
Abstract
Progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) are neurodegenerative tauopathies with neuronal and glial lesions composed of tau that is composed predominantly of isomers with four repeats in the microtubule-binding domain (4R tau). The brain regions vulnerable to pathology in PSP and CBD overlap, but there are differences, particularly with respect to distribution of neuronal loss, the relative abundance of neuronal and glial lesions, the morphologic features of glial lesions, and the frequency of comorbid pathology. Both PSP and CBD have a wide spectrum of clinical manifestations, including disorders of movement and cognition. Recognition of phenotypic diversity in PSP and CBD may improve antemortem diagnostic accuracy, which tends to be very good for the most common presentation of PSP (Richardson syndrome), but poor for the most characteristic presentation of CBD (corticobasal syndrome: CBS). Development of molecular and imaging biomarkers may improve antemortem diagnostic accuracy. Currently, multidisciplinary symptomatic and supportive treatment with pharmacological and non-pharmacological strategies remains the standard of care. In the future, experimental therapeutic trials will be important to slow disease progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Progressive supranuclear palsy
- Corticobasal syndrome
- Corticobasal degeneration
- 4R tauopathies
- Epidemiology
- Clinical criteria
- Neuropathology
- Etiology
- Biomarkers
- Treatment
Introduction
The two most common clinicopathologic subtypes of frontotemporal lobar degeneration (FTLD) are characterized by TDP-43 or tau pathology [1]. Tau is a microtubule-associated protein important for stability and functional properties of microtubules. The gene that encodes tau protein (MAPT) is located on chromosome 17, and it undergoes alternative splicing of exons 2, 3, and 10 to generate six isoforms of tau [2]. Alternative splicing of exon 10 generates two major classes of tau protein that contain either three (3R) or four (4R) ≈30-amino acid repeats in the microtubule-binding domain of tau. Neurodegenerative tauopathies can be subclassified based upon the predominant type of tau that accumulates in cellular lesions [3]. Pick’s disease, a rare frontotemporal dementia with lobar cortical atrophy and neuronal Pick bodies, is characterized by tau composed predominantly of 3R tau, while neurofibrillary tangles that characterize the pathology in Alzheimer’s disease and chronic traumatic encephalopathy are composed of a mixture of 3R and 4R tau with distinct ultrastructural properties [4, 5]. Disorders associated with 4R tau are clinically and pathologically heterogeneous and include aging-related disorders, such as aging-related tau astrogliopathy (ARTAG) [6] and argyrophilic grain disease (AGD) [3, 7]. The most common of the neurodegenerative 4R tauopathies are progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), which is the focus of this chapter.
Progressive Supranuclear Palsy
PSP was described by Steele, Richardson, and Olszewski in a small autopsy series of patients with postural instability, vertical supranuclear gaze palsy, facial and cervical dystonia, as well as dementia. Despite some clinical variability, they shared distinctive pathologic features, including argyrophilic neurofibrillary tangles in select subcortical and brainstem nuclei. [8]. With the advent of tau biochemistry and molecular biology, the pathologic features of PSP have been expanded to include not only neuronal lesions but also glial lesions [3, 9]. The clinical syndromes associated with the characteristic tau pathology of PSP have also expanded from the original descriptions and is described later in the chapter.
Epidemiology of Progressive Supranuclear Palsy
The prevalence of PSP is thought to be approximately 6/100,000 patients [10,11,12,13]; however, there is a growing understanding that PSP pathology is associated with multiple clinical phenotypes, suggesting that the above figure may require revision. Increased awareness of this fact led to increased age-adjusted prevalence estimates in Europe (8.8–10.8/100,000 patients) [11, 14]. Of note, age-adjusted prevalence estimates from the same city in Japan (Yonogo) adjusted to the census of the earlier study increased from 5.8/100,000 patients in 1999 to 17/100,000 patients in 2010 [15, 16]. This is, in part, due to identification of more phenotypes, since the previous studies used the National Institute of Neurologic Disease and Stroke and Society for PSP (NINDS-SPSP) criteria that only identified the classical PSP phenotype (also named PSP-Richardson syndrome [PSP-RS]) .
Clinical Features of Progressive Supranuclear Palsy
In addition to the typical presentation described by Richardson and colleagues (PSP-RS), other phenotypes associated with PSP pathology have been described, including an extrapyramidal disorder mimicking Parkinson’s disease (PSP-P), corticobasal syndrome (PSP-CBS), dementia with predominantly frontal characteristics (PSP-F), dementia with speech and language disturbances (PSP-SL), and others. Consequently, the newest clinical criteria for PSP, supported by the International Parkinson and Movement Disorder Society (MDS-PSP criteria), include a wider clinical spectrum [17]. Typical age of onset of PSP is in the seventh decade of life [17,18,19], and average survival is 5–6 years; however, certain phenotypes are associated with much longer disease durations [19, 20].
Several criteria for PSP were proposed based upon clinical case series [21,22,23,24], but the first widely used criteria that were based on autopsy-confirmed cases was reported by Litvan et al. [18] and supported by the NINDS-SPSP. The NINDS-SPSP criteria outlined several core features of PSP-RS. Mandatory features included a gradually progressive disorder with age of onset 40 years of age or later, presence of vertical supranuclear gaze palsy, and/or postural instability with falls within the first year of disease. Both features had to be present for a diagnosis of “probable PSP,” and only vertical supranuclear gaze palsy or slowing of saccades and postural instability with falls within the first year of disease was consistent with a diagnosis of “possible PSP.”
Regarding vertical supranuclear gaze palsy, restricted downward gaze has been considered most specific for PSP because restricted upward gaze can be seen to a lesser degree in aging [25], Parkinson’s disease [26], and other conditions [27,28,29,30,31] like severely restricted upward gaze and slowing of vertical saccades. At more advanced stages, horizontal supranuclear gaze palsy may develop, as well [32]. Vertical supranuclear gaze palsy may be preceded by subtle ocular motor abnormalities, including loss of vertical optokinetic nystagmus [33], “stair casing,” and the “round the house sign” [34], where horizontal saccadic excursions interrupt vertical eye movements. Other ocular motor movement abnormalities include hypometric saccades, breakdown of smooth pursuit, and square wave jerks [35]. Loss of vergence is observed early and may contribute to frequent complaints of diplopia [36]. Other eye findings include blepharospasm and eyelid-opening apraxia [37], although these are not usually early features.
Early loss of postural reflexes and falls are common and often an early complaint in PSP-RS, usually occurring within the first year of illness. Falls tend to be backwards, but it can occur in any direction and may be compounded by freezing of gait. Falls can result in significant morbidity due to lacerations, fractures, or intracerebral bleeding [32, 38].
While these features define the core clinical features of PSP-RS, a number of other clinical features are often observed. Parkinsonism manifested by symmetric akinesia and rigidity with an axial predominance is common. Neck stiffness with retrocollis has been described in early descriptions of PSP, but it is rare [8]. Facial dystonia produces the so-called PSP stare, with decreased blink rate, furrowed and raised eyebrows, and a look of surprise. Inappropriate laughter and crying episodes are often observed (pseudobulbar affect). Early hypokinetic and spastic dysarthria is a secondary feature, which can progress to anarthria in severe cases [39]. Dysphagia occurs relatively early, and it is frequently implicated as a cause of death due to aspiration pneumonia [40, 41]. Cognitive manifestations associated with PSP overlap with corticobasal syndrome and frontotemporal dementia (FTD). The clinical course of PSP is relentless and nearly always is associated with a frontal-subcortical-type dementia.
PSP-RS phenotype is the clinical syndrome most likely to have PSP pathology at autopsy. Because of this, the NINDS-SPSP criteria proved to be specific for PSP pathology [42, 43], but to have relatively low sensitivity [43,44,45]. This is because PSP pathology can present with other clinical syndromes, and eye movement abnormalities seen in PSP often occur later in the course of the disease and sometimes not at all [19, 20, 46,47,48,49,50,51,52,53,54,55,56,57]. In one autopsy series, 76% of pathologically confirmed PSP had a clinical syndrome other than PSP-RS [58].
The most common clinical PSP variant mimics idiopathic Parkinson’s disease (PSP-P) and makes up about one-third of pathologically confirmed cases [46, 59,60,61,62]. These patients have asymmetric resting tremor and asymmetric appendicular bradykinesia and rigidity, making the distinction between PSP-P and Parkinson’s disease challenging [46, 59, 60, 62, 63]. As many as one-third of these patients will respond to levodopa and show greater than 30% reduction in the Unified Parkinson’s Disease Rating Scale [46, 64,65,66,67]. Some also develop levodopa-induced dyskinesias [46]. Most PSP patients have minimal or no response to levodopa therapy, and if a response occurs, it is typically mild and not sustained [20, 24, 68]. Robust and prolonged response to levodopa therapy is an exclusionary criterion for PSP and makes Parkinson’s disease a more likely diagnosis [17]. It can be 3–4 years into the disease course before supranuclear gaze palsy is present to aid in refining the diagnosis in PSP-P [19, 62]. PSP-P patients also have a longer disease duration than PSP-RS, with an average survival of 10–15 years [19, 46, 62].
Other syndromes have been described in autopsy-confirmed PSP. Some present with impulsivity and behavioral changes, including apathy, impulsivity, and social inappropriateness akin to behavioral-variant frontotemporal dementia (PSP-F) [53, 69, 70]. Others present with progressive non-fluent aphasia or apraxia of speech (PSP-SL) [48, 52, 70, 71]. About 10% have a corticobasal syndrome with asymmetrical dystonia, myoclonus, apraxia, and cortical sensory loss (PSP-CBS) [55, 56, 70, 72]. Another rare presentation, but one that is highly predictive of PSP pathology, is pure akinesia with gait freezing (PSP-PAGF) [47, 73, 74]. Early presentations currently considered to be “suggestive” of PSP in MDS-PSP criteria are isolated postural instability (PSP-PI) [19, 75] and isolated oculomotor dysfunction (PSP-OM) [19, 20]. The most uncommon presentations are progressive cerebellar ataxia (PSP-C) [51, 76, 77] and primary lateral sclerosis (PSP-PLS) [50, 57]. It is important to note that while some patients present with discrete syndromes, it is common for considerable overlap, and patients also acquire new signs and symptoms as the disease progresses. Regardless of the initial syndrome, most patients develop vertical supranuclear gaze palsy and postural instability, which are core features of PSP-RS, that make diagnosis obvious, but these may occur only later in the disease course in some of the PSP clinical variants [19].
Recognition of the spectrum of clinical heterogeneity in PSP, led the MDS-PSP criteria to incorporate a broader set of symptoms and signs, as well as levels of certainty that would be associated with PSP pathology [17]. These criteria are more sensitive, but they are less specific than the NINDS-SPSP criteria [78, 79]. The implementation of “multiple allocation extinction” rules (MAX rules) have been necessary to help disentangle patients who may be classified into more than one clinical MDS-PSP category [79]. Even so, these MAX rules may fail to separate up to 40% of patients with PSP-P and PSP-RS overlap syndromes [80]. These issues highlight the ongoing need for specific biomarkers to improve diagnostic accuracy of PSP during life.
