin, M., Pinto-Grau, M., Burke, T., Bede, P., Rooney, J., O'Sullivan, M., et al. (2017). Identifying behavioural changes in ALS: Validation of the Beaumont Behavioural inventory (BBI). Amyotroph Lateral Scler Frontotemporal Degener, 18(1–2), 68–73. https://doi.org/10.1080/21678421.2016.1248976.
Finegan, E., Chipika, R. H., Li Hi Shing, S., Doherty, M. A., Hengeveld, J. C., Vajda, A., Donaghy, C., McLaughlin, R. L., Pender, N., Hardiman, O., & Bede, P. (2019a). The clinical and radiological profile of primary lateral sclerosis: A population-based study. Journal of Neurology, 266(11), 2718–2733. https://doi.org/10.1007/s00415-019-09473-z.
Finegan, E., Chipika, R. H., Li Hi Shing, S., Hardiman, O., & Bede, P. (2019b). Pathological crying and laughing in motor neuron disease: Pathobiology, screening, intervention. Frontiers in Neurology, 10, 260. https://doi.org/10.3389/fneur.2019.00260.
Finegan, E., Chipika, R. H., Shing, S. L. H., Hardiman, O., & Bede, P. (2019c). Primary lateral sclerosis: A distinct entity or part of the ALS spectrum? Amyotroph Lateral Scler Frontotemporal Degener, 20(3–4), 133–145. https://doi.org/10.1080/21678421.2018.1550518.
Finegan, E., Hi Shing, S. L., Chipika, R. H., McKenna, M. C., Doherty, M. A., Hengeveld, J. C., Vajda, A., Donaghy, C., McLaughlin, R. L., Hutchinson, S., Hardiman, O., & Bede, P. (2020a). Thalamic, hippocampal and basal ganglia pathology in primary lateral sclerosis and amyotrophic lateral sclerosis: Evidence from quantitative imaging data. Data in Brief, 29, 105115. https://doi.org/10.1016/j.dib.2020.105115.
Finegan, E., Li Hi Shing, S., Chipika, R. H., Doherty, M. A., Hengeveld, J. C., Vajda, A., Donaghy, C., Pender, N., McLaughlin, R. L., Hardiman, O., & Bede, P. (2019d). Widespread subcortical grey matter degeneration in primary lateral sclerosis: A multimodal imaging study with genetic profiling. Neuroimage Clin, 24, 102089. https://doi.org/10.1016/j.nicl.2019.102089.
Finegan, E., Li Hi Shing, S., Siah, W. F., Chipika, R. H., Chang, K. M., McKenna, M. C., Doherty, M. A., Hengeveld, J. C., Vajda, A., Donaghy, C., Hutchinson, S., McLaughlin, R. L., Hardiman, O., & Bede, P. (2020b). Evolving diagnostic criteria in primary lateral sclerosis: The clinical and radiological basis of “probable PLS”. Journal of the Neurological Sciences, 417, 117052. https://doi.org/10.1016/j.jns.2020.117052.
Finegan, E., Siah, W. F., Shing, S. L. H., Chipika, R. H., Chang, K. M., McKenna, M. C., Doherty, M. A., Hengeveld, J. C., Vajda, A., Donaghy, C., Hutchinson, S., McLaughlin, R. L., Hardiman, O., & Bede, P. (2020c). Imaging and clinical data indicate considerable disease burden in ‘probable’ PLS: Patients with UMN symptoms for 2-4 years. Data in Brief, 106247, 106247. https://doi.org/10.1016/j.dib.2020.106247.
Fischl, B. (2012). FreeSurfer. Neuroimage, 62(2), 774–781. https://doi.org/10.1016/j.neuroimage.2012.01.021.
Fischl, B., & Dale, A. M. (2000). Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proceedings of the National Academy of Sciences, 97(20), 11050–11055. https://doi.org/10.1073/pnas.200033797.
Floeter, M. K., Katipally, R., Kim, M. P., Schanz, O., Stephen, M., Danielian, L., Wu, T., Huey, E. D., & Meoded, A. (2014). Impaired corticopontocerebellar tracts underlie pseudobulbar affect in motor neuron disorders. Neurology, 83(7), 620–627. https://doi.org/10.1212/wnl.0000000000000693.
