
Abstract
Aim
To investigate whether primary lateral sclerosis (PLS) represents part of the amyotrophic lateral sclerosis–frontotemporal dementia (ALS–FTD) spectrum of diseases.
Methods
Comprehensive assessment was taken on 21 patients with PLS and results were compared to patients diagnosed with pure motor ALS (n = 27) and ALS–FTD (n = 12). Clinical features, Addenbrooke’s Cognitive Examination (ACE) scores, Motor Neuron Disease Behaviour (Mind-B) scores, motor disability on the ALS functional rating scale (ALSFRS) and survival times were documented. Motor cortex excitability was evaluated using transcranial magnetic stimulation (TMS).
Results
Global cognition was impaired in PLS (mean total ACE score 82.5 ± 13.6), similar to ALS–FTD (mean total ACE score 76.3 ± 7.7, p > 0.05) while behavioural impairments were not prominent. TMS revealed that resting motor threshold (RMT) was significantly higher in PLS (75.5 ± 6.2) compared ALS–FTD (50.1 ± 7.2, p < 0.001) and ALS (62.3 ± 12.6, p = 0.046). Average short-interval intracortical inhibition (SICI) was similar in all three patient groups. The mean survival time was longest in PLS (217.4 ± 22.4 months) and shortest in ALS–FTD (38.5 ± 4.5 months, p = 0.002). Bulbar onset disease (β = − 0.45, p = 0.007) and RMT (β = 0.54, p = 0.001) were independent predictors of global cognition while motor scores (β = 0.47, p = 0.036) and SICI (β = 0.58, p = 0.006) were significantly associated with ALSFRS.
Conclusion
The cognitive profile in PLS resembles ALS–FTD, without prominent behavioural disturbances. A higher RMT in PLS than ALS and ALS–FTD is consistent with differential cortical motor neuronal abnormalities and more severe involvement of corticospinal axons while SICI, indicative of inhibitory interneuronal dysfunction was comparable with ALS and ALS–FTD. Overall, while these findings support the notion that PLS lies on the ALS–FTD spectrum, the mechanisms underlying slow disease progression are likely to be distinct in PLS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary lateral sclerosis (PLS) is a late onset, slowly progressive spastic motor disorder with, by definition, absence of lower motor neuron involvement [1, 2]. While early descriptions of PLS emphatically argued for normal intellectual function as part of the diagnosis, there is accumulating evidence of cognitive dysfunction and neuroimaging changes overlapping with ALS [3,4,5,6,7]. One of the first systematic neuropsychological evaluations in PLS [3] found predominant deficits in executive function, alongside memory and verbal learning deficits. More recent work comparing cognitive and behavioural deficits in PLS with ALS [4] found evidence of executive dysfunction, verbal learning and association memory alongside behavioural changes.
Pseudobulbar affect (PBA) is common to PLS and ALS [8] and coincides with executive dysfunction [9]. In addition to white matter changes in the corticospinal tract and callosal fibres, patients with PLS and ALS who develop PBA, show further changes in white matter underlying the frontotemporal cortex, transverse pontine fibres as well as cerebellar white matter, suggesting common involvement of descending corticopontocerebellar pathways [10].
Pathologically, Betz cell involvement is a common feature in both ALS and PLS, with the latter showing more prominent changes [11,12,13]. TDP-43 pathology, considered a key feature in ALS and a pathological link between ALS and FTD [14, 15], has been observed in some PLS cases [16]. Rare concurrent descriptions of PLS and FTD have reported TDP-43 pathology with a language predominant cognitive syndrome [17]. Intriguingly, TDP-43 pathology has been described in cases of semantic dementia, which characteristically progresses more slowly that other FTD variants, alongside a clinical picture identical to PLS [16].
Motor cortical atrophy is associated with cognitive dysfunction in ALS [18, 19]. While the relationship of motor cortical atrophy with cognition in PLS is unclear, involvement of motor callosal fibres and corticospinal tracts coincides with cognitive deficits in PLS [5, 6].
Functional assessment of the motor cortex using TMS techniques has demonstrated cortical hyperexcitability as an early and prominent feature of ALS [20]. The RMT, which is indicative of Betz cell function, and SICI, which is indicative of inhibitory interneuronal function, are both decreased early on in ALS, indicating cortical hyperexcitability [21]. The evolution of motor cortical excitability changes in ALS is less well understood, with some suggestion of an initial progression of hyperexcitability followed by cortical inexcitability, which may be reflective of greater loss of corticospinal projection neurons [21, 22]. Limited evaluation of PLS indicates a higher than normal RMT and relative cortical inexcitability [2, 23], which can differentiate PLS from other upper motor neuron (UMN) disorders [24].
While it is now well established that ALS and FTD lie on opposite ends of the frontal neurodegeneration spectrum, whether PLS forms part of this spectrum remains unclear.
