
Abstract
The unique adaptation of Eucalyptus benthamii to low temperatures coupled to fast growth and versatile wood quality has made it a valued plantation species in frost-prone areas worldwide, but little is known on its quantitative genetic parameters for key industrial traits. We used GBLUP additive (GA), additive-dominant (GAD), single-step (HBLUP), and pedigree-based predictive models to estimate lignin, extractives, carbohydrates, and wood density at age 4 and tree volume at age 6. By capturing hidden relatedness and correcting pedigree errors, SNP data disentangled non-additive from additive variance providing more realistic estimates of narrow-sense heritability than pedigrees, and more accurate predictions of trait values. Predictive abilities (PAs) ranged from 0.12 for volume (pedigree-based model) to 0.44 for wood density (models H, GA, and GAD). Considerable dominance variance was seen for all traits, growth was the trait most influenced by it, resulting in PAs 48.9% higher when this effect is considered, a result with important consequences both for clonal propagation and overall selection efficiency (Seff). Using a HBLUP model, phenotypes of non-genotyped trees increased PAs by increasing sample size and provided realized relationships with reduced genotyping cost. In a recurrent selection program, the preclusion of progeny testing provides an increase in Seff between 232% and 299%. In a clonal selection program, the elimination of both progeny and initial clonal trial may increase Seff between 134% and 277%. Increasing selection intensity by genomic prediction resulted in an additional impact on Seff. This study provides groundwork to implement genomic selection in E. benthamii breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Eucalyptus native to Australia, Indonesia, and nearby islands includes the most widely planted hardwood tree species in the tropical and subtropical world. Due to the outstanding natural plasticity found across species, plantations can be established in various edaphoclimatic zones. Currently, a set of “big nine” Eucalyptus species (E. camaldulensis, E. grandis, E. tereticornis, E. globulus, E. nitens, E. urophylla, E. saligna, E. dunnii, and E. pellita) are extensively planted due to their high productivity, rapid growth, production of commercially important wood for various uses, and great adaptive ability (Harwood 2011).
Eucalyptus benthamii Maiden et Cambage, commonly known as “Camden White Gum,” is a subtropical species with restricted distribution between latitudes 33° S and 34° S in the coastal region of the Australian state of New South Wales, at an altitude between 50 and 800 m. Its climatic requirements include an average annual temperature between 13 °C and 17 °C and annual precipitation between 900 and 1.400 mm. E. benthamii is a species with low to moderate natural regeneration ability, now considered vulnerable to extinction (Kjaer et al. 2004; Butcher et al. 2005; Han et al. 2020). Its genetic base has been drastically reduced due to anthropic actions, such as the introduction of other species, increased agricultural activities, urban expansion, flooding, and fires (Hall and Brooker 1973; Kjaer et al. 2004; Butcher et al. 2005). The main interest in the species is due to its unique ability to withstand frosts growing in subtropical regions with an average annual temperature of up to 14.5 °C and withstand absolute minimum temperatures between −6 °C and −10 °C (Swain 1996; Lin et al. 2003; Paludzyszyn Filho et al. 2006). This unique adaptation has made E. benthamii a highly valued species to expand eucalypt plantations, as it combines rapid growth, with the possibility of growing in frost-prone areas in subtropical high-altitude areas worldwide.
Breeding efforts of E. benthamii are recent when compared to other mainstream species of the genus. To exploit its high potential for frost tolerance, breeding programs are currently ongoing in subtropical regions, such as southern Brazil, southeastern USA, Uruguay, Argentina, Chile, and China, using both E. benthamii as a pure species or combined with other species, such as E. dunni, with the objective of introgressing frost tolerance in hybrid combinations (Butcher et al. 2005; Resende and Assis 2008; Fonseca et al. 2010; Brondani et al. 2011; Arnold et al. 2015; Yu and Gallagher 2015). The long breeding time required to obtain superior genotypes is, however, a significant hurdle (Kageyama and Vencovsky 1983; Pereira et al. 1997; De Gonçalves et al. 1998; Grattapaglia et al. 2018) that justifies the high expectations toward the application of genomic selection (GS) in breeding programs of perennial forest and fruit trees. This approach has many advantages over traditional breeding and selection methods, mainly due to the possibility of considerable time savings, increased selection intensity, and equal or possibly higher selection accuracy at much earlier ages (Resende et al. 2012; Grattapaglia 2014).