Neuropathology of Progressive Supranuclear Palsy
The external appearance of PSP at postmortem evaluation depends upon the clinical syndrome. PSP-RS may have no significant cortical atrophy or mild atrophy affecting the dorsolateral frontal lobe. PSP-F and PSP-CBS usually have more marked frontal atrophy, especially affecting the superior frontal gyrus, while PSP-SL may have more significant frontal atrophy, especially affecting the peri-Sylvian inferior frontal gyrus. Asymmetry, which is not often assessed with research protocols that evaluate only one side of the brain for histology, can be notable in PSP-SL and PSP-CBS. PSP-PLS has focal atrophy affecting the precentral gyrus; it can be asymmetrical as well. The most striking macroscopic finding in PSP-RS (and PSP-P) is midbrain atrophy (Fig. 1a) with loss of neuromelanin pigment on transverse sections of the brainstem (Fig. 1d). The subthalamic nucleus invariably has atrophy (Fig. 1b), and there is also atrophy of the superior cerebellar peduncle (Fig. 1e) and atrophy of the hilus of the cerebellar dentate nucleus (Fig. 1c). Atrophy of subthalamic nucleus and midbrain is usually less severe in PSP-F and PSP-CBS, and often very severe in PSP-PAGF. In the latter, atrophy is frequently accompanied by similar changes in the globus pallidus and with reddish-brown discoloration due to deposition of iron pigment (pallido-nigro-luysial “pigment-spheroid degeneration” [81]).

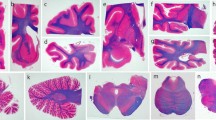
Macroscopic findings in PSP . (a) A sagittal section of the brainstem shows marked atrophy of the midbrain (arrows). (b) A coronal section of the diencephalon shows marked atrophy of the subthalamic nucleus (arrowheads). (c) A section of the cerebellum at the level of the middle cerebellar peduncle shows marked atrophy and discoloration of the dentate nucleus of the cerebellum (arrow). (d) A transverse section of the midbrain shows atrophy and marked neuromelanin pigment loss in the substantia nigra (asterisk). (e) A transverse section of the pons shows marked atrophy of the superior cerebellar peduncle (arrowheads)
Histopathologic findings in PSP are similar in the various subtypes. The clinicopathologic subtypes differ in the relative distribution of the neuronal loss and gliosis, and in the density of tau pathology [82]. There are no distinctive cellular pathologies in PSP clinicopathologic variants. The major histopathologic lesions in PSP are neurofibrillary tangles, which often have a globose shape in vulnerable subcortical nuclei, such as the subthalamic nucleus (Fig. 2a) and substantia nigra (Fig. 2b). The morphology and distribution of tangles in PSP is different from the most common disorder with neurofibrillary tangles, Alzheimer’s disease (AD), in that subcortical and brainstem nuclei are preferentially affected. The tangles are positive for phospho-tau (Fig. 2d). Using antibodies specific to tau isoforms, the tangles in PSP preferentially accumulate 4R tau (not shown). Tau immunohistochemistry also shows distinctive glial pathology in PSP, including tufted astrocytes (Figs. 2d and 3e) and oligodendroglial coiled bodies (Fig. 2f). Tufted astrocytes are most frequent in neocortex, neostriatum, and midbrain tectum. Coiled bodies are widespread in affected cerebral white matter and vulnerable subcortical fiber tracts in the basal telencephalon, diencephalon, brain stem, and cerebellum. A common neurodegenerative change in the cerebellar dentate nucleus that is not associated with tau pathology is the presence of irregularly swollen cell processes around apical dendrites and cell bodies of cerebellar dentate nucleus neurons (Fig. 2c), a process referred to as grumose degeneration [83]. Glial pathology is increasingly recognized to play a significant role in pathogenesis of neurodegenerative disease, and in PSP microgliosis and astrogliosis parallels the systems affected by neurodegeneration [84], with little evidence to suggest that it precedes tau pathology.

Microscopic findings in PSP . (a) An H&E stained section of the subthalamic nucleus shows severe neuronal loss and astrocytosis, with neurofibrillary tangles (arrow) in residual neurons. (b) An H&E stained section of the substantia nigra shows neuronal loss and gliosis with extraneuronal neuromelanin pigment and globose neurofibrillary tangles (arrowheads). (c) An H&E stained section of the cerebellar dentate nucleus shows granular eosinophilic swollen cell processes (arrowhead), obscuring the outlines of the neuron, findings characteristic of grumose degeneration (arrow). (d) Phospho-tau immunohistochemistry of the caudate nucleus shows a globose neurofibrillary tangle (arrowhead) and a tufted astrocyte (arrow). (e) Phospho-tau immunohistochemistry of the caudate nucleus shows several tufted astrocytes (arrows) with morphologic heterogeneity. (f) Phospho-tau immunohistochemistry of the internal capsule shows oligodendroglial coiled bodies (arrowhead). All images are of same magnification, bar in (f) is 20 μm

Macroscopic findings in CBD . (a) The medial surface of left hemibrain shows atrophy of the superior frontal gyrus (asterisk indicates area of greatest pathology) and focal atrophy of the corpus callosum (arrows). (b) A coronal section of the brain at the level of the fornix shows marked enlargement of the frontal horn of the lateral ventricle (large asterisk). There is also atrophy and discoloration of the globus pallidus (small asterisk). (c) A coronal section of the diencephalon and anterior medial temporal lobe shows no hippocampal atrophy and minimal-to-no atrophy of the subthalamic nucleus (arrowheads). (d) A section of the cerebellum at the level of the middle cerebellar peduncle shows no atrophy and normal myelin in the hilus of the dentate nucleus (arrow). (e) A transverse section of the pons shows no atrophy of the superior cerebellar peduncle (arrowheads). (f) A transverse section of the midbrain shows mild atrophy and marked neuromelanin pigment loss in the substantia nigra (asterisk)
Corticobasal Degeneration
The term corticobasal degeneration was coined by Gibb, Luthert and Marsden [85] to describe the pathology of a rare disorder associated with cognitive and motor features affecting the neocortex and basal ganglia. The clinically defined corticobasal syndrome (CBS) is characterized by progressive cognitive decline associated with asymmetrical rigidity, dystonia, myoclonus, and alien-limb phenomenon. Early autopsy studies reported focal cortical atrophy and swollen achromatic neurons (“ballooned neurons” [86]), as well as neuronal loss in the substantia nigra and cerebellar dentate nucleus—“corticodentatonigral degeneration with neuronal achromasia” [87]. These descriptions did not recognize the tau pathology in CBD because neuronal lesions in CBD are weakly positive or negative with traditional silver impregnation methods. It was not until the early 1990s that widespread tau pathology in CBD was shown to be distinct from Alzheimer’s disease, using immunohistochemistry and ultrastructural methods [88,89,90]. The pathognomonic astrocytic lesion of CBD (“astrocytic plaques”) was described in 1995 [91].
Epidemiology of Corticobasal Degeneration
Like PSP , pathologically confirmed CBD has a range of clinical presentations, and CBS may not be the most common. Moreover, the pathologic substrate of CBS is mixed, with PSP being as common as CBD [56, 92], but other disorders, particularly atypical presentations of Alzheimer’s disease, can also present with CBS [56, 85, 93,94,95,96,97,98]. Estimates of prevalence of CBD are inherently flawed. For these reasons, the term corticobasal syndrome (CBS) is now preferred to refer to the clinical presentation described earlier, whereas corticobasal degeneration (CBD) is reserved for the neuropathological diagnosis. The incidence of CBD is estimated to be 0.62–0.92/100,000 [93, 99,100,101].
Clinical Features of Corticobasal Degeneration Presenting as Corticobasal Syndrome
The onset of CBS is typically in the sixth or seventh decade of life, with a mean survival of about 7 years from diagnosis [93, 99,100,101]. The motor manifestations of CBS include an asymmetric parkinsonism manifested predominantly by rigidity and bradykinesia [93]. While asymmetry in parkinsonian features is common in Parkinson’s disease, the asymmetry in CBS can be striking. There is frequently additional dystonic posturing of the limb. Superimposed may be ideomotor and limb-kinetic apraxia [55, 99, 102]. Alien-limb phenomenon affecting the arm or leg has been described and often results in an unawareness of a levitating hand or leg due to feeling the limb alien, and more rarely, intermanual conflict [103]. Myoclonus is often present, and it may affect limbs or, rarely, the face [99, 104]. Myoclonus is worsened by action, posture, or stimuli [55, 99, 104]. At times, myoclonus can be difficult to differentiate from tremor, although the quality of myoclonic tremor is jerky rather than the smooth oscillatory tremor observed in Parkinson’s disease and other parkinsonian disorders [105]. Postural instability and falls are common, but usually later in the disease course than in PSP, unless the symptoms start in lower extremities [93]. Parkinsonism associated with CBS may benefit from levodopa therapy, but improvement in symptoms is rare and levodopa-induced dyskinesias are also rare [55]. Sustained and robust levodopa responsiveness is an exclusionary criterion to the diagnosis of CBS [93, 106].
Several cognitive features and other signs referable to higher-order cortical function are common in CBS. As previously mentioned, apraxia is a core feature. Ideomotor apraxia is usually one of the first disease features. Some patients develop orobuccal apraxia or apraxia of eyelid opening [99, 104, 107]. Cortical sensory loss with astereognosis and agraphesthesia are frequently observed [108, 109]. Visual neglect may be seen, and it is related to parietal lobe dysfunction [95, 107, 110]. A progressive non-fluent aphasia is also described in CBS, with occasional overlay of apraxia of speech from frontal lobe dysfunction [95, 104, 107, 111]. Other features of frontal lobe dysfunction, such as apathy and disinhibition, are common and early [55, 93].
The clinical presentation of autopsy-confirmed CBD is varied, with some presenting with a cognitive syndrome, and some primarily with a motor phenotype. Other neurodegenerative disorders, PSP and Alzheimer’s disease in particular, can present with CBS. Unlike PSP, these initial presentations may not necessarily coalesce into a common phenotype over time, making diagnostics even more challenging. Concomitantly, the clinical diagnosis of CBS has relatively poor predictive value for CBD pathology at autopsy compared to other neurodegenerative disorders. The sensitivity of clinical findings predicting CBD at autopsy is between 26% and56%. The majority of these studies were performed using older criteria; recently, more specific criteria have not been fully vetted [55, 59, 70, 95]. Current clinical criteria for CBD define a gradual progressive disorder with insidious onset and several possible phenotypes, including CBS, a frontal behavioral-spatial syndrome, a variant of primary non-fluent aphasia, and a PSP syndrome. The clinical syndrome of probable CBS is defined as having two of the following signs: limbs with asymmetric rigidity and akinesia, limb dystonia or limb myoclonus, and two of the following signs and symptoms: orobuccal or limb apraxia, cortical sensory deficits, or alien-limb phenomena. Possible clinical CBS involves having one limb with rigidity or akinesia, limb dystonia, or limb myoclonus with one of the above supportive features. A frontal behavioral spatial syndrome is described with the attendant cognitive features. Non-fluent primary progressive aphasia and a PSP phenotype are recognized but considered as possible CBD. Patients with a PSP clinical syndrome must have at least one additional symptom or sign (limb rigidity/akinesia, limb dystonia or myoclonus, apraxia, and cortical sensory loss) [93].