Floeter, M. K., & Wu, T. (2020). Longitudinal evaluation of upper motor neuron burden scales in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener, 1–7. https://doi.org/10.1080/21678421.2020.1790609.
Gazulla, J., Ferrer, I., Izquierdo-Alvarez, S., Alvarez, S., Sánchez-Alcudia, R., Bestué-Cardiel, M., Seral, M., Benavente, I., Sierra-Martínez, E., & Berciano, J. (2019). Hereditary primary lateral sclerosis and progressive nonfluent aphasia. Journal of Neurology, 266(5), 1079–1090. https://doi.org/10.1007/s00415-019-09235-x.
Good, C. D., Johnsrude, I. S., Ashburner, J., Henson, R. N., Friston, K. J., & Frackowiak, R. S. (2001). A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage, 14(1 Pt 1), 21–36.
Gordon, P., Cheng, B., Katz, I., Pinto, M., Hays, A., Mitsumoto, H., & Rowland, L. (2006). The natural history of primary lateral sclerosis. Neurology, 66(5), 647–653.
Grace, G. M., Orange, J. B., Rowe, A., Findlater, K., Freedman, M., & Strong, M. J. (2011). Neuropsychological functioning in PLS: A comparison with ALS. Canadian Journal of Neurological Sciences, 38(1), 88–97.
Grace, J., & Malloy, P. F. (2002). Frontal systems behavior scale. Florida: Psychological Assessment Resources.
Grollemund, V., Pradat, P. F., Querin, G., Delbot, F., Le Chat, G., Pradat-Peyre, J. F., & Bede, P. (2019). Machine learning in amyotrophic lateral sclerosis: Achievements, pitfalls, and future directions. Frontiers in Neuroscience, 13, 135. https://doi.org/10.3389/fnins.2019.00135.
Ikemoto, S., & Panksepp, J. (1999). The role of nucleus accumbens dopamine in motivated behavior: A unifying interpretation with special reference to reward-seeking. Brain Research. Brain Research Reviews, 31(1), 6–41. https://doi.org/10.1016/s0165-0173(99)00023-5.
Kobayashi, Z., Tsuchiya, K., Arai, T., Yokota, O., Yoshida, M., Shimomura, Y., Kondo, H., Haga, C., Asaoka, T., Onaya, M., Ishizu, H., Akiyama, H., & Mizusawa, H. (2010). Clinicopathological characteristics of FTLD-TDP showing corticospinal tract degeneration but lacking lower motor neuron loss. Journal of the Neurological Sciences, 298(1–2), 70–77. https://doi.org/10.1016/j.jns.2010.08.013.
Le Forestier, N., Maisonobe, T., Piquard, A., Rivaud, S., Crevier-Buchman, L., Salachas, F., et al. (2001). Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain, 124(Pt 10), 1989–1999.
Machts, J., Loewe, K., Kaufmann, J., Jakubiczka, S., Abdulla, S., Petri, S., Dengler, R., Heinze, H. J., Vielhaber, S., Schoenfeld, M. A., & Bede, P. (2015). Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology, 85(15), 1301–1309. https://doi.org/10.1212/wnl.0000000000002017.
Moore, S. R., Gresham, L. S., Bromberg, M. B., Kasarkis, E. J., & Smith, R. A. (1997). A self report measure of affective lability. Journal of Neurology, Neurosurgery, and Psychiatry, 63(1), 89–93.
Muller, H. P., Agosta, F., Gorges, M., Kassubek, R., Spinelli, E. G., Riva, N., et al. (2018). Cortico-efferent tract involvement in primary lateral sclerosis and amyotrophic lateral sclerosis: A two-Centre tract of interest-based DTI analysis. Neuroimage Clin, 20, 1062–1069. https://doi.org/10.1016/j.nicl.2018.10.005.
Müller, H. P., Gorges, M., Kassubek, R., Dorst, J., Ludolph, A. C., & Kassubek, J. (2018). Identical patterns of cortico-efferent tract involvement in primary lateral sclerosis and amyotrophic lateral sclerosis: A tract of interest-based MRI study. Neuroimage: Clinical, 18, 762–769. https://doi.org/10.1016/j.nicl.2018.03.018.
Muller, H. P., Unrath, A., Huppertz, H. J., Ludolph, A. C., & Kassubek, J. (2012). Neuroanatomical patterns of cerebral white matter involvement in different motor neuron diseases as studied by diffusion tensor imaging analysis. Amyotrophic Lateral Sclerosis, 13(3), 254–264. https://doi.org/10.3109/17482968.2011.653571.