Generally, ABLUP, or pedigree-BLUP, is the standard methodology in predicting genetic values from an expected relationship between individuals (Crossa et al. 2010). The method was first suggested by Fisher (1918) and consists of using the pedigree to calculate the expected relatedness matrix (A) to predict genetic values based on the infinitesimal additive model. However, the expected relatedness, especially in open-pollinated progenies of species with mixed mating, not only incorporates a high level of uncertainty regarding the effective number of pollen parents involved, but also does not account for the relatedness already existing in the population, generally referred to as “cryptic or hidden relatedness”. The hidden relatedness can affect the accuracy of estimated genetic parameters, genotypic values, and predictive abilities and inflate the additive variance (Squillace 1974; Namkoong et al. 1988; Tambarussi et al. 2018; Klápště et al. 2018).
With the advent of accessible genome-wide genotyping technologies, the GBLUP method is materialized, providing a more precise approach to estimating relatedness among individuals (Grattapaglia et al. 2018). Differently from the ABLUP that uses a numerator-based relationship matrix A, GBLUP makes use of a relationship matrix estimated from genotypes collected for large numbers of genome-wide markers. This matrix is often called G matrix or GRM for Genomic Relationship Matrix and represents the marker-inferred proportion of the genome that two individuals share, frequently called the effectively realized relatedness (VanRaden 2008). The use of genomic relationships can improve the prediction accuracy, as shown in several studies with forest species (Muñoz et al. 2014; Gamal El-Dien et al. 2015, 2018; Cappa et al. 2017; Resende et al. 2017; Tan et al. 2018).
Besides capturing the realized additive relationships, molecular marker data also allow the inclusion of non-additive effects in genomic predictions. The inclusion of the dominance effect in genomic prediction models has been suggested and used in previous studies (Vitezica et al. 2013; Muñoz et al. 2014; Aliloo et al. 2016; Xiang et al. 2016; Tan et al. 2017). The presence of significant dominance variance for a trait has shown that the inclusion of this effect tends to improve the accuracy of heritability and predictive ability when compared to genomic models containing only the additive effect, with important consequences on the correct estimation of genetic gain (Denis and Bouvet 2011; Wellmann and Bennewitz 2012; Zeng et al. 2013; Muñoz et al. 2014; Nishio and Satoh 2014; Duenk et al. 2017; De Almeida Filho et al. 2019). Furthermore, the single-step GBLUP (HBLUP), originally proposed in animal breeding (Legarra et al. 2009; Misztal et al. 2009; Aguilar et al. 2010; Christensen and Lund 2010), has provided a powerful additional approach for tree breeding (Cappa et al. 2017, 2019; Ratcliffe et al. 2017; Klápště et al. 2018). This method entails genotyping only part of the individuals in a trial but making inferences about all individuals tested. This has become an exciting approach because genotyping still involves a significant investment not accessible to all tree breeding programs, despite the considerable cost reductions that took place in recent years (Ratcliffe et al. 2017; Klápště et al. 2018).
We were interested in assessing the prospects of including the use of genomic data to accelerate E. benthamii breeding currently carried out by open-pollinated population advancement. To that end, we set out to compare the relative performances of pedigree-based, additive, additive-dominant, and single-step models for volume growth and a suite of industrially relevant wood quality traits in an open-pollinated progeny trial of E. benthamii. Genomic relationships were estimated using high-quality genome-wide SNP data generated with the fixed content SNP chip EuCHIP60K. Additionally, we used the estimates of genomic prediction ability obtained with the different models to evaluate the relative impact of breeding cycle reduction and increased selection intensity on the overall selection efficiency in E. benthamii breeding.
Material and methods
Experimental material and phenotyping
The target study population was an open-pollinated progeny trial of E. benthamii located at Otacílio Costa, Santa Catarina state, Brazil, planted in October 2010, following a randomized complete block design with 20 replicates of 81 families in single-tree plots, totaling 1620 trees in the trial. Open-pollinated seeds used to establish the trial were collected from select trees in two progeny trials at two different locations of Telêmaco Borba (Paraná state) with genetic material originally introduced from Kedumba Valley, NSW, Australia.
The trees were measured at age 4 years for diameter at breast height and total height. Using these two traits, volume growth was estimated. A sample of 780 individuals from 77 families was selected for volume using REML/BLUP methodology by the following mixed model:
where y is the phenotypic measure of the evaluated trait; β is the vector of fixed effect associated with the block replication; a is the random vector of genetic additive individual effects; e is the random residual effect; X and Z are the incidence matrices for the respective effects.