The current study aimed to describe clinical, neuropsychological and motor cortical excitability characteristics in PLS to investigate if PLS forms part of a wider ALS–FTD spectrum [25]. Additionally, it explored the relationship between motor cortical excitability and cognitive function. The motivation behind this was to explore if the nature and degree of motor cortical excitability changes were a surrogate marker of disease severity.
Materials and methods
Patients
All patients with a clinical diagnosis of PLS (n = 21), assessed between 2005 and 2017 were identified from clinical referrals to a specialist multidisciplinary neurodegenerative clinical service at ForeFront/Brain and Mind Centre, as part of the NHMRC Sydney Health Partners Advanced Healthcare and Clinical Translation Centre.
Ethical approval was granted by the Human Research Ethics Committees of the South-Eastern Sydney Local Health District and the Western Sydney Local Health District. All patients or their nominated next of kin gave written informed consent for participation in the research.
The diagnosis of PLS was established using the Pringle criteria [2]. Specifically, the PLS diagnostic criteria included: (1) the presence of UMN signs with absence of focal muscle atrophy, fasciculations and electromyography (EMG) evidence of denervation for at least 3 years after symptom onset; (2) age of onset > 40 years; and (3) exclusion of mimic disorders by laboratory and imaging evaluation.
Brain and spinal cord magnetic resonance imaging excluded alternative pathologies such as structural, metabolic or demyelinating lesions. Biochemical and immunological testing was normal. In addition, cerebrospinal fluid analysis was unremarkable, as was genetic testing for the spastin gene mutation.
As disease control groups, patients with a diagnosis of ALS–FTD (n = 12) and pure motor ALS (n = 27), prospectively assessed between 2014 and 2017, were recruited from the ForeFront/Brain and Mind Centre clinical service. All ALS patients in the study (n = 27) had probable or definite ALS, as per the revised El Escorial and Awaji criteria [26]. The presence of cognitive and behavioural abnormalities was assessed in the ALS group as per the consensus criteria for these impairments in ALS [27]. All patients with a diagnosis of ALS–FTD satisfied the consensus criteria for ALS–FTD [27]. Briefly, as listed in these criteria, a diagnosis of ALS–FTD was defined by:
-
1.
Evidence of progressive deterioration in cognitive and/or behavioural function.
AND
-
2.
Presence of at least three characteristic behavioural and/or cognitive symptoms of FTD.
OR
-
3.
Presence of at least two of the characteristic behavioural and/or cognitive symptoms of FTD with loss of insight and/or psychotic symptoms.
OR
-
4.
Presence of language impairment meeting criteria for FTD syndromes.
Clinical assessment
All patients underwent detailed clinical, cognitive and functional assessments. Limb power was assessed using the Medical Research Council (MRC) grading system [28]. Subscores for the MRC grading system were divided into upper limb, lower limb and abductor pollicis brevis (APB) scores, yielding a maximum possible MRC sum score (MRCSS) of 90. The degree of upper motor neuron (UMN) dysfunction was assessed using a previously described UMN score [29].
Clinical staging was ascertained using the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R) [30]. The maximum possible score on ALSFRS-R was 48 with a lower score indicating greater motor disability. Progression rate was defined as follows [31, 32]:
Cognitive and behavioural assessment
Cognitive assessment was performed using the Addenbrooke’s Cognitive Examination (ACE) [33]. Score on the ACE ranged from 0 to 100 with subdomain scores being assigned out of 18 for attention/orientation, 14 for fluency, 26 for memory, 26 for language and 16 for visuospatial function. Letter fluency was used as an additional measure of executive function with a cut-off of 9 used to define impairment as defined in the consensus criteria [27]. A cut value of 88 was used for defining abnormal scores as per previously published studies [33].
Behavioural symptoms were assessed using a previously validated Motor Neuron Disease Behaviour (Mind-B) scale [34]. Mind-B assesses three behavioural domains based on frequency and severity of symptoms. Subdomain scores were assigned out of 16 for disinhibition, 12 for apathy and 8 for stereotypy, with a maximum possible total Mind-B score being 36. A lower score indicated greater dysfunction, with a total score of below 33, as previously defined, being used to indicate an abnormality.
Motor cortical function
To determine cortical function, a threshold tracking transcranial magnetic stimulation (TT-TMS) paradigm was used. Magnetic stimulation of the motor cortex was delivered using a 90-mm circular coil placed on the patient’s scalp [35]. The coil position was adjusted to a suitable point on the vertex from where a stable motor-evoked potential (MEP) was recorded from the APB muscle in the dominant hand at rest, with the smallest TMS stimulus. Stimuli were delivered by two magnetic stimulators connected via a BiStim (The Magstim Company Ltd., Whitlands, Carmarthenshire, UK) system.