There are multiple exclusion criteria that, if present, make CBD a less likely cause of the clinical presentation. The most important are the presence of genetic mutations in GRN, FUS, TARDBP, PSEN1/2, and APP genes. Another exclusionary criterion is a cerebrospinal fluid (CSF) Aβ42/tau ratio consistent with Alzheimer’s disease [112]. Classic 4–6 Hz parkinsonian resting tremor, hallucinations, dysautonomia, cerebellar signs, the presence of both upper and motor neuron signs, or the semantic or logopenic variants of primary progressive aphasia are also considered exclusionary; they are more likely to indicate Parkinson’s disease, dementia with Lewy bodies, multiple systems atrophy, ALS, or FTLD . Lastly, because there are occasional reports of fulminant presentations of CBD [113, 114], imaging consistent with Creutzfeldt-Jakob disease is also exclusionary.
Neuropathology of Corticobasal Degeneration
The external appearance of the CBD brain at postmortem evaluation depends upon the clinical syndrome. For patients presenting with CBS or frontotemporal dementia syndromes, there is usually focal atrophy, especially affecting the medial superior frontal gyrus (Fig. 3a). Language-predominant syndromes often have inferior frontal gyrus (peri-Sylvian) atrophy. There is often atrophy of the corpus callosum (Fig. 3a), which tends to parallel the distribution and severity of the focal cortical pathology. Atrophy can be asymmetrical, but this is often difficult to assess at autopsy, given that half the brain is usually frozen for research purposes. Some cases, particularly patients with long tract signs, may have atrophy that extends to the motor cortex. Coronal sections frequently show enlargement of the frontal horn of the lateral ventricle (Fig. 3b). The most common finding in the basal ganglia is atrophy and reddish-brown discoloration of the globus pallidus (Fig. 2b). Unlike PSP, there is usually no significant atrophy of the subthalamic nucleus (Fig. 3c). Similarly, the hilus of the cerebellar dentate nucleus (Fig. 3d) and the superior cerebellar peduncle (Fig. 3e) do not have atrophy. Similar to PSP, there is usually loss of neuromelanin pigment in the substantia nigra (Fig. 3f).
Microscopic examination of atrophic cortical sections shows neuronal loss with superficial spongiosis, gliosis, and usually achromatic or ballooned neurons, which are readily detected with routine histology stains, such as hematoxylin-and-eosin (Fig. 4a). Ballooned neurons are found in middle and lower cortical layers of affected neocortices and have diffuse phospho-tau immunoreactivity (Fig. 4d), as well as intense immunoreactivity with antibodies to alpha-B-crystallin, a small heat-shock protein (not shown), and for neurofilament.

Microscopic findings in CBD . (a) An H&E stained section of superior frontal gyrus shows ballooned neurons (arrow). (b) An H&E stained section of the subthalamic nucleus shows mild neuronal loss, but more marked gliosis. (c) An H&E stained section of the substantia nigra shows focal neuronal loss (extraneuronal neuromelanin—asterisk) and several neurons with so-called corticobasal bodies (arrowheads). (d) Phospho-tau immunohistochemistry of the superior frontal gyrus shows many neuropil threads and a ballooned neuron with diffuse cytoplasmic tau immunoreactivity (arrow). (e) Phospho-tau immunohistochemistry of the caudate nucleus shows an astrocytic plaque (asterisk). (f) Phospho-tau immunohistochemistry of the subthalamic nucleus shows morphologic heterogeneity of neuronal inclusions (arrowheads). Panels a and c–f are of same magnification, bar in (f) is 20 μm. Panel (b) is a lower magnification, bar is 50 μm
In addition to ballooned neurons, the neocortex and neostriatum in CBD have widespread deposition of tau in both neurons and glia [3, 9]. Glial inclusions are found in both oligodendroglia and astrocytes. The astrocytic lesions have a characteristic plaque-like morphology (“astrocytic plaques” [91]) (Fig. 4e) that is morphologically distinct from tufted astrocytes of PSP. The pathologic feature that best discriminates PSP from CBD is pervasive thread-like cell processes in affected gray and white matter in CBD, to the extent that the difference can be seen by examining the slide with the naked eye (Fig. 5).

Comparison of tau burden in PSP and CBD. Sections of the neostriatum in PSP and CBD, immunostained under the same conditions with a sensitive phospho-tau antibody (CP13 from Peter Davies, Feinstein Institute, Long Island, NY), show a clear distinction between PSP and CBD, due to dense tau pathology, mostly thread-like processes (not visible at this magnification), in CBD
The subthalamic nucleus often has at least mild neuronal loss and gliosis (Fig. 4b), but it is rarely as severe as in PSP. Similarly, the substantia nigra has neuronal loss in CBD, but it can be mild (Fig. 4c). Neurons in the substantia nigra may have so-called corticobasal bodies [85] (Fig. 4c). Cortical neurons in atrophic areas have pleomorphic tau-immunoreactive lesions. In some neurons, tau is densely packed into small irregular inclusion bodies. In other neurons, the inclusions are more diffuse (“pre-tangles”). Neurofibrillary lesions in subcortical nuclei, such as the subthalamic nucleus, also typically have marked morphologic heterogeneity (Fig. 4f), while those in the locus ceruleus and substantia nigra can resemble globose neurofibrillary tangles (Fig. 4c).
Pathogenesis of Progressive Supranuclear Palsy and Corticobasal Degeneration
There is no single cause of PSP or CBD, but several environmental and genetic factors have been investigated. The Environmental Genetic PSP (ENGENE-PSP) study found that lower educational attainment, exposure to well water and industrial wastes, and firearm use were related to higher risk of developing PSP [115, 116]. These findings are also supported by a cluster of PSPs that emerged in northern France in an area of high industrial waste contamination [117]. Consumption of high levels of annonacin, a mitochondrial complex I inhibitor, found in the paw-paw fruit was associated with developing PSP or other atypical parkinsonian syndromes in studies in the Caribbean island of Guadeloupe [118, 119]. There may be a slight male predominance within PSP patients [22, 46], and one study documented that increased estrogen exposure in women may be protective against developing PSP [120]. Environmental exposures have not been evaluated in CBD to date.
MAPT mutations may lead to either PSP or CBD [121,122,123,124]. Mutations in this gene can also lead to frontotemporal dementia, FTLD with parkinsonism, or primary progressive aphasia [125]. The H1/H1 genotype elevates the risk for developing PSP and CBD [17, 126, 127]. One genome-wide association study in a large cohort of pathologically validated PSP patients additionally identified genetic risk variants at the MOBP, STX6, and EIF2AK3 loci [128]. MOBP , which encodes for myelin oligodendrocyte-binding protein, is also implicated in CBD and highlights potential importance of white matter [121, 129]. STX6 encodes for a SNARE protein implicated in fusing vesicles in the Golgi network [130]. EIF2AK3 encodes for a protein responsible for inhibiting protein synthesis in the face of excess endoplasmic reticulum stress [131, 132]. These genes have been validated in a second genome-wide association study, which additionally identified SLCO1A2 and DUSP10 as other genomic loci of interest [133].
Oxidative stress and inflammation can also be demonstrated in PSP and CBD. Mitochondrial enzymatic activity is decreased in both brain tissue and also in skeletal muscle in PSP patients [134,135,136,137,138,139,140]. Higher IL-1β and other inflammatory cytokines are found in the brains and CSF of PSP patients and lead to microglial activation [141, 142], which has been implicated in tau deposition [84]. Superoxide dismutase and glutathione, essential antioxidants, are often seen to be elevated in PSP brain tissue, possibly as a defense mechanism [139, 143].
Recent data suggest that misfolded tau oligomers are capable of acting as a template and induce further misfolding of normal monomeric tau leading to larger and larger aggregates, causing cellular damage and ultimately death and likely leading to spreading of disease in a ‘prion-like’ manner. In vivo animal studies using preformed fibrils [144, 145], human diseased brain homogenates [146], and other techniques [147, 148] have shown distal spread of tau pathology via trans-synaptic spread [149, 150]. There may be specific “strains” of tau capable of seeding unique tau pathologies [147, 151, 152].
Biomarkers in Progressive Supranuclear Palsy and Corticobasal Degeneration
The clinicopathologic overlap between PSP and CBD and other neurodegenerative diseases makes the discovery of sensitive and specific biomarkers for these diseases of paramount importance.
Magnetic Resonance Imaging
PSP is well described to be associated with several features on structural magnetic resonance imaging (MRI) . Most recognized is the presence of midbrain atrophy, resulting in the “hummingbird sign” best seen on the mid-sagittal section (Fig. 6) [153], as well as “morning glory sign [154]”, or “Mickey Mouse sign [155]”. In one study of an autopsy series of pathologically confirmed cases with PSP, multiple systems atrophy (MSA), or Parkinson’s disease (PD), 16/22 (72.7%) of PSP cases were able to be correctly identified by a radiologist reviewing conventional MRI that had been performed during life, and the presence of a hummingbird sign or morning glory sign was 100% specific but was 68.4% sensitive [156]. One study, however, that included different clinical variants of PSP found midbrain atrophy to be a feature of the Richardson syndrome variant, but midbrain atrophy was not found to be a biomarker of PSP pathology [157]. The superior cerebellar peduncle is also frequently atrophied in PSP and, consequently, several different ratios comparing brain stem, pons, superior cerebellar peduncle, and middle cerebellar peduncle measurements have been studied to differentiate PSP from other parkinsonian diseases and from healthy controls. A frequent problem with these measurements is that they are often insensitive, and the radiologic signs will only manifest at later stages of the disease after neurodegeneration has progressed to the point of causing these recognizable patterns [158,159,160,161,162,163]. A more specific technique to assess the superior cerebellar peduncle is with diffusion tensor imaging (DTI). One DTI study did find the superior cerebellar peduncle to be able to accurately distinguish PSP from normal controls [164]. It is unclear whether atrophy of the superior cerebellar peduncle is a feature of PSP pathology or a feature of Richardson syndrome. Another technique that has also been studied in PSP is resting-state functional magnetic resonance imaging (fMRI). Resting-state fMRI studies have demonstrated disrupted thalamocortical connectivity in PSP [165, 166].