Murphy, M. J., Grace, G. M., Tartaglia, M. C., Orange, J. B., Chen, X., Rowe, A., Findlater, K., Kozak, R. I., Freedman, M., Strong, M. J., & Lee, T. Y. (2008). Cerebral haemodynamic changes accompanying cognitive impairment in primary lateral sclerosis. Amyotrophic Lateral Sclerosis, 9(6), 359–368.
Nasseroleslami, B., Dukic, S., Broderick, M., Mohr, K., Schuster, C., Gavin, B., McLaughlin, R., Heverin, M., Vajda, A., Iyer, P. M., Pender, N., Bede, P., Lalor, E. C., & Hardiman, O. (2019). Characteristic increases in EEG connectivity correlate with changes of structural MRI in amyotrophic lateral sclerosis. Cerebral Cortex, 29(1), 27–41. https://doi.org/10.1093/cercor/bhx301.
Newsom-Davis, I. C., Abrahams, S., Goldstein, L. H., & Leigh, P. N. (1999). The emotional lability questionnaire: A new measure of emotional lability in amyotrophic lateral sclerosis. Journal of the Neurological Sciences, 169(1–2), 22–25.
O'Callaghan, C., Bertoux, M., & Hornberger, M. (2013). Beyond and below the cortex: The contribution of striatal dysfunction to cognition and behaviour in neurodegeneration. Journal of Neurology, Neurosurgery, and Psychiatry, 85, 371–378. https://doi.org/10.1136/jnnp-2012-304558.
Olney, R. K., Murphy, J., Forshew, D., Garwood, E., Miller, B. L., Langmore, S., Kohn, M. A., & Lomen-Hoerth, C. (2005). The effects of executive and behavioral dysfunction on the course of ALS. Neurology, 65(11), 1774–1777. https://doi.org/10.1212/01.wnl.0000188759.87240.8b.
Omer, T., Finegan, E., Hutchinson, S., Doherty, M., Vajda, A., McLaughlin, R. L., Pender, N., Hardiman, O., & Bede, P. (2017). Neuroimaging patterns along the ALS-FTD spectrum: A multiparametric imaging study. Amyotroph Lateral Scler Frontotemporal Degener, 18(7–8), 611–623. https://doi.org/10.1080/21678421.2017.1332077.
Patenaude, B., Smith, S. M., Kennedy, D. N., & Jenkinson, M. (2011). A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage, 56(3), 907–922.
Pinto-Grau, M., Burke, T., Lonergan, K., McHugh, C., Mays, I., Madden, C., Vajda, A., Heverin, M., Elamin, M., Hardiman, O., & Pender, N. (2017). Screening for cognitive dysfunction in ALS: Validation of the Edinburgh cognitive and Behavioural ALS screen (ECAS) using age and education adjusted normative data. Amyotroph Lateral Scler Frontotemporal Degener, 18(1–2), 99–106. https://doi.org/10.1080/21678421.2016.1249887.
Piquard, A., Le Forestier, N., Baudoin-Madec, V., Delgadillo, D., Salachas, F., Pradat, P. F., et al. (2006). Neuropsychological changes in patients with primary lateral sclerosis. Amyotrophic Lateral Sclerosise, 7(3), 150–160.
Pringle, C. E., Hudson, A. J., Munoz, D. G., Kiernan, J. A., Brown, W. F., & Ebers, G. C. (1992). Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain, 115(Pt 2), 495–520.
Querin, G., El Mendili, M. M., Bede, P., Delphine, S., Lenglet, T., Marchand-Pauvert, V., & Pradat, P. F. (2018). Multimodal spinal cord MRI offers accurate diagnostic classification in ALS. Journal of Neurology, Neurosurgery, and Psychiatry, 89(11), 1220–1221. https://doi.org/10.1136/jnnp-2017-317214.
Quinn, C., Edmundson, C., Dahodwala, N., & Elman, L. (2020). Reliable and efficient scale to assess upper motor neuron disease burden in amyotrophic lateral sclerosis. Muscle & Nerve, 61(4), 508–511. https://doi.org/10.1002/mus.26764.