The resting motor threshold (RMT) was defined as the single stimulus intensity required to achieve and maintain a target motor-evoked potential of 0.2 mV (± 20%) [36]. A previously validated paired-pulse threshold tracking TMS (TT-TMS) paradigm [21, 35] was applied to measure changes in RMT at various interstimulus intervals (ISI) to yield cortical excitability measures: short-interval intracortical inhibition (SICI) and intracortical facilitation (ICF) [35, 37]. SICI and ICF were defined as the percentage increase and decrease respectively, in a test stimulus, following a conditioning stimulus, required to generate a target MEP response (0.2 mV ± 20%) at pre-specified interstimulus intervals. Short-interval intracortical inhibition (SICI) was defined as the percentage increase in test stimulus intensity required to achieve the target motor-evoked potential of 0.2 mV at ISI’s of 1, 1.5, 2, 2.5, 3, 3.5, 4, 5 and 7 ms, whilst intra cortical facilitation (ICF) was measured at ISIs of 10, 15, 20, 25 and 30 ms. Average SICI was calculated as the mean of SICI values recorded at each interstimulus interval from 1 to 7 ms. Peak SICI was the highest SICI value recorded between 1 and 7 ms. Average ICF was calculated as the mean of ICF values recorded at each interstimulus interval from 10 to 30 ms. Inhibition (SICI) and facilitation (ICF) were calculated as the increase and decrease in intensity using the following equation, respectively:
The TMS protocol was delivered using the QTRACS software (©Institute of Neurology, Queen Square, UK). Motor responses were amplified (1 mV/V) and band-pass filtered (2 Hz–2 kHz) using a purpose-built amplifier. HumBug 50/60 Noise Eliminator (Quest Scientific; North Vancouver, BC, Canada®) was used to remove the mains frequency contamination inline and subsequently, the amplified signals were digitized using a 16-bit data acquisition system (NI-USB6251; National Instruments; Austin, TX, USA®).
Following the paired-pulse threshold tracking protocol, the maximum MEP was recorded after three single-stimuli at 150% resting motor threshold intensity. Maximal cortical silent period (CSP), defined as the maximum duration of electrical silence following a motor-evoked potential that interfered with ongoing EMG activity, was recorded while patients performed weak voluntary contraction [38]. Three single-stimuli at 150% resting motor threshold intensity were administered with resultant silent period measurements averaged to determine the maximum cortical silent period. The duration of the silent period was measured from motor-evoked potential onset to the return of EMG activity.
In the patients where the motor cortex was inexcitable, i.e. no MEP was recordable with a 100% stimulus intensity, cortical excitability parameters were excluded from the analysis.
All therapies including riluzole administration were recorded at the time of the TMS evaluation. No centrally acting medications were taken 24 h prior to administration of TMS. In addition, all patients were studied while at rest and encouraged to remain relaxed. If the study data quality was degraded by patient movement, the protocol was recommenced, and the initial data discarded.
Statistical analysis
Statistical analysis was performed using the statistical package SPSS 24® for Mac. Kolmogorov–Smirnov test of normality was applied. One way analysis of variance (ANOVA) testing for group comparisons, followed by post hoc Tukey’s tests were used for normally distributed variables (age, years of education, ALSFRS-R, FVC, MRC score, UMN score, progression rate, ACE score, RMT, SICI, and CSP). Non-parametric comparisons were made using the Kruskal–Wallis test followed by post hoc Mann–Whitney U tests, corrected for multiple comparisons (p < 0.01 for significance), for variables that were not normally distributed (Mind-B scores, fluency and ICF). Categorical variables were compared using the Chi-square tests for group comparisons, followed by post hoc Fisher’s exact tests. Two-sided p values were obtained and considered significant when < 0.05.
Combining all three groups, a multivariable regression analysis was performed using major baseline clinical features, and key motor cortical function measures, using a hierarchical and enter block regression model, to determine independent factors associated with global cognition measured as the total ACE score. Similarly, predictors of motor disability on the ALSFRS-R scale and behavioural symptom burden on MiND-B scores were studied. Disease duration was used as a regressor in the models to account for variation in motor cortical changes with disease progression [22].
Estimated survival times, including censored observations, were compared across the three groups using Kaplan–Meier curves.
Results
Demographics and baseline characteristics
Demographic features and clinical baseline features are summarized in Table 1. Consistent with the known natural course of PLS, disease progression was slower (progression rate = 0.18 ± 0.1) compared to the other two groups (0.57 ± 0.5 in ALS–FTD, p < 0.05 and 0.38 ± 0.4 in ALS, p < 0.005). High MRI T2 signal intensity in the corticospinal tract (internal capsule) was evident in one PLS patient, but all other investigations excluded an alternative diagnosis in this case.
Cognitive and behavioural profile
Global cognitive function measured on the ACE (mean total ACE score 82.5 ± 13.6, which is below the cut-off value of 88) was impaired in PLS. Also, total ACE was significantly different (p < 0.001) between the three groups, with PLS (p < 0.001 for group comparisons) showing similar deficits to ALS–FTD (mean total ACE score 76.3 ± 7.7 in ALS–FTD, p > 0.05 for PLS > ALS–FTD; ALS > ALS–FTD, p < 0.005), but significant deficits when compared with pure motor ALS (mean total ACE score 93.4 ± 3.9, PLS < ALS, p < 0.001) (Fig. 1). Subdomain scores of the ACE are summarized in Table 2.