MRI scan in autopsy-confirmed PSP and CBD. MRI scan in PSP shows the classic hummingbird sign on sagittal MRI, while asymmetric atrophy of the posterior frontal cortex is seen in CBD
Fewer MRI studies have been performed in CBD, but the most frequently cited sign is asymmetric cortical atrophy, affecting the parietal and frontal lobes (Fig. 6) [167,168,169,170,171]. Corpus callosum atrophy is also cited occasionally. Regrettably, neither of these features are specific for CBD to fully differentiate it from other pathologies that cause CBS clinical phenotypes [70, 167, 172]. In addition, symmetric cortical atrophy has been described in autopsy-confirmed cases of CBD [173]. Research studies have utilized voxel-based morphometry to try to distinguish CBD from Alzheimer’s disease and other neurodegenerative diseases that present with CBS. These studies have found distinguishing features at the group level [174, 175]. No biomarker exists to distinguish CBD from other neurodegenerative diseases at the single subject level.
Given the prominent white matter degeneration that is common to these conditions, diffusion tensor imaging and white matter volumetric measurements may show more degeneration in PSP and CBD than atypical AD or FTLD TDP-43 that may have overlapping presentations [176,177,178,179].
DaTscan
A DaTscan is used to detect dopamine transporters on dopamine neurons. DaTscans are typically utilized to differentiate Parkinson’s disease from essential tremor. However, DaTscans have been performed in PSP and CBS patients and show a reduction in dopamine transporter receptors. Unfortunately, this finding is nonspecific and can also be seen in other parkinsonian disorders, for example, MSA .
Positron Emission Tomography
The most common PET scan is the fluorodeoxyglucose (FDG)-PET scan, which utilizes radioactive glucose to assess for functional integrity of neocortical regions. FDG-PET findings in PSP and CBS tend to mirror findings on MRI. In PSP, hypometabolism is observed in the premotor cortex as well as the midbrain, the latter when present is known as the pimple sign of PSP [180] (Fig. 7). In CBS and CBD, the FDG-PET scan reveals asymmetric frontal and/or parietal hypometabolism (Fig. 7). There are less than a handful of studies on FDG-PET in autopsy-confirmed PSP, CBD, and other 4R tauopathies. One such study found parietal hypometabolism in CBD and premotor hypometabolism in PSP [181]. Several tracers are currently under investigation that bind to the tau proteins, including 18F-5105, 18F-FDDNP, 18F-THK523, 11C-PBB3, and others [182]. 18F-Flortaucipir (formerly AV-1451 and T807) is the most researched tau tracer to date and appears to bind avidly to paired helical filaments in 3R/4R tauopathies, such as AD [183], and exhibits retention patterns in amnestic AD consistent with Braak tau staging [184, 185] and in posterior cortical regions in posterior cortical atrophy patients [186, 187]. However, 18F-Flortaucipir retention appears to be less robust in 4R tauopathies [183, 188, 189]. Increased retention in the basal ganglia and midbrain can be demonstrated in PSP (Fig. 8), but there is off-site binding, which makes individual patient-level distinctions at early stage difficult [184, 190,191,192,193]. Similarly, in CBS, mild increases in retention in cortical regions can be demonstrated (Fig. 8) that correlate with postmortem tau findings [194], although this has been reported to occur predominantly in CBS patients who presented with a motor speech disorder [195]. PET tracers targeting activated microglia (11C-(R) PK11195) may aid in assessing inflammation associated with neurodegeneration in PSP and CBD [196, 197].

FDG-PET in autopsy-confirmed PSP and CBD. FDG-PET in PSP shows the classic “pimple sign” (hypometabolism of the midbrain) on mid-sagittal section. Also seen is mild hypometabolism of medial prefrontal and supplementary motor cortex. In CBD, asymmetric frontoparietal hypometabolism is observed on the lateral view

Flortaucipir PET in autopsy-confirmed PSP and CBD. Flortaucipir PET (AV-1451) in PSP shows increased uptake in the midbrain (substantia nigra) and dentate nucleus of the cerebellum. In a case of CBD that presented with progressive speech apraxia, flortaucipir PET demonstrates asymmetric increased uptake in premotor neocortex
Biofluid Biomarkers
CSF tau species, including measures of total tau (t-tau) and phosphorylated tau (p-tau) tend not to be elevated in PSP [198,199,200]. One study reported that a ratio of certain tau fragments may aid in distinguishing PSP from healthy controls and other conditions [201], but the findings could not be replicated [202]. CSF neurofilament light chain (NfL) is an intermediate filament, which can be measured from CSF and is a nonspecific measure of neuronal injury [203], but it shows elevation in PSP, CBD, and other parkinsonian syndromes that can aid in differentiating PSP or CBD from Parkinson’s disease [200, 204,205,206,207]. The sensitivity of the next-generation single-molecule-array assays has made blood-based NfL measurements possible now as well [208, 209]. Real-time quaking-induced conversion (RT-QuIC) is an emerging assay that was originally developed to aid in diagnosis of Creutzfeldt-Jakob Disease (CJD), where a biologic sample is placed in wells containing monomeric proteins and a fluorescent marker and through polymerization encouraged by sequential shaking steps, can show the presence or absence of a pathologic “seed” from the patient sample. This technique has been adapted to detect alpha-synuclein [210], 3R/4R tau species [211], 3R tau species [212], and a 4R tauopathy assay is under development as well [213], which may offer molecularly specific aid in diagnosis in the near future.
Treatment of Progressive Supranuclear Palsy and Corticobasal Degeneration
Current treatment strategies for both PSP and CBS are supportive and symptomatic as no disease-modulating therapies are currently available for either condition.
Parkinsonism
Levodopa preparation may still be trialed to treat the parkinsonism associated with PSP and CBS. In one study of pathologically confirmed PSP patients, approximately one-third of PSP patients showed a significant improvement (> 30% improvement in the Unified Parkinson’s Disease Rating Scale) [46], which is a response rate that has been reported in other studies as well [64,65,66,67]. Doses of over 1 gm/day of levodopa for 1 month are proposed to elicit responses. Often, however, responses to levodopa are very mild in PSP and CBS, if present at all, and typically wane over time [20, 24, 55, 68, 99, 214]. Dopamine agonists have been trialed in PSP but are generally less effective than levodopa and are more likely to cause side effects [65, 215, 216]. Smaller studies documented improvement in parkinsonism using amantadine or amitriptyline in PSP, but caution is warranted because of possible anticholinergic side effects, including cognitive and psychiatric disturbances, dry mouth, or difficulty with urination [65, 217,218,219].
Ocular Symptoms
Zolpidem showed mild improvements in saccadic speed in one small study of patients with PSP, but those findings have not been replicated [220,221,222]. Botulinum toxin may be used to treat blepharospasm and eyelid-opening apraxia, but high doses are often required to achieve benefits [223, 224]. Artificial tears and ophthalmic ointments may be used to treat dry eyes, and sunglasses may be of use to aid in photosensitivity symptoms. Alternating an eye patch is useful for double vision, and, occasionally, prism lenses may be fashioned, if the deficits are fixed.
Spasticity, Dystonia, and Myoclonus
Muscle relaxants such as baclofen, tizanadine , and cyclobenzaprine may be considered, but they must be carefully weighed against their possible side effects of somnolence [225]. Botulinum toxin may be used for the disabling focal dystonia of the limbs or neck that occurs in both conditions [223, 225, 226]. Clonazepam or levetiracetam can treat the myoclonus associated with CBS as can valproate [214, 227, 228].
Sialorrhea
Again, botulinum toxin may be used to treat sialorrhea [229], as can medications including glycopyrrolate or 1% atropine drops placed sublingually, although the latter, if not carefully applied, can be absorbed systemically and cause anticholinergic side effects [230].
Memory Impairments
Acetylcholinesterase inhibitors such as donepezil, rivastigmine, or galantamine may offer some mild improvement in memory function, but studies showed that it may worsen gait and dysphagia in PSP and worsen behavioral symptoms in FTD, so it should be used with caution [227, 231, 232]. No studies of memantine in autopsy-confirmed CBD have been performed, but multiple studies of memantine for memory dysfunction in FTD have failed to show benefits [233, 234].
Mood Changes
Selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors may be used to treat depression and anxiety, but they are not helpful for the apathy that can accompany PSP or CBS [227]. Dextromethorphan-quinidine is an effective treatment for pseudobulbar affect as are antidepressants [235].
Nonpharmacological Therapies
PSP and CBS patients benefit from multidisciplinary care from providers knowledgeable about these conditions. Physical therapy decreases the likelihood of falls and improves global functioning [227, 236,237,238]. Weighted walkers are often recommended to aid in safer ambulation. Speech therapy may be employed to strengthen vocal muscles but to also provide strategies for more effective communication [239, 240]. Swallowing evaluations are essential if the patient complains of dysphagia or frequent coughing during meals as food consistency or eating habits may be modified. Safety inspections of the home may be helpful and can often be done my occupational therapists who can suggest changes and modifications to promote safety. Social workers are often needed to aid in utilization of resources that may be available to these patients. Lastly, palliative care consultants can help to manage transitions to less aggressive modalities of care and to promote symptom management and navigate end-of-life decision-making in a way that aids in both the patients and the families’ quality of life [241].
Experimental Therapies for Progressive Supranuclear Palsy and Corticobasal Degeneration
Although there are no current disease-modulating treatment for PSP or CBD, several medications are under investigation, many of which target the tau protein by different mechanisms: by decreasing production, stabilizing microtubules, promoting immune system clearance, or modifying post-translational changes.
Tau in PSP and CBD commonly undergoes post-translational phosphorylation and acetylation [242]; unfortunately, trials of the GSK-3β kinase inhibitors lithium, valproate, and Tideglusib failed to show efficacy or were stopped due to poor tolerability [243]. Salsalate inhibits tau acetylation in animal models and is currently under early investigation (NCT02422485) [244]. O-Glc-NAC modification and caspase-mediated cleavage are other potential therapeutic targets [245, 246]
The microtubule-stabilizing agent davunetide failed to show efficacy in a phase IIb/III trial [247], and the taxane derivative TPI-287 inducted anaphylactic reactions, which necessitated trial stoppage [248]. Other compounds still under investigation that are thought to work through this mechanism include epothilone-D and methylene blue [249, 250].
Anti-inflammatory medications have been trialed in PSP, including rasagiline, CoQ10, and riluzole, but studies have failed to show efficacy [251,252,253], although there was significant benefit in a shorter trial using CoQ10 [254].
Tau immunotherapy is actively under investigation. Specifically, in PSP, the BIIB092 antibody product, directed against the N terminus of extracellular tau [255], showed promise in early trials [256, 257], but a phase II study failed to show efficacy (PASSPORT NCT03068468) [258]. Similarly, ABBV-8E12 had favorable early safety results and good target engagement [259, 260] but failed to show efficacy in larger trials. While these results are discouraging, a number of questions remain regarding this strategy, namely if proper epitopes of tau were selected [261, 262], if oligomeric species or intracellular tau should be prioritized although it is technically more challenging [184, 262,263,264,265,266,267], or if alternative delivery systems may increase blood–brain barrier penetration of antibody products and improve efficacy [184].
Gene therapy through small interfering RNA (siRNA) or antisense oligonucleotides are currently being investigated in animal models of tauopathies [268,269,270] and may be of future use in PSP and CBD.