Salameh, J. S., Patel, N., Zheng, S., & Cauley, K. A. (2013). Focal absence of diffusion tensor tracts from primary motor cortex in primary lateral sclerosis. European Journal of Neurology, 20(4), e63–e64. https://doi.org/10.1111/ene.12093.
Schuster, C., Elamin, M., Hardiman, O., & Bede, P. (2016). The segmental diffusivity profile of amyotrophic lateral sclerosis associated white matter degeneration. European Journal of Neurology, 23(8), 1361–1371. https://doi.org/10.1111/ene.13038.
Schuster, C., Hardiman, O., & Bede, P. (2016). Development of an automated MRI-based diagnostic protocol for amyotrophic lateral sclerosis using disease-specific pathognomonic features: A quantitative disease-state classification study. PLoS One, 11(12), e0167331. https://doi.org/10.1371/journal.pone.0167331.
Schuster, C., Kasper, E., Machts, J., Bittner, D., Kaufmann, J., Benecke, R., Teipel, S., Vielhaber, S., & Prudlo, J. (2013). Focal thinning of the motor cortex mirrors clinical features of amyotrophic lateral sclerosis and their phenotypes: A neuroimaging study. Journal of Neurology, 260(11), 2856–2864.
Smith, S. M., Jenkinson, M., Johansen-Berg, H., Rueckert, D., Nichols, T. E., Mackay, C. E., Watkins, K. E., Ciccarelli, O., Cader, M. Z., Matthews, P. M., & Behrens, T. E. (2006). Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. Neuroimage, 31(4), 1487–1505. https://doi.org/10.1016/j.neuroimage.2006.02.024.
Statland, J. M., Barohn, R. J., Dimachkie, M. M., Floeter, M. K., & Mitsumoto, H. (2015). Primary lateral sclerosis. Neurologic Clinics, 33(4), 749–760.
Suh, S. I., Song, I. C., & Koh, S. B. (2006). Primary lateral sclerosis with MR diffusion tensor image and tract tracking. American Journal of Physical Medicine & Rehabilitation, 85(11), 863–864. https://doi.org/10.1097/01.phm.0000242651.30244.a4.
Tu, S., Menke, R. A. L., Talbot, K., Kiernan, M. C., & Turner, M. R. (2019). Cerebellar tract alterations in PLS and ALS. Amyotroph Lateral Scler Frontotemporal Degener, 20(3–4), 281–284. https://doi.org/10.1080/21678421.2018.1562554.
Turner, M. R., Barohn, R. J., Corcia, P., Fink, J. K., Harms, M. B., Kiernan, M. C., Ravits, J., Silani, V., Simmons, Z., Statland, J., van den Berg, L. H., Delegates of the 2nd International PLS Conference, & Mitsumoto, H. (2020). Primary lateral sclerosis: consensus diagnostic criteria. Journal of Neurology, Neurosurgery, and Psychiatry, 91(4), 373–377. https://doi.org/10.1136/jnnp-2019-322541.
Turner, M. R., Hammers, A., Al-Chalabi, A., Shaw, C. E., Andersen, P. M., Brooks, D. J., & Leigh, P. N. (2007). Cortical involvement in four cases of primary lateral sclerosis using [(11)C]-flumazenil PET. Journal of Neurology, 254(8), 1033–1036.
Tzarouchi, L. C., Kyritsis, A. P., Giannopoulos, S., Astrakas, L. G., Diakou, M., & Argyropoulou, M. I. (2011). Voxel-based diffusion tensor imaging detects pyramidal tract degeneration in primary lateral sclerosis. British Journal of Radiology, 84(997), 78–80.
van der Graaff, M. M., Lavini, C., Akkerman, E. M., Majoie Ch, B., Nederveen, A. J., Zwinderman, A. H., et al. (2010). MR spectroscopy findings in early stages of motor neuron disease. Ajnr: American Journal of Neuroradiology, 31(10), 1799–1806.
van der Graaff, M. M., Sage, C. A., Caan, M. W., Akkerman, E. M., Lavini, C., Majoie, C. B., et al. (2011). Upper and extra-motoneuron involvement in early motoneuron disease: A diffusion tensor imaging study. Brain, 134(Pt 4), 1211–1228.