Cognition and behaviour in PLS, ALS and ALS–FTD. Cognitive and behavioural scores across the three patient groups showing that PLS patients had a similar cognitive profile to ALS–FTD but did not have prominent behavioural deficits. The solid line represents normal cut-off value (88) for total ACE and the broken line represents cut-off (33) for total MiND-B score. *p < 0.05 for PLS < ALS and ALS–FTD < ALS. **p < 0.05 for ALS–FTD < ALS
Comparing the neuropsychological profile between PLS and ALS–FTD patients, cognitive profiles were largely similar, except the attention subscore on the ACE (PLS 17 ± 1.2, ALS–FTD 14.6 ± 3.4, p = 0.047) (Table 2). Behavioural impairment was significantly worse in ALS–FTD (total Mind-B scores: PLS 32.1 ± 5.6, ALS–FTD 22.4 ± 6.8, p = 0.005) than in PLS. In relation to subdomains of behavioural deficits, disinhibition and apathy were similar between PLS and ALS–FTD (both p > 0.01), while stereotypy was not seen in PLS (stereotypy score: PLS 7.1 ± 1.5, ALS–FTD 4.3 ± 1.9, p = 0.005).
Compared with ALS, the PLS patients had significant impairments across all domains except the attention subscore (Table 2).
Motor cortical function
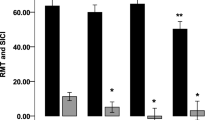
The proportion of patients were found to have a relatively inexcitable motor cortex was significantly higher in PLS (65%, p < 0.001) compared to ALS–FTD (8.3%) and ALS (3.7%) patients. Mean RMT was significantly higher in PLS (75.5 ± 6.2%) compared to ALS–FTD (50.1 ± 7.2%, p < 0.001) and ALS (62.3 ± 12.6%, p = 0.046). Average SICI was similarly reduced in PLS, ALS–FTD and ALS; while ICF was significantly lower in PLS (− 10.1 ± 9.1, p = 0.002) compared to ALS–FTD (0.38 ± 14.2) as shown in Fig. 2.

Corticomotor excitability parameters in PLS, ALS and ALS–FTD. Mean RMT was significantly greater in PLS than both ALS and ALS–FTD groups; while mean ICF was significantly lower (more negative implying greater interneuronal excitability) in PLS compared to the other two patient groups. SICI reduction was comparable between the three groups. *p < 0.05 for PLS > ALS–FTD as well as PLS > ALS. **p < 0.05 for PLS < ALS–FTD as well as PLS < ALS
This was a significant relationship between RMT and disease duration (Pearson’s R = 0.433, p = 0.001), across the entire cohort.
Cognitive function and motor disability
On multivariable regression modelling (Table 3), bulbar onset disease (β = − 0.45, p = 0.007) and RMT (β = 0.54, p = 0.001) were independently associated total ACE score across the three groups. In relation to motor disability (Table 4), MRC sum score (β = 0.47, p = 0.036) and average SICI (β = 0.58, p = 0.006) were independently associated with total ALSFRS-R score. No significant associations for MiND-B scores were found using the same multivariate model in this cohort (p > 0.05 for overall model).
Survival analysis
Mean survival time varied significantly between the three groups (p = 0.002), being longest in PLS (217.4 ± 22.4 months), shortest in ALS–FTD (38.5 ± 4.5 months) and intermediate in ALS (66.7 ± 9.9 months) (Fig. 3).

Survival in PLS, ALS and ALS–FTD. Kaplan–Meier survival curves showing months of survival from symptom onset for the patients with PLS, ALS–FTD and pure motor ALS. Mean survival time was longest in PLS (217.4 ± 22.4 months), shortest in ALS–FTD (38.5 ± 4.5 months) and intermediate in ALS (66.7 ± 9.9 months); p = 0.002 for difference on survival times between the three groups
Discussion
The present study has identified neuropsychological deficits in PLS, intermediate between ALS and ALS–FTD. The cognitive profile in PLS seems largely indistinguishable from ALS–FTD, but the frontal behavioural features that are classic for bvFTD are not prominent in PLS (Fig. 1). Differences were also established in motor cortical function in PLS, that represented part of a spectrum of change. Additionally, the relationship between cortical motor change and cognitive function, independent of disease duration and motor features, provides preliminary evidence regarding cortical excitability as a potential marker of disease severity.
Mean total score on the ACE, as a global cognitive measure, was abnormal (< 88) in the PLS cohort [39]. The most prominent deficit in PLS was a reduction in fluency, although deficits were also evident across other domains including memory, language and visuospatial function. Previous descriptions of cognitive deficits in PLS also found a predominance of frontal lobe dysfunction [3] raising the possibility that PLS may lie on frontal degenerative disorders spectrum.