References
Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM et al (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122(2):137–153
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12(6):609–622
Dickson DW (2004) Sporadic tauopathies: Pick’s disease, corticobasal degeneration, progressive suprnauclear palsy and argyrophilic grain disease. In: Esiri MM, Lee VMY, Trojanowski JQ (eds) The neuropathology of dementia, 2nd edn. Cambridge University Press, Cambridge/New York, pp 227–256
Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R et al (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568(7752):420–423
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ et al (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547(7662):185–190
Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al (2016) Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131(1):87–102
Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M et al (2002) Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 61(6):547–556
Steele JC, Richardson JC, Olszewski J (1964) Progressive supranuclear palsy. A heterogenous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10:333–359
Dickson DW, Hauw J-J, Agid Y, Litvan I (2011) Progressive supranuclear palsy and corticobasal degeneration. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Wiley-Blackwell/International Society of Neuropathology, Chichester/West Sussex, p xvii, 477 p.
Nath U, Ben-Shlomo Y, Thomson R, Morris HR, Wood N, Lees A et al (2001) The prevalence of progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome) in the UK. Brain 124(7):1438–1449
Coyle-Gilchrist IT, Dick KM, Patterson K, Rodríquez PV, Wehmann E, Wilcox A et al (2016) Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 86(18):1736–1743
Schrag A, Ben-Shlomo Y, Quinn N (1999) Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 354(9192):1771–1775
Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW et al (2017) Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord 32(7):995–1005
Fleury V, Brindel P, Nicastro N, Burkhard PR (2018) Descriptive epidemiology of parkinsonism in the Canton of Geneva, Switzerland. Parkinsonism Relat Disord 54:30–39
Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K (2004) Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord: Off J Mov Disord Soc 19(10):1239–1240
Takigawa H, Ikeuchi T, Aiba I, Morita M, Onodera O, Shimohata T et al (2016) Japanese longitudinal biomarker study in PSP and CBD (JALPAC): a prospective multicenter PSP/CBD cohort study in Japan. Parkinsonism Relat Disord 22:e120–e1e1
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE et al (2017) Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord:n/a–n/a
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC et al (1996) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47(1):1–9
Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ et al (2014) The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord: Off J Mov Disord Soc 29(14):1758–1766
Litvan I, Mangone CA, McKee A, Verny M, Parsa A, Jellinger K et al (1996) Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 60(6):615–620
Maher E, Lees A (1986) The clinical features and natural history of the Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 36(7):1005
Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC (1988) Prevalence and natural history of progressive supranuclear palsy. Neurology 38(7):1031
Litvan I, Agid Y (1992) Progressive supranuclear palsy: clinical and research approaches. Oxford University Press, New York
Collins S, Ahlskog J, Parisi JE, Maraganore D (1995) Progressive supranuclear palsy: neuropathologically based diagnostic clinical criteria. J Neurol Neurosurg Psychiatry 58(2):167–173
Chamberlain W (1971) Restriction in upward gaze with advancing age. Am J Ophthalmol 71(1):341–346
Vidailhet M, Rivaud S, Gouider-Khouja N, Pillon B, Bonnet AM, Gaymard B et al (1994) Eye movements in parkinsonian syndromes. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 35(4):420–426
Gibb W, Esiri M, Lees A (1987) Clinical and pathological features of diffuse cortical Lewy body disease (Lewy body dementia). Brain 110(5):1131–1153
Grant MP, Cohen M, Petersen RB, Halmagyi GM, McDougall A, Tusa RJ et al (1993) Abnormal eye movements in Creutzfeldt–Jakob disease. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 34(2):192–197
Paulson H, Subramony S (2003) Spinocerebellar Ataxia 3—Machado-Joseph disease (SCA3). In: Genetics of movement disorders. Elsevier, pp 57–69
Brüggemann N, Wandinger KP, Gaig C, Sprenger A, Junghanns K, Helmchen C et al (2016) Dystonia, lower limb stiffness, and upward gaze palsy in a patient with IgLON5 antibodies. Mov Disord 31(5):762–764
Adams C, McKeon A, Silber MH, Kumar R (2011) Narcolepsy, REM sleep behavior disorder, and supranuclear gaze palsy associated with Ma1 and Ma2 antibodies and tonsillar carcinoma. Arch Neurol 68(4):521–524
Boeve BF (2012) Progressive supranuclear palsy. Parkinsonism Relat Disord 18:S192–S1S4
Garbutt S, Riley D, Kumar A, Han Y, Harwood M, Leigh R (2004) Abnormalities of optokinetic nystagmus in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 75(10):1386–1394
Quinn N (1996) The “round the houses” sign in progressive supranuclear palsy. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 40(6):951
Lal V, Truong D (2019) Eye movement in movement disorders. Clin Parkinsonism Relat Disord 1:54–63
Kitthaweesin K, Riley DE, Leigh RJ (2002) Vergence disorders in progressive supranuclear palsy. Ann New York Acad Sci 956(1):504–507
Yoon WT, Chung EJ, Lee SH, Kim BJ, Lee WY (2005) Clinical analysis of blepharospasm and apraxia of eyelid opening in patients with parkinsonism. J Clin Neurol 1(2):159–165
Williams DR, Watt HC, Lees AJ (2006) Predictors of falls and fractures in bradykinetic rigid syndromes: a retrospective study. J Neurol Neurosurg Psychiatry 77(4):468–473
Kluin KJ, Foster NL, Berent S, Gilman S (1993) Perceptual analysis of speech disorders in progressive supranuclear palsy. Neurology 43(3 Part 1):563
Müller J, Wenning GK, Verny M, McKee A, Chaudhuri KR, Jellinger K et al (2001) Progression of dysarthria and dysphagia in postmortem-confirmed parkinsonian disorders. Arch Neurol 58(2):259–264
Papapetropoulos S, Singer C, McCorquodale D, Gonzalez J, Mash DC (2005) Cause, seasonality of death and co-morbidities in progressive supranuclear palsy (PSP). Parkinsonism Relat Disord 11(7):459–463
Litvan I, Hauw J, Bartko J, Lantos P, Daniel S, Horoupian D et al (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55(1):97–105
Respondek G, Roeber S, Kretzschmar H, Troakes C, Al-Sarraj S, Gelpi E et al (2013) Accuracy of the National Institute for Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Mov Disord: Off J Mov Disord Soc 28(4):504–509
Osaki Y, Ben-Shlomo Y, Lees AJ, Daniel SE, Colosimo C, Wenning G et al (2004) Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord 19(2):181–189
Birdi S, Rajput AH, Fenton M, Donat JR, Rozdilsky B, Robinson C et al (2002) Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Mov Disord: Off J Mov Disord Soc 17(6):1255–1264
Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L et al (2005) Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 128(Pt 6):1247–1258
Williams DR, Holton JL, Strand K, Revesz T, Lees AJ (2007) Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord: Off J Mov Disord Soc 22(15):2235–2241
Boeve B, Dickson D, Duffy J, Bartleson J, Trenerry M, Petersen R (2003) Progressive nonfluent aphasia and subsequent aphasic dementia associated with atypical progressive supranuclear palsy pathology. Eur Neurol 49(2):72–78
Boeve BF, Maraganore D, Parisi JE, Ahlskog J, Graff-Radford N, Caselli RJ et al (1999) Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 53(4):795
Josephs KA, Katsuse O, Beccano-Kelly DA, Lin W-L, Uitti RJ, Fujino Y et al (2006) Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 65(4):396–405
Koga S, Josephs KA, Ogaki K, Labbé C, Uitti RJ, Graff-Radford N et al (2016) Cerebellar ataxia in progressive supranuclear palsy: an autopsy study of PSP-C. Mov Disord 31(5):653–662
Josephs KA, Duffy JR (2008) Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol 21(6):688–692
Hassan A, Parisi JE, Josephs KA (2012) Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase 18(6):478–488
Han HJ, Kim H, Park JH, Shin HW, Kim GU, Kim DS et al (2010) Behavioral changes as the earliest clinical manifestation of progressive supranuclear palsy. J Clin Neurol (Seoul, Korea) 6(3):148–151
Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR et al (2010) Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 133(7):2045–2057
Ling H, De Silva R, Massey L, Courtney R, Hondhamuni G, Bajaj N et al (2014) Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol 40(2):149–163
Nagao S, Yokota O, Nanba R, Takata H, Haraguchi T, Ishizu H et al (2012) Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci 323(1–2):147–153
Respondek G, Höglinger G (2016) The phenotypic spectrum of progressive supranuclear palsy. Parkinsonism Relat Disord 22:S34–SS6
Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ (2002) The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 125(Pt 4):861–870
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55(3):181–184
Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ et al (2007) Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 130(6):1566–1576
Williams DR, Lees AJ (2010) What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord 25(3):357–362
Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E et al (2014) Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 83(5):406–412
Richardson J, Steele J, Olszewski J (1963) Supranuclear opthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of “heterogeneous system degeneration”. Trans Am Neurol Assoc 88:25
Nieforth KA, Golbe LI (1993) Retrospective study of drug response in 87 patients with progressive supranuclear palsy. Clin Neuropharmacol 16(4):338–346
Tan E, Chan L, Wong M (2003) Levodopa-induced oromandibular dystonia in progressive supranuclear palsy. Clin Neurol Neurosurg 105(2):132–134
Lang AE (2005) Treatment of progressive supranuclear palsy and corticobasal degeneration. Mov Disord: Off J Mov Disord Soc 20(S12):S83–S91
Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I et al (2003) Movement disorders society scientific issues committee report: SIC task force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov Disord: Off J Mov Disord Soc 18(5):467–486
Donker Kaat L, Boon AJ, Kamphorst W, Ravid R, Duivenvoorden HJ, van Swieten JC (2007) Frontal presentation in progressive supranuclear palsy. Neurology 69(8):723–729
Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR et al (2006) Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66(1):41–48
Mochizuki A, Ueda Y, Komatsuzaki Y, Tsuchiya K, Arai T, Shoji S (2003) Progressive supranuclear palsy presenting with primary progressive aphasia--clinicopathological report of an autopsy case. Acta Neuropathol 105(6):610–614
Tsuboi Y, Josephs KA, Boeve BF, Litvan I, Caselli RJ, Caviness JN et al (2005) Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord 20(8):982–988