Verde, F., Zaina, G., Bodio, C., Borghi, M. O., Soranna, D., Peverelli, S., Ticozzi, N., Morelli, C., Doretti, A., Messina, S., Maderna, L., Colombrita, C., Gumina, V., Tiloca, C., Meroni, P. L., Zambon, A., Ratti, A., & Silani, V. (2020). Cerebrospinal fluid phosphorylated neurofilament heavy chain and chitotriosidase in primary lateral sclerosis. Journal of Neurology, Neurosurgery, and Psychiatry, jnnp-2020-324059. https://doi.org/10.1136/jnnp-2020-324059.
Verstraete, E., Turner, M. R., Grosskreutz, J., Filippi, M., & Benatar, M. (2015). Mind the gap: The mismatch between clinical and imaging metrics in ALS. Amyotroph Lateral Scler Frontotemporal Degener, 16(7–8), 524–529. https://doi.org/10.3109/21678421.2015.1051989.
Wakana, S., Caprihan, A., Panzenboeck, M. M., Fallon, J. H., Perry, M., Gollub, R. L., Hua, K., Zhang, J., Jiang, H., Dubey, P., Blitz, A., van Zijl, P., & Mori, S. (2007). Reproducibility of quantitative tractography methods applied to cerebral white matter. Neuroimage, 36(3), 630–644. https://doi.org/10.1016/j.neuroimage.2007.02.049.
Yunusova, Y., Plowman, E. K., Green, J. R., Barnett, C., & Bede, P. (2019). Clinical measures of bulbar dysfunction in ALS. Frontiers in Neurology, 10, 106. https://doi.org/10.3389/fneur.2019.00106.
Zhai, P., Pagan, F., Statland, J., Butman, J. A., & Floeter, M. K. (2003). Primary lateral sclerosis: A heterogeneous disorder composed of different subtypes? Neurology, 60(8), 1258–1265.
Zigmond, A. S., & Snaith, R. P. (1983). The hospital anxiety and depression scale. Acta Psychiatrica Scandinavica, 67(6), 361–370.
Acknowledgements
We acknowledge all patients with primary lateral sclerosis and all healthy controls who have kindly participated in this research study. Without their contribution, this study would have not been possible. This work was sponsored by the Spastic Paraplegia Foundation, Inc. (SPF). Professor Peter Bede is also supported by the Health Research Board (HRB EIA-2017-019), the EU Joint Programme – Neurodegenerative Disease Research (JPND), the Andrew Lydon scholarship, the Irish Institute of Clinical Neuroscience (IICN), the Iris O’Brien Foundation, and the Research Motor Neuron (RMN-Ireland) Foundation. Professor Russell L. McLaughlin and Ms. Jennifer C. Hengeveld are supported by Science Foundation Ireland (17/CDA/4737). The genetic aspects of this study were supported in part by a research grant from Science Foundation Ireland (SFI) under Grant Number 16/RC/3948, co-funded under the European Regional Development Fund and by FutureNeuro industry partners. The sponsors of the authors had no role in the study design, analyses or the interpretation of our research findings.
Funding sources
Spastic Paraplegia Foundation, Inc. (SPF), Health Research Board (HRB EIA-2017-019), the EU Joint Programme – Neurodegenerative Disease Research (JPND), the Andrew Lydon scholarship, the Irish Institute of Clinical Neuroscience (IICN), the Iris O’Brien Foundation, the Research Motor Neuron (RMN-Ireland) Foundation, Science Foundation Ireland (17/CDA/4737 and 16/RC/3948), European Regional Development Fund and FutureNeuro.
Author information
Authors and Affiliations
Contributions
Imaging analyses: EF, SLHS, RC, KMC, MCMcK, PB.
Genetic analyses: MD, JH, AV, RMcL.
Clinical characterization: EF, OH, NP, SH, CD.
Drafting the paper: EF, PB.
Corresponding author
Ethics declarations
Conflict of interest
none declared.
Ethical approval
The study was approved by the Ethics Committee of Beaumont Hospital, Dublin, Ireland and all participants provided written informed consent prior to inclusion.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 652 kb)
Rights and permissions
About this article

Cite this article
Finegan, E., Shing, S.L.H., Chipika, R.H. et al. Extra-motor cerebral changes and manifestations in primary lateral sclerosis. Brain Imaging and Behavior 15, 2283–2296 (2021). https://doi.org/10.1007/s11682-020-00421-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11682-020-00421-4