Predictors of global cognition were explored in this study, combining the three patient groups, to identify common mechanisms leading to cognitive decline across the ALS FTD spectrum. The motivation to investigate cortical motor neuronal function in the context of cognition comes from emerging evidence that these changes may represent more widespread cortical dysfunction. Motor cortical excitability changes have been demonstrated in FTD [40], although are less prominent than those in ALS and may help differentiate FTD from Alzheimer’s disease [41].
Findings from the current study indicate that bulbar onset disease and lower resting motor threshold, indicative of greater cortical output cell excitability predict poorer cognitive function. Notably, bulbar onset disease was associated with poorer cognitive function. While it is possible that cognitive deficit may be overestimated in the patients who had bulbar onset, this is unlikely to have been a major factor in this study cohort, given that the PLS patients who had cognitive deficits had overwhelmingly limb onset disease (95%). Also, in the ALS–FTD group, which had the most prominent cognitive deficits, half of the patients had limb onset and half had bulbar onset disease. The more likely explanation for the association of bulbar onset disease and greater cognitive deficits is a greater level of cortical involvement with bulbar onset disease.
Average RMT was higher in PLS, yet there was greater cognitive impairment compared to pure ALS. Importantly, RMT was a predictor of cognition, independent of disease duration, combining the three patient groups. Given that a majority of patients with PLS had a relatively inexcitable motor cortex, a meaningful multivariable analysis in the PLS group per se is not feasible from this data. One possibility is that a lower threshold may occur early on in the disease course in PLS and evolve to inexcitability. However, this is difficult to document since a longer disease duration is part of the diagnostic criteria in PLS and longitudinal studies may be challenging. Alternatively, it remains possible that RMT may not reflect cognitive symptom burden in PLS per se and this cannot be reliably ascertained from the current dataset.
High RMT and relative motor cortex inexcitability in PLS are likely to be reflective of overall burden of motor cortical change. Interestingly, interneuronal function captured by the SICI and ICF, appear reduced in PLS to a similar degree as ALS and ALS–FTD. Current physiological understanding of SICI generation incorporates modulation of predominantly GABAergic synaptic neurotransmission in the inhibitory interneurons [42]. The finding of a reduced SICI in line with previous studies [43] is in keeping with previous limited evidence from PET imaging of GABAergic receptor dysfunction in PLS [44]. ICF is more negative in PLS (Fig. 2) indicating greater facilitation and thus a greater level of cortical excitability at the interneuronal level in PLS. In keeping with the previously published data, this study confirms that SICI is a marker of motor disease burden (Table 4). This study raises some interesting possibilities regarding why the RMT relates to cognition while SICI does not. Physiological studies indicate that the RMT is reflective of cortical output cell function and thus, reflects neuronal membrane excitability dependent on glutaminergic pathways [21, 42]. On the other hand, SICI is regarded as a marker of interneuronal circuits which provide an index of GABAergic neurotransmission [42]. This neurochemical difference in the physiological origins of these two markers may be one potential explanation why the RMT is reduced alongside cognitive impairment. Also, interneuronal circuits ultimately impact on Betz cell function which is reflected in the RMT. Thus, the presence of decreased RMT as well as SICI may be reflective of a more aggressive disease process.
A theoretical consideration regarding TMS measures is that they are upper limb based and potentially, a lack of UL involvement in PLS may be driving some of the differences. However, this is unlikely to have been a significant factor in the current dataset. As shown in Table 1, the MRC UL subscores were not significantly different between the three patient groups, which would argue against the lack of UL involvement driving the group differences. Analysis of ALSFRS subscores would be an alternative way of quantifying upper limb involvement. However, in the current PLS dataset, the ALSFRS subscores were not available for all the patients making it difficult to make a meaningful comparison based on the ALSFRS per se.
The time course of SICI evolution in ALS remains undetermined at present. Previous studies indicate that RMT is lower in early disease [22] and may increase as the disease progresses as a reflection of Betz cell dysfunction. In this cohort, SICI was not found to be related to disease duration (Pearson’s correlation coefficient 0.04, p > 0.05). However, a high RMT may technically preclude measurement of SICI which somewhat limits a direct interpretation of this finding. The predictive value of SICI in the multivariate model for a lower ALSFRS is likely to indicate that cortical excitability may be predictive of an aggressive primary disease process rather than reflective of disease course per se. This is further supported by the fact that the ALSFRS itself, does not show linear progression with disease course in ALS [45]. Longitudinal studies may help clarify some of these issues.
Neuropathological studies in PLS demonstrate prominent changes in Betz cells in the primary motor cortex and prefrontal cortex [2, 46]. Betz cell loss is also seen in ALS, though to a lesser degree, and inter neuronal loss is prominent in ALS [12]. Animal models of ALS suggest that pathological changes appear in motor neurons before the pathology spreads to the interneurons [47]. Neuroimaging studies show a reduction in N-acetylaspartate/creatinine ratio, hypometabolism on fluorodeoxyglucose-PET and reduction in C11 flumazenil binding in patterns similar to neuropathological changes in ALS [44, 48, 49]. Given the markedly different disease progression in PLS compared to ALS and ALS–FTD, it is likely that evolution of cortical changes is distinct in PLS, despite a comparable degree of disease pathology.