Compta Y, Valldeoriola F, Tolosa E, Rey MJ, Martí MJ, Valls-Solé J (2007) Long lasting pure freezing of gait preceding progressive supranuclear palsy: a clinicopathological study. Mov Disord 22(13):1954–1958
Facheris MF, Maniak S, Scaravilli F, Schüle B, Klein C, Pramstaller PP (2008) Pure akinesia as initial presentation of PSP: a clinicopathological study. Parkinsonism Relat Disord 14(6):517–519
Kurz C, Ebersbach G, Respondek G, Giese A, Arzberger T, Höglinger GU (2016) An autopsy-confirmed case of progressive supranuclear palsy with predominant postural instability. Acta Neuropathol Commun 4(1):120
Kanazawa M, Shimohata T, Toyoshima Y, Tada M, Kakita A, Morita T et al (2009) Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord: Off J Mov Disord Soc 24(9):1312–1318
Kanazawa M, Tada M, Onodera O, Takahashi H, Nishizawa M, Shimohata T (2013) Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia. Parkinsonism Relat Disord 19(12):1149–1151
Ali F, Martin PR, Botha H, Ahlskog JE, Bower JH, Masumoto JY et al (2019) Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Mov Disord 34(8):1144–1153
Ali F, Botha H, Whitwell JL, Josephs KA (2019) Utility of the movement disorders society criteria for progressive supranuclear palsy in clinical practice. Mov Disord Clin Pract 6(6):436–439
Shoeibi A, Litvan I, Juncos JL, Bordelon Y, Riley D, Standaert D et al (2019) Are the International Parkinson disease and Movement Disorder Society progressive supranuclear palsy (IPMDS-PSP) diagnostic criteria accurate enough to differentiate common PSP phenotypes? Parkinsonism Relat Disord. In Press
Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW (2008) Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain 131(Pt 2):460–472
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23(4):394–400
Ishizawa K, Lin WL, Tiseo P, Honer WG, Davies P, Dickson DW (2000) A qualitative and quantitative study of grumose degeneration in progressive supranuclear palsy. J Neuropathol Exp Neurol 59(6):513–524
Ishizawa K, Dickson DW (2001) Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol 60(6):647–657
Gibb WR, Luthert PJ, Marsden CD (1989) Corticobasal degeneration. Brain 112(Pt 5):1171–1192
Dickson DW, Yen SH, Suzuki KI, Davies P, Garcia JH, Hirano A (1986) Ballooned neurons in select neurodegenerative diseases contain phosphorylated neurofilament epitopes. Acta Neuropathol 71(3–4):216–223
Rebeiz JJ, Kolodny EH, Richardson EP Jr (1967) Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 92:23–26
Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH et al (1994) Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol 145(6):1496–1508
Mori H, Nishimura M, Namba Y, Oda M (1994) Corticobasal degeneration: a disease with widespread appearance of abnormal tau and neurofibrillary tangles, and its relation to progressive supranuclear palsy. Acta Neuropathol 88(2):113–121
Uchihara T, Mitani K, Mori H, Kondo H, Yamada M, Ikeda K (1994) Abnormal cytoskeletal pathology peculiar to corticobasal degeneration is different from that of Alzheimer’s disease or progressive supranuclear palsy. Acta Neuropathol 88(4):379–383
Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146(6):1388–1396
Kouri N, Murray ME, Hassan A, Rademakers R, Uitti RJ, Boeve BF et al (2011) Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain 134(Pt 11):3264–3275
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B et al (2013) Criteria for the diagnosis of corticobasal degeneration. Neurology 80(5):496–503
Boeve BF (2011) The multiple phenotypes of corticobasal syndrome and corticobasal degeneration: implications for further study. J Mol Neuro: MN 45(3):350–353
Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ et al (2011) Clinicopathological correlations in corticobasal degeneration. Ann Neurol 70(2):327–340
Schneider J, Watts R, Gearing M, Brewer R, Mirra S (1997) Corticobasal degeneration neuropathologic and clinical heterogeneity. Neurology 48(4):959–968
Hu WT, Rippon GW, Boeve BF, Knopman DS, Petersen RC, Parisi JE et al (2009) Alzheimer’s disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord: Off J Mov Disord Soc 24(9):1375–1379
Chand P, Grafman J, Dickson D, Ishizawa K, Litvan I (2006) Alzheimer’s disease presenting as corticobasal syndrome. Mov Disord: Off J Mov Disord Soc 21(11):2018–2022
Wenning GK, Litvan I, Jankovic J, Granata R, Mangone CA, McKee A et al (1998) Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry 64(2):184–189
Litvan I, Agid Y, Goetz C, Jankovic J, Wenning GK, Brandel JP et al (1997) Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 48(1):119–125
Togasaki DM, Tanner CM (2000) Epidemiologic aspects. Adv Neurol 82:53–59
Tsuchiya K, Murayama S, Mitani K, Oda T, Arima K, Mimura M et al (2005) Constant and severe involvement of Betz cells in corticobasal degeneration is not consistent with pyramidal signs: a clinicopathological study of ten autopsy cases. Acta Neuropathol 109(4):353–366
Josephs KA, Rossor MN (2004) The alien limb. Pract Neurol 4(1):44–45
Grimes DA, Lang AE, Bergeron CB (1999) Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology 53(9):1969–1974
Kompoliti K, Goetz C, Boeve BF, Maraganore D, Ahlskog J, Marsden C et al (1998) Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol 55(7):957–961
Boeve BF, Lang AE, Litvan I (2003) Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 54(S5):S15–SS9
Murray R, Neumann M, Forman M, Farmer J, Massimo L, Rice A et al (2007) Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 68(16):1274–1283
Jacobs DH, Adair JC, Macauley B, Gold M, Rothi LJG, Heilman KM (1999) Apraxia in corticobasal degeneration. Brain Cogn 40(2):336–354
Reich SG, Grill SE (2009) Corticobasal degeneration. Curr Treat Options Neurol 11(3):179
Spotorno N, McMillan CT, Powers JP, Clark R, Grossman M (2014) Counting or chunking? Mathematical and heuristic abilities in patients with corticobasal syndrome and posterior cortical atrophy. Neuropsychologia 64:176–183
Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE et al (2006) Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 129(Pt 6):1385–1398
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC et al (2009) Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65(4):403–413
Ling H, Gelpi E, Davey K, Jaunmuktane Z, Mok KY, Jabbari E et al Fulminant corticobasal degeneration: a distinct variant with predominant neuronal tau aggregates. Acta Neuropathol:1–18
Rodriguez-Porcel F, Lowder L, Rademakers R, Ravenscroft T, Ghetti B, Hagen MC et al (2016) Fulminant corticobasal degeneration: Agrypnia excitata in corticobasal syndrome. Neurology 86(12):1164–1166
Litvan I, Lees PS, Cunningham CR, Rai SN, Cambon AC, Standaert DG et al (2016) Environmental and occupational risk factors for progressive supranuclear palsy: case-control study. Mov Disord 31(5):644–652
Kelley KD, Checkoway H, Hall DA, Reich SG, Cunningham C, Litvan I (2018) Traumatic brain injury and firearm use and risk of progressive supranuclear palsy among veterans. Front Neurol 9:474
Caparros-Lefebvre D, Golbe LI, Deramecourt V, Maurage CA, Huin V, Buee-Scherrer V et al (2015) A geographical cluster of progressive supranuclear palsy in northern France. Neurology 85(15):1293–1300
Caparros-Lefebvre D, Sergeant N, Lees A, Camuzat A, Daniel S, Lannuzel A et al (2002) Guadeloupean parkinsonism: a cluster of progressive supranuclear palsy-like tauopathy. Brain 125(Pt 4):801–811
Lannuzel A, Ruberg M, Michel PP (2008) Atypical parkinsonism in the Caribbean island of Guadeloupe: etiological role of the mitochondrial complex I inhibitor annonacin. Mov Disord: Off J Mov Disord Soc 23(15):2122–2128
Park HK, Ilango S, Charriez CM, Checkoway H, Riley D, Standaert DG et al (2018) Lifetime exposure to estrogen and progressive supranuclear palsy: environmental and genetic PSP study. Mov Disord 33(3):468–472
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A et al (2015) Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun 6:7247
Rohrer JD, Paviour D, Vandrovcova J, Hodges J, De Silva R, Rossor MN (2011) Novel L284R MAPT mutation in a family with an autosomal dominant progressive supranuclear palsy syndrome. Neurodegener Dis 8(3):149–152
Ogaki K, Li Y, Takanashi M, Ishikawa K-I, Kobayashi T, Nonaka T et al (2013) Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord 19(1):15–20
Ahmed S, Fairen MD, Sabir MS, Pastor P, Ding J, Ispierto L et al (2019) MAPT p.V363I mutation: a rare cause of corticobasal degeneration. Neurology Genetics 5(4):e347
Boeve BF, Hutton M (2008) Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch Neurol 65(4):460–464
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J et al (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8(4):711–715
Houlden H, Baker M, Morris H, MacDonald N (2001) Pickering–Brown S, Adamson J, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56(12):1702–1706
Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang L-S, Klei L et al (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43(7):699
Yokoyama JS, Karch CM, Fan CC, Bonham LW, Kouri N, Ross OA et al (2017) Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol 133(5):825–837
Wendler F, Tooze S (2001) Syntaxin 6: the promiscuous behaviour of a SNARE protein. Traffic 2(9):606–611
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5(5):897–904
Yuan SH, Hiramatsu N, Liu Q, Sun XV, Lenh D, Chan P et al (2018) Tauopathy-associated PERK alleles are functional hypomorphs that increase neuronal vulnerability to ER stress. Hum Mol Genet 27(22):3951–3963
Sanchez-Contreras MY, Kouri N, Cook CN, Serie DJ, Heckman MG, Finch NA et al (2018) Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegener 13(1):37
Albers DS, Augood SJ, Martin DM, Standaert DG, Vonsattel JPG, Beal MF (1999) Evidence for oxidative stress in the subthalamic nucleus in progressive supranuclear palsy. J Neurochem 73(2):881–884
Albers DS, Augood SJ, Park LC, Browne SE, Martin DM, Adamson J et al (2000) Frontal lobe dysfunction in progressive supranuclear palsy: evidence for oxidative stress and mitochondrial impairment. J Neurochem 74(2):878–881
Albers DS, Swerdlow RH, Manfredi G, Gajewski C, Yang L, Parker WD Jr et al (2001) Further evidence for mitochondrial dysfunction in progressive supranuclear palsy. Exp Neurol 168(1):196–198
Park LC, Albers DS, Xu H, Lindsay JG, Beal MF, Gibson GE (2001) Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J Neurosci Res 66(5):1028–1034
Albers DS, Beal MF (2002) Mitochondrial dysfunction in progressive supranuclear palsy. Neurochem Int 40(6):559–564
Cantuti-Castelvetri I, Keller-McGandy CE, Albers DS, Beal MF, Vonsattel J-P, Standaert DG et al (2002) Expression and activity of antioxidants in the brain in progressive supranuclear palsy. Brain Res 930(1–2):170–181
Martinelli P, Scaglione C, Lodi R, Iotti S, Barbiroli B (2000) Deficit of brain and skeletal muscle bioenergetics in progressive supranuclear palsy shown in vivo by phosphorus magnetic resonance spectroscopy. Mov Disord 15(5):889–893
Fernandez-Botran R, Ahmed Z, Crespo FA, Gatenbee C, Gonzalez J, Dickson DW et al (2011) Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism Relat Disord 17(9):683–688
Starhof C, Winge K, Heegaard NHH, Skogstrand K, Friis S, Hejl A (2018) Cerebrospinal fluid pro-inflammatory cytokines differentiate parkinsonian syndromes. J Neuroinflammation 15(1):305
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F et al (1994) Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 36(3):348–355
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM-Y (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33(3):1024–1037
Clavaguera F, Lavenir I, Falcon B, Frank S, Goedert M, Tolnay M (2013) “Prion-like” templated misfolding in tauopathies. Brain Pathol 23(3):342–349
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci 110(23):9535–9540
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82(6):1271–1288
Probst A, Götz J, Wiederhold K, Tolnay M, Mistl C, Jaton A et al (2000) Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol 99(5):469–481
Dujardin K, Defebvre L, Duhamel A, Lecouffe P, Rogelet P, Steinling M et al (2004) Cognitive and SPECT characteristics predict progression of Parkinson’s disease in newly diagnosed patients. J Neurol 251(11):1383–1392
Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M et al (2014) Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol 127(2):299–301
Nishimura M, Namba Y, Ikeda K, Oda M (1992) Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett 143(1–2):35–38
Yamada T, McGeer P, McGeer E (1992) Appearance of paired nucleated, tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett 135(1):99–102
Kato N, Arai K, Hattori T (2003) Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci 210(1–2):57–60
Adachi M, KAWANAMI T, OHSHIMA H, Sugai Y, Hosoya T (2004) Morning glory sign: a particular MR finding in progressive supranuclear palsy. Magn Reson Med Sci 3(3):125–132
Massey LA, Micallef C, Paviour DC, O’sullivan SS, Ling H, Williams DR et al (2012) Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 27(14):1754–1762
Massey LA, Micallef C, Paviour DC, O’Sullivan SS, Ling H, Williams DR et al (2012) Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord: Off J Mov Disord Soc 27(14):1754–1762
Whitwell JL, Jack CR Jr, Parisi JE, Gunter JL, Weigand SD, Boeve BF et al (2013) Midbrain atrophy is not a biomarker of progressive supranuclear palsy pathology. Eur J Neurol 20(10):1417–1422
Massey LA, Jager HR, Paviour DC, O’Sullivan SS, Ling H, Williams DR et al (2013) The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology 80(20):1856–1861
Quattrone A, Nicoletti G, Messina D, Fera F, Condino F, Pugliese P et al (2008) MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology 246(1):214–221
Moller L, Kassubek J, Sudmeyer M, Hilker R, Hattingen E, Egger K et al (2017) Manual MRI morphometry in parkinsonian syndromes. Mov Disord: Off J Mov Disord Soc 32(5):778–782
Nigro S, Arabia G, Antonini A, Weis L, Marcante A, Tessitore A et al (2017) Magnetic Resonance Parkinsonism Index: diagnostic accuracy of a fully automated algorithm in comparison with the manual measurement in a large Italian multicentre study in patients with progressive supranuclear palsy. Eur Radiol 27(6):2665–2675
Zanigni S, Calandra-Buonaura G, Manners DN, Testa C, Gibertoni D, Evangelisti S et al (2016) Accuracy of MR markers for differentiating Progressive Supranuclear Palsy from Parkinson’s disease. NeuroImage Clinical 11:736–742
Hussl A, Mahlknecht P, Scherfler C, Esterhammer R, Schocke M, Poewe W et al (2010) Diagnostic accuracy of the magnetic resonance Parkinsonism index and the midbrain-to-pontine area ratio to differentiate progressive supranuclear palsy from Parkinson’s disease and the Parkinson variant of multiple system atrophy. Mov Disord: Off J Mov Disord Soc 25(14):2444–2449
Whitwell JL, Master AV, Avula R, Kantarci K, Eggers SD, Edmonson HA et al (2011) Clinical correlates of white matter tract degeneration in progressive supranuclear palsy. Arch Neurol 68(6):753–760
Whitwell JL, Avula R, Master A, Vemuri P, Senjem ML, Jones DT et al (2011) Disrupted thalamocortical connectivity in PSP: a resting-state fMRI, DTI, and VBM study. Parkinsonism Relat Disord 17(8):599–605
Gardner RC, Boxer AL, Trujillo A, Mirsky JB, Guo CC, Gennatas ED et al (2013) Intrinsic connectivity network disruption in progressive supranuclear palsy. Ann Neurol 73(5):603–616
Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC et al (2008) Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 29(2):280–289
Gröschel K, Hauser T-K, Luft A, Patronas N, Dichgans J, Litvan I et al (2004) Magnetic resonance imaging-based volumetry differentiates progressive supranuclear palsy from corticobasal degeneration. NeuroImage 21(2):714–724
Hauser RA, Murtaugh FR, Akhter K, Gold M, Olanow C (1996) Magnetic resonance imaging of corticobasal degeneration. J Neuroimaging 6(4):222–226
Koyama M, Yagishita A, Nakata Y, Hayashi M, Bandoh M, Mizutani T (2007) Imaging of corticobasal degeneration syndrome. Neuroradiology 49(11):905–912
Boxer AL, Geschwind MD, Belfor N, Gorno-Tempini ML, Schauer GF, Miller BL et al (2006) Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol 63(1):81–86
Josephs KA, Tang-Wai DF, Edland SD, Knopman DS, Dickson DW, Parisi JE et al (2004) Correlation between antemortem magnetic resonance imaging findings and pathologically confirmed corticobasal degeneration. Arch Neurol 61(12):1881–1884
Hassan A, Whitwell JL, Boeve BF, Jack CR Jr, Parisi JE, Dickson DW et al (2010) Symmetric corticobasal degeneration (S-CBD). Parkinsonism Relat Disord 16(3):208–214
Josephs KA, Whitwell JL, Boeve BF, Knopman DS, Petersen RC, Hu WT et al (2010) Anatomical differences between CBS-corticobasal degeneration and CBS-Alzheimer’s disease. Mov Disord: Off J Mov Disord Soc 25(9):1246–1252
Whitwell JL, Jack CR Jr, Boeve BF, Parisi JE, Ahlskog JE, Drubach DA et al (2010) Imaging correlates of pathology in corticobasal syndrome. Neurology 75(21):1879–1887
McMillan CT, Boyd C, Gross RG, Weinstein J, Firn K, Toledo JB et al (2016) Multimodal imaging evidence of pathology-mediated disease distribution in corticobasal syndrome. Neurology 87(12):1227–1234
Whitwell JL, Jack CR, Parisi JE, Knopman DS, Boeve BF, Petersen RC et al (2011) Imaging signatures of molecular pathology in behavioral variant frontotemporal dementia. J Mol Neurosci 45(3):372
Whitwell JL, Josephs KA (2011) Neuroimaging in frontotemporal lobar degeneration--predicting molecular pathology. Nat Rev Neurol 8(3):131–142
McMillan CT, Irwin DJ, Avants BB, Powers J, Cook PA, Toledo JB et al (2013) White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry 84(9):949–955
Botha H, Whitwell JL, Madhaven A, Senjem ML, Lowe V, Josephs KA (2014) The pimple sign of progressive supranuclear palsy syndrome. Parkinsonism Relat Disord 20(2):180–185
Zalewski N, Botha H, Whitwell JL, Lowe V, Dickson DW, Josephs KA (2014) FDG-PET in pathologically confirmed spontaneous 4R-tauopathy variants. J Neurol 261(4):710–716
Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC (2015) Tau imaging: early progress and future directions. Lancet Neurol 14(1):114–124
Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG et al (2015) Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 78(5):787–800
Passamonti L, Vazquez Rodriguez P, Hong YT, Allinson KS, Williamson D, Borchert RJ et al (2017) 18F-AV-1451 positron emission tomography in Alzheimer’s disease and progressive supranuclear palsy. Brain 140(3):781–791
Pontecorvo MJ, Devous Sr MD, Navitsky M, Lu M, Salloway S, Schaerf FW et al (2017) Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140(3):748–763
Nasrallah IM, Chen YJ, Hsieh M-K, Phillips JS, Ternes K, Stockbower GE et al (2018) 18F-Flortaucipir PET/MRI correlations in nonamnestic and amnestic variants of Alzheimer disease. J Nucl Med 59(2):299–306
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL et al (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139(5):1551–1567
Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE et al (2016) An autoradiographic evaluation of AV-1451 tau PET in dementia. Acta Neuropathol Commun 4(1):58
Bevan Jones WR, Cope TE, Passamonti L, Fryer TD, Hong YT, Aigbirhio F et al (2016) [18F]AV-1451 PET in behavioral variant frontotemporal dementia due to MAPT mutation. Ann Clin Trans Neurol 3(12):940–947
Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS et al (2017) Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord: Off J Mov Disord Soc 32(1):134–140
Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hagerstrom D et al (2017) Increased basal ganglia binding of 18 F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord: Off J Mov Disord Soc 32(1):108–114
Whitwell JL, Lowe VJ, Tosakulwong N, Weigand SD, Senjem ML, Schwarz CG et al (2017) [18 F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord: Off J Mov Disord Soc 32(1):124–133
Whitwell JL, Tosakulwong N, Botha H, Ali F, Clark HM, Duffy JR et al (2020) Brain volume and flortaucipir analysis of progressive supranuclear palsy clinical variants. NeuroImage: Clinical 25:102152
McMillan CT, Irwin DJ, Nasrallah I, Phillips JS, Spindler M, Rascovsky K et al (2016) Multimodal evaluation demonstrates in vivo (18)F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 132(6):935–937
Ali F, Whitwell JL, Martin PR, Senjem ML, Knopman DS, Jack CR et al (2018) [(18)F] AV-1451 uptake in corticobasal syndrome: the influence of beta-amyloid and clinical presentation. J Neurol 265(5):1079–1088
Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord: Off J Mov Disord Soc 21(1):89–93
Gerhard A, Watts J, Trender-Gerhard I, Turkheimer F, Banati RB, Bhatia K et al (2004) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov Disord: Off J Mov Disord Soc 19(10):1221–1226
Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S et al (2001) Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 101(2):167–173
Urakami K, Wada K, Arai H, Sasaki H, Kanai M, Shoji M et al (2001) Diagnostic significance of tau protein in cerebrospinal fluid from patients with corticobasal degeneration or progressive supranuclear palsy. J Neurol Sci 183(1):95–98
Hall S, Öhrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F et al (2012) Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol 69(11):1445–1452
Borroni B, Malinverno M, Gardoni F, Alberici A, Parnetti L, Premi E et al (2008) Tau forms in CSF as a reliable biomarker for progressive supranuclear palsy. Neurology 71(22):1796–1803
Kuiperij HB, Borroni B, Verbeek MM, Gardoni F, Malinverno M, Padovani A et al (2011) Tau forms in CSF as a reliable biomarker for progressive supranuclear palsy. Neurology 76(16):1443
Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T et al (2018) Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol:1
Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ, Andreasson U et al (2017) Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 88(10):930–937
Holmberg B, Rosengren L, Karlsson JE, Johnels B (1998) Increased cerebrospinal fluid levels of neurofilament protein in progressive supranuclear palsy and multiple-system atrophy compared with Parkinson’s disease. Mov Disord: Off J Mov Disord Soc 13(1):70–77
Marques TM, van Rumund A, Oeckl P, Kuiperij HB, Esselink RA, Bloem BR et al (2019) Serum NFL discriminates Parkinson disease from atypical parkinsonisms. Neurology 92(13):e1479–e1e86