The administration of riluzole in patients with PLS is likely related to the fact that the diagnosis of PLS becomes evident over time. While this dataset does not incorporate all psychoactive medications which may have been taken by the patients at the time of their TMS evaluation, the administration of riluzole, which may modify cortical excitability [50], may be an indirect indicator of the fact that the clinical phenotype in the PLS cohort, at least in the initial period may have been largely similar to ALS.
The findings from the present study support a distinctive cognitive profile without prominent behavioural features in PLS. Previous rare case reports of PLS concurrent with FTD indicate a predominantly language domain involvement in these cases [17]. Predominant language domain involvement has also been observed in more recent descriptions of large ALS–FTD cohorts [51]. Thus, it is reasonable to propose that PLS is likely to lie on a wider ALS–FTD spectrum.
The lack of prominent behavioural features in PLS may point to potentially differential spread of disease pathology. Behavioural symptoms in ALS [18, 19] and FTD [52, 53] relate to atrophy in orbitofrontal and insular cortices alongside involvement of limbic structures. Interestingly, these regions do not seem to be involved in PLS imaging studies [54]. It is, thus, likely that the pathological spread of disease is more towards dorsolateral frontal cortices which relate more to frontal cognitive functions than the core behavioural features. This study focused on core behavioural features most frequently affected in FTD, but did not address the entire spectrum of behavioural changes that may be seen. Thus, it remains possible that other behavioural domains may be affected in PLS [4], which is a potential limitation of this study. PBA which is a common behavioural manifestation in PLS and ALS as mentioned in the introduction was not documented in this patient cohort, a further potential limitation. While this study utilised the ACE and MiND-B for documenting cognition and behaviour as validated previously [55], novel tools such as the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) [56] specifically developed for evaluating cognitive and behavioural function in ALS populations were precluded from this study given the time scale over which the PLS cohort was followed up.
In terms of therapeutic aspects, the administration of riluzole in patients with PLS is likely related to complexity of the diagnosis of PLS, which typically becomes evident over time. Administration of riluzole remains a consideration for patients diagnosed with conditions in the larger spectrum of diseases involving the motor neurone, although it is accepted that clinical trial data related to efficacy may remain lacking. Separately, defining a significant change would be made particularly complex in PLS given the long survival. Specifically, the survival analyses from this study demonstrate longest survival times in PLS, intermediate in ALS and shortest in ALS–FTD. These survival characteristics are in keeping with previous studies in ALS [57] and PLS [2, 46].
Conclusion
There are significant cognitive deficits in PLS which resemble ALS–FTD, without prominent behavioural disturbances typical of the FTD spectrum disorders. Cortical motor threshold predicts poorer global cognitive function, independent of motor features and disease duration, while dysfunction of inhibitory interneuronal circuits predicts motor disability, across ALS, ALS–FTD and PLS. Overall, while these findings support the notion that PLS represents part of an ALS–FTD spectrum, the pathophysiological mechanisms leading to slow disease progression and longer survival are likely to be distinct in PLS.
References
Le Forestier N, Maisonobe T, Piquard A et al (2001) Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain 124:1989–1999
Pringle CE, Hudson AJ, Munoz DG et al (1992) Primary lateral sclerosis: clinical features, neuropathology and diagnostic criteria. Brain 115:495–520
Caselli RJ, Smith BE, Osborne D (1995) Primary lateral sclerosis: a neuropsychological study. Neurology 45:2005–2009
Grace GM, Orange JB, Rowe A et al (2011) Neuropsychological functioning in PLS: a comparison with ALS. Can J Neurol Sci 38:88–97
Canu E, Agosta F, Galantucci S et al (2013) Extramotor damage is associated with cognition in primary lateral sclerosis patients. PLoS ONE 8:e82017–e82018. https://doi.org/10.1371/journal.pone.0082017
Agosta F, Galantucci S, Riva N et al (2013) Intrahemispheric and interhemispheric structural network abnormalities in PLS and ALS. Hum Brain Mapp 35:1710–1722. https://doi.org/10.1002/hbm.22286
Agosta F, Canu E, Inuggi A et al (2014) Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiol Aging 35:916–925. https://doi.org/10.1016/j.neurobiolaging.2013.09.041
Gallagher JP (1989) Pathologic laughter and crying in ALS: a search for their origin. Acta Neurol Scand 80:114–117. https://doi.org/10.1111/j.1600-0404.1989.tb03851.x
Abrahams S, Goldstein LH, Al-Chalabi A et al (1997) Relation between cognitive dysfunction and pseudobulbar palsy in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 62:464–472. https://doi.org/10.1136/jnnp.62.5.464
Floeter MK, Katipally R, Kim MP et al (2014) Impaired corticopontocerebellar tracts underlie pseudobulbar affect in motor neuron disorders. Neurology 83:620–627. https://doi.org/10.1212/WNL.0000000000000693
Pringle CE, Hudson AJ, Munoz DG et al (1992) Primary lateral sclerosis—clinical-features, neuropathology and diagnostic-criteria. Brain 115:495–520. https://doi.org/10.1093/brain/115.2.495
Nihei K, McKee AC, Kowall NW (1993) Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients. Acta Neuropathol 86:55–64. https://doi.org/10.1007/BF00454899
Kiernan MC, Vucic S, Cheah BC et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955. https://doi.org/10.1016/S0140-6736(10)61156-7
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Turner MR, Hardiman O, Benatar M et al (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 12:310–322. https://doi.org/10.1016/S1474-4422(13)70036-X
Dickson DW, Josephs KA, Amador-Ortiz C (2007) TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol 114:71–79. https://doi.org/10.1007/s00401-007-0234-5
Josephs KA, Dickson DW (2007) Frontotemporal lobar degeneration with upper motor neuron disease/primary lateral sclerosis. Neurology 69:1800–1801. https://doi.org/10.1212/01.wnl.0000277270.99272.7e
Agosta F, Ferraro PM, Riva N et al (2016) Structural brain correlates of cognitive and behavioral impairment in MND. Hum Brain Mapp 37:1614–1626. https://doi.org/10.1002/hbm.23124
Mioshi E, Lillo P, Yew B et al (2013) Cortical atrophy in ALS is critically associated with neuropsychiatric and cognitive changes. Neurology 80:1117–1123. https://doi.org/10.1212/WNL.0b013e31828869da
Menon P, Geevasinga N, Yiannikas C et al (2015) Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol 14:478–484. https://doi.org/10.1016/S1474-4422(15)00014-9
Vucic S, Ziemann U, Eisen A et al (2013) Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights. J Neurol Neurosurg Psychiatry 84:1161–1170. https://doi.org/10.1136/jnnp-2012-304019
Mills KR (2003) The natural history of central motor abnormalities in amyotrophic lateral sclerosis. Brain 126:2558–2566. https://doi.org/10.1093/brain/awg260
Kuipers-Upmeijer J, de Jager AE, Hew JM et al (2001) Primary lateral sclerosis: clinical, neurophysiological, and magnetic resonance findings. J Neurol Neurosurg Psychiatry 71:615–620
Geevasinga N, Menon P, Sue CM et al (2015) Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur J Neurol 22:826–858. https://doi.org/10.1111/ene.12669
Burrell JR, Halliday GM, Kril JJ et al (2016) The frontotemporal dementia-motor neuron disease continuum. Lancet 388:919–931. https://doi.org/10.1016/S0140-6736(16)00737-6
Costa J, Swash M, de Carvalho M (2012) Awaji criteria for the diagnosis of amyotrophic lateral sclerosis. Arch Neurol 69:1410–1417. https://doi.org/10.1001/archneurol.2012.254
Strong MJ, Abrahams S, Goldstein LH et al (2017) Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 18:153–174. https://doi.org/10.1080/21678421.2016.1267768
O’Brien M (2010) Aids to the examination of the peripheral nervous system. Saunders Limited, Philadelphia
Turner MR, Cagnin A, Turkheimer FE et al (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 15:601–609. https://doi.org/10.1016/j.nbd.2003.12.012
Cedarbaum JM, Stambler N, Malta E et al (1999) The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci 169:13–21. https://doi.org/10.1016/S0022-510X(99)00210-5
Kimura F, Fujimura C, Ishida S et al (2006) Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 66:265–267. https://doi.org/10.1212/01.wnl.0000194316.91908.8a
Labra J, Menon P, Byth K et al (2016) Rate of disease progression: a prognostic biomarker in ALS. J Neurol Neurosurg Psychiatry 87:628–632. https://doi.org/10.1136/jnnp-2015-310998
Hsieh S, Schubert S, Hoon C et al (2013) Validation of the Addenbrooke’s cognitive examination III in frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord 36:242–250. https://doi.org/10.1159/000351671
Mioshi E, Caga J, Lillo P et al (2014) Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology 82:149–155. https://doi.org/10.1212/WNL.0000000000000023
Vucic S, Kiernan MC (2006) Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129:2436–2446. https://doi.org/10.1093/brain/awl172