Sako W, Murakami N, Izumi Y, Kaji R (2015) Neurofilament light chain level in cerebrospinal fluid can differentiate Parkinson’s disease from atypical parkinsonism: evidence from a meta-analysis. J Neurol Sci 352(1–2):84–87
Rojas JC, Karydas A, Bang J, Tsai RM, Blennow K, Liman V et al (2016) Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 3(3):216–225
Kuhle J, Barro C, Andreasson U, Derfuss T, Lindberg R, Sandelius A et al (2016) Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med 54(10):1655–1661
Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B et al (2018) Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun 6(1):7
Kraus A, Saijo E, Metrick MA 2nd, Newell K, Sigurdson CJ, Zanusso G et al (2019) Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol 137(4):585–598
Saijo E, Ghetti B, Zanusso G, Oblak A, Furman JL, Diamond MI et al (2017) Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol 133(5):751–765
Saijo E, Metrick MA, Koga S, Parchi P, Litvan I, Spina S et al (2020) 4-repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol 139(1):63–77
Riley D, Lang A, Ae L, Resch L, Ashby P, Hornykiewicz O et al (1990) Cortical-basal ganglionic degeneration. Neurology 40(8):1203
Jackson JA, Jankovic J, Ford J (1983) Progressive supranuclear palsy: clinical features and response to treatment in 16 patients. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 13(3):273–278
Stowe R, Ives N, Clarke CE, Ferreira J, Hawker RJ, Shah L et al (2008) Dopamine agonist therapy in early Parkinson’s disease. Cochrane Database Syst Rev (2):CD006564
Kompoliti K, Goetz C, Litvan I, Jellinger K, Verny M (1998) Pharmacological therapy in progressive supranuclear palsy. Arch Neurol 55(8):1099–1102
Engel PA (1996) Treatment of progressive supranuclear palsy with amitriptyline: therapeutic and toxic effects. J Am Geriatr Soc 44(9):1072–1074
Rajrut A, Uitti R, Fenton M, George D (1997) Amantadine effectiveness in multiple system atrophy and progressive supranuclear palsy. Parkinsonism Relat Disord 3(4):211–214
Daniele A, Moro E, Bentivoglio AR (1999) Zolpidem in progressive supranuclear palsy. N Engl J Med 341(7):543–544
Cotter C, Armytage T, Crimmins D (2010) The use of zolpidem in the treatment of progressive supranuclear palsy. J Clin Neurosci 17(3):385–386
Mayr BJ, Bonelli RM, Niederwieser G, Költringer P, Reisecker F (2002) Zolpidem in progressive supranuclear palsy. Eur J Neurol 9(2):184–185
Müller J, Wenning G, Wissel J, Seppi K, Poewe W (2002) Botulinum toxin treatment in atypical parkinsonian disorders associated with disabling focal dystonia. J Neurol 249(3):300–304
Piccione F, Mancini E, Tonin P, Bizzarini M (1997) Botulinum toxin treatment of apraxia of eyelid opening in progressive supranuclear palsy: report of two cases. Arch Phys Med Rehabil 78(5):525–529
Vanek Z, Jankovic J (2001) Dystonia in corticobasal degeneration. Mov Disord: Off J Mov Disord Soc 16(2):252–257
Polo KB, Jabbari B (1994) Botulinum toxin-A improves the rigidity of progressive supranuclear palsy. Ann Neurol: Off J Am Neurol Assoc Child Neurol Soc 35(2):237–239
Boeve BF, Josephs KA, Drubach DA (2008) Current and future management of the corticobasal syndrome and corticobasal degeneration. Handb Clin Neurol 89:533–548
Kovács T, Farsang M, Vitaszil E, Barsi P, Györke T, Szirmai I et al (2009) Levetiracetam reduces myoclonus in corticobasal degeneration: report of two cases. J Neural Transm 116(12):1631
Gómez-Caravaca MT, Cáceres-Redondo MT, Huertas-Fernández I, Vargas-González L, Carrillo F, Carballo M et al (2015) The use of botulinum toxin in the treatment of sialorrhea in parkinsonian disorders. Neurol Sci 36(2):275–279
Hyson HC, Johnson AM, Jog MS (2002) Sublingual atropine for sialorrhea secondary to parkinsonism: a pilot study. Mov Disord: Off J Mov Disord Soc 17(6):1318–1320
Litvan I, Phipps M, Pharr VL, Hallett M, Grafman J, Salazar A (2001) Randomized placebo-controlled trial of donepezil in patients with progressive supranuclear palsy. Neurology 57(3):467–473
Mendez MF, Shapira JS, McMurtray A, Licht E (2007) Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiatry 15(1):84–87
Boxer AL, Knopman DS, Kaufer DI, Grossman M, Onyike C, Graf-Radford N et al (2013) Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 12(2):149–156
Boxer AL, Lipton AM, Womack K, Merrilees J, Neuhaus J, Pavlic D et al (2009) An open label study of memantine treatment in three subtypes of frontotemporal lobar degeneration. Alzheimer Dis Assoc Disord 23(3):211
Pattee GL, Wymer JP, Lomen-Hoerth C, Appel SH, Formella AE, Pope LE (2014) An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin 30(11):2255–2265
Clerici I, Ferrazzoli D, Maestri R, Bossio F, Zivi I, Canesi M et al (2017) Rehabilitation in progressive supranuclear palsy: effectiveness of two multidisciplinary treatments. PLoS One 12(2):e0170927
Zampieri C, Di Fabio RP (2008) Balance and eye movement training to improve gait in people with progressive supranuclear palsy: quasi-randomized clinical trial. Phys Ther 88(12):1460–1473
Steffen TM, Boeve BF, Mollinger-Riemann LA, Petersen CM (2007) Long-term locomotor training for gait and balance in a patient with mixed progressive supranuclear palsy and corticobasal degeneration. Phys Ther 87(8):1078–1087
Henry M, Meese M, Truong S, Babiak M, Miller B, Gorno-Tempini M (2013) Treatment for apraxia of speech in nonfluent variant primary progressive aphasia. Behav Neurol 26(1–2):77–88
Farrajota L, Maruta C, Maroco J, Martins IP, Guerreiro M, De Mendonca A (2012) Speech therapy in primary progressive aphasia: a pilot study. Dement Geriatr Cogn Disord Extra 2(1):321–331
Wiblin L, Lee M, Burn D (2017) Palliative care and its emerging role in multiple system atrophy and progressive supranuclear palsy. Parkinsonism Relat Disord 34:7–14
Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 33(1):95–130
Leclair-Visonneau L, Rouaud T, Debilly B, Durif F, Houeto JL, Kreisler A et al (2016) Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin Neurol Neurosurg 146:35–39
Min S-W, Chen X, Tracy TE, Li Y, Zhou Y, Wang C et al (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21(10):1154
Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K et al (2012) Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol 8(4):393–399
Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC (2016) Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci U S A 113(52):15120–15125
Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS et al (2014) Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol 13(7):676–685
Boxer AMZ, Tsai R, Koestler M, Rojas J, Ljubenkov P, Rosen H et al (2017) A phase 1B, randomized, double-blind, placebo-controlled, sequential cohort, dose-ranging study of the safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary efficacy of TPI 287 (abeotaxane) in patients with primary four repeat tauopathies: corticobasal syndrome or progressive supranuclear palsy; or the secondary tauopathy, Alzheimer’s disease. J Prev Alz Dis 4(4):282–428
Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS et al (2012) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci Off J Soc Neurosci 32(11):3601–3611
Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM et al (2015) Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimer’s Disease: JAD 44(2):705–720
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN (2009) Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 132(Pt 1):156–171
Apetauerova D, Scala SA, Hamill RW, Simon DK, Pathak S, Ruthazer R et al (2016) CoQ10 in progressive supranuclear palsy: a randomized, placebo-controlled, double-blind trial. Neurology(R) Neuroimmunol Neuroinflammation 3(5):e266
Nuebling G, Hensler M, Paul S, Zwergal A, Crispin A, Lorenzl S (2016) PROSPERA: a randomized, controlled trial evaluating rasagiline in progressive supranuclear palsy. J Neurol 263(8):1565–1574
Stamelou M, Reuss A, Pilatus U, Magerkurth J, Niklowitz P, Eggert KM et al (2008) Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Mov Disord: Off J Mov Disord Soc 23(7):942–949
Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T et al (2015) Human secreted tau increases amyloid-beta production. Neurobiol Aging 36(2):693–709
Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M (2018) A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimer’s & Dement: Transl Res Clin Interv 4:746–755
Boxer AL, Qureshi I, Ahlijanian M, Grundman M, Golbe LI, Litvan I et al (2019) Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol 18(6):549–558
Dam T, Boxer A, Golbe LI, Höglinger G, Morris HR, Litvan I et al (2018) Efficacy and safety of BIIB092 in patients with progressive supranuclear palsy: passport phase 2 study design (P6. 073). AAN Enterprises
Budur K, West T, Braunstein JB, Fogelman I, Bordelon YM, Litvan I et al (2017) Results of a phase 1, single ascending dose, placebo-controlled study of ABBV-8E12 in patients with progressive supranuclear palsy and phase 2 study design in early Alzheimer’s disease. Alzheimers Dement 13(7):P599–P600
West T, Hu Y, Verghese P, Bateman R, Braunstein J, Fogelman I et al (2017) Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 4(04):236–241
Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM (2011) Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem 118(4):658–667
Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H et al (2011) Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem 286(39):34457–34467
Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G et al (2015) Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One 10(5):e0125614
Ittner A, Bertz J, Suh LS, Stevens CH, Gotz J, Ittner LM (2015) Tau-targeting passive immunization modulates aspects of pathology in tau transgenic mice. J Neurochem 132(1):135–145
Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Kayed R (2014) Specific targeting of tau oligomers in htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis 40(s1):S97–S111
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V et al (2012) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2:700
Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K (2011) Engineering the variable region of therapeutic IgG antibodies. MAbs 3(3):243–252
Xu H, Rosler TW, Carlsson T, de Andrade A, Fiala O, Hollerhage M et al (2014) Tau silencing by siRNA in the P301S mouse model of tauopathy. Curr Gene Ther 14(5):343–351
Sud R, Geller ET, Schellenberg GD (2014) Antisense-mediated exon skipping decreases tau protein expression: a potential therapy for tauopathies. Mol Ther Nucleic Acids 3:e180
Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF et al (2016) Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron 90(5):941–947
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Coughlin, D.G., Dickson, D.W., Josephs, K.A., Litvan, I. (2021). Progressive Supranuclear Palsy and Corticobasal Degeneration. In: Ghetti, B., Buratti, E., Boeve, B., Rademakers, R. (eds) Frontotemporal Dementias . Advances in Experimental Medicine and Biology, vol 1281. Springer, Cham. https://doi.org/10.1007/978-3-030-51140-1_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-51140-1_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-51139-5
Online ISBN: 978-3-030-51140-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)