Vucic S, Howells J, Trevillion L, Kiernan MC (2006) Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve 33:477–486. https://doi.org/10.1002/mus.20481
Fisher RJ, Nakamura Y, Bestmann S et al (2002) Two phases of intracortical inhibition revealed by transcranial magnetic threshold tracking. Exp Brain Res 143:240–248. https://doi.org/10.1007/s00221-001-0988-2
Cantello R, Gianelli M, Civardi C, Mutani R (1992) Magnetic brain stimulation: the silent period after the motor evoked potential. Neurology 42:1951–1959
Mathuranath PS, Nestor PJ, Berrios GE et al (2000) A brief cognitive test battery to differentiate Alzheimer’s disease and frontotemporal dementia. Neurology 55:1613–1620
Burrell JR, Kiernan MC, Vucic S, Hodges JR (2011) Motor neuron dysfunction in frontotemporal dementia. Brain 134:2582–2594. https://doi.org/10.1093/brain/awr195
Benussi A, Di Lorenzo F, Dell’Era V et al (2017) Transcranial magnetic stimulation distinguishes Alzheimer disease from frontotemporal dementia. Neurology 89:665–672. https://doi.org/10.1212/WNL.0000000000004232
Vucic S, Kiernan MC (2017) Transcranial magnetic stimulation for the assessment of neurodegenerative disease. Neurotherapeutics 14:91–106. https://doi.org/10.1007/s13311-016-0487-6
Menon P, Geevasinga N, Yiannikas C, Howells J (2015) Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet 14:478–484. https://doi.org/10.1016/S1474-4422(15)00014-9
Turner MR, Hammers A, Al-Chalabi A et al (2007) Cortical involvement in four cases of primary lateral sclerosis using [(11)C]-flumazenil PET. J Neurol 254:1033–1036. https://doi.org/10.1007/s00415-006-0482-7
Gordon PH, Cheng B, Salachas F et al (2010) Progression in ALS is not linear but is curvilinear. J Neurol 257:1713–1717. https://doi.org/10.1007/s00415-010-5609-1
Singer MA, Statland JM, Wolfe GI, Barohn RJ (2007) Primary lateral sclerosis. Muscle Nerve 35:291–302. https://doi.org/10.1002/mus.20728
Hossaini M, Cardona Cano S, van Dis V et al (2011) Spinal inhibitory interneuron pathology follows motor neuron degeneration independent of glial mutant superoxide dismutase 1 expression in SOD1-ALS mice. J Neuropathol Exp Neurol 70:662–677. https://doi.org/10.1097/NEN.0b013e31822581ac
Turner MR, Agosta F, Bede P et al (2012) Neuroimaging in amyotrophic lateral sclerosis. Biomark Med 6:319–337. https://doi.org/10.2217/bmm.12.26
Van Laere K, Vanhee A, Verschueren J et al (2014) Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol 71:553–561. https://doi.org/10.1001/jamaneurol.2014.62
Vucic S, Lin CS-Y, Cheah BC et al (2013) Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 136:1361–1370. https://doi.org/10.1093/brain/awt085
Saxon JA, Thompson JC, Jones M et al (2017) Examining the language and behavioural profile in FTD and ALS–FTD. J Neurol Neurosurg Psychiatry 88:675–680. https://doi.org/10.1136/jnnp-2017-315667
Woolley JD, Gorno-Tempini M-L, Seeley WW et al (2007) Binge eating is associated with right orbitofrontal–insular–striatal atrophy in frontotemporal dementia. Neurology 69:1424–1433. https://doi.org/10.1212/01.wnl.0000277461.06713.23
Ahmed RM, Latheef S, Bartley L et al (2015) Eating behavior in frontotemporal dementia: peripheral hormones vs hypothalamic pathology. Neurology 85:1310–1317. https://doi.org/10.1212/WNL.0000000000002018
Tartaglia MC, Laluz V, Rowe A et al (2009) Brain atrophy in primary lateral sclerosis. Neurology 72:1236–1241. https://doi.org/10.1212/01.wnl.0000345665.75512.f9
Hsieh S, Caga J, Leslie FVC et al (2016) Cognitive and behavioral symptoms in ALSFTD: detection, differentiation, and progression. J Geriatr Psychiatry Neurol 29:3–10. https://doi.org/10.1177/0891988715598232
Niven E, Newton J, Foley J et al (2015) Validation of the edinburgh cognitive and behavioural amyotrophic lateral sclerosis screen (ECAS): a cognitive tool for motor disorders. Amyotroph Lateral Scler Frontotemporal Degener 16:172–179. https://doi.org/10.3109/21678421.2015.1030430
Shibuya K, Park SB, Geevasinga N et al (2016) Motor cortical function determines prognosis in sporadic ALS. Neurology 87:513–520. https://doi.org/10.1212/WNL.0000000000002912
Funding
This work was supported by funding to ForeFront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council (NHMRC) (APP1037746) and the Australian Research Council (ARC) Centre of Excellence in Cognition and its Disorders Memory Program (CE11000102). SA was funded by the Ellison-Cliffe travelling fellowship from the Royal Society of Medicine, UK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Agarwal, S., Highton-Williamson, E., Caga, J. et al. Primary lateral sclerosis and the amyotrophic lateral sclerosis–frontotemporal dementia spectrum. J Neurol 265, 1819–1828 (2018). https://doi.org/10.1007/s00415-018-8917-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8917-5