To obtain wood quality traits, from all 780 trees, two wood cores were extracted at breast height (1.3 m) at age 4 years using an increment borer with 12 mm diameter. The first core was used to measure basic wood density (BWD) using the direct relation between dry weight (Wdry) and volume at full saturation (Vs): BWD = Wdry/Vs. The second core was ground by a Wiley mill and the sawdust was graded using 40 and 60 mesh sieves and stored in a room with controlled temperature and humidity (23 °C and 50% respectively) and further used for chemical analysis. The following traits were measured: extractive content (EXT) (TAPPI TA of the P and P 2000), total lignin content (LIG) (Goldschmid 1971; Gomide and Demuner 1986), and the sum of cellulose and hemicellulose content, here referred to as carbohydrates content (CBO) (Wallis et al. 1996).
Using data for the 780 measured trees, prediction models for all wood chemical and quality traits were built using near-infrared spectroscopy (NIRS). These models were then used to predict the wood traits for the remaining alive individuals in the progeny trial (476). At age 6 years, an additional measurement of diameter at breast height and total height was carried out for all trees in the trial, and volume growth was estimated (VOL6).
Genotypic material and quality control
The 780 measured trees were genotyped using the Illumina Infinum EUChip60K for Eucalyptus (Silva-Junior et al. 2015) at Geneseek (Lincoln, NE, USA). Genomic DNA was extracted from fresh leaves using a rapid optimized protocol for high phenolics content tissue (Inglis et al. 2018). Data quality control was carried out using PLINK (Purcell et al. 2007). The data were filtered to remove SNP markers with a call rate ≤ 95% and individual samples with a call rate ≤ 90%. Samples that did not have phenotype measurements for all evaluated traits were also removed. Some outlier samples possibly due to non-sampling errors (i.e., error that occurs during data collection, causing the data to differ from the true values) were also removed using a PCA (principal component analysis) using a LD-pruned (r2 < 0.2) subset of markers. Another filtering step removed monomorphic SNPs keeping SNPs with a minor allele frequency (MAF) > 0. In the end, data for 15,293 SNPs and 671 individuals remained and were used in subsequent analyses.
Statistical modeling
The relationship matrix based on pedigree information (A) was computed using the expected genetic relatedness between individuals considering the ancestors that we were able to track down, i.e., 24 grandmothers for 53 of the 77 genotyped families.
With information from SNP markers, the additive genomic relationship matrix (G) was obtained according to VanRaden (2008):
where Z is the centered matrix of SNP encoded as 0, 1, and 2 and pi is the allele frequency of individuals for the i SNP locus. The resulting matrix was checked and confirmed to be positive semi-definite, as it is a common prior for genomic analysis.
The dominance relationship matrix (D) was estimated according to Vitezica et al. (2013):
where S is the matrix codified as −2(1 − pi)2 for the alternative homozygote, 2pi(1 − pi) for the heterozygote, and −2pi2 for the reference homozygote; pi is the same as stated in the estimation of the G matrix.
The hybrid single-step matrix (H) that includes expected relatedness (A) and realized genomic relatedness (G) was obtained using the approach of Legarra et al. (2009) and Aguilar et al. (2010):
where matrices Agg, Ann, and Agn are submatrices of A, and contain the relatedness between genotyped individuals (Agg), non-genotyped (Ann), and between genotyped and non-genotyped individuals (Agn) (Christensen 2012). Ga is the genomic relationship matrix (G) adjusted with the following expression:
where β and α are obtained by solving the following system of equations:
This adjustment has the function of rescaling G in order to obtain Ga, so that the mean of its diagonal equals the diagonal mean of Agg, as well as the mean of all Ga and Agg elements.
The G matrix, accounting for the additive and additive-dominant GBLUP models, contains only genotyped individuals. The matrix H, in turn, contemplates genotyped and non-genotyped individuals, that is, the entire progeny trial, generating different sample sizes between these models. So, in order to allow the comparisons between the performance of the models against the standard infinitesimal additive model, the A model (ABLUP) was adjusted with two datasets: one containing only genotyped individuals (Ag model), and another contemplating all individuals (At model).
Fitting of both A models was carried out with the rrBLUP package (Endelman 2011), with the following linear mixed model:
where y is the phenotypic measure of the evaluated trait; β is the vector of fixed effect associated with the block replication; u is the random effect vector associated with the additive effect; e is the random residual effect; X and Za are the incidence matrices for the respective effects. It was assumed that \( a\sim N\left(0,A{\sigma}_a^2\right) \).
The subsequent models were adjusted using Bayesian regression modeling as particular cases of Reproducing Kernel Hilbert Spaces (RKHS) using the statistical package BGLR (Pérez and De Los 2014).
For the GBLUP model that considers only the additive effect (GA), the genomic relationship matrix (G) was provided as a kernel matrix, in order to fit the following mixed model:
where y is the phenotypic measure of the evaluated trait; β is the vector of fixed effect associated with the block replication; a is the random effect vector associated with the additive effect; e is the random residual effect; X and Za are the incidence matrices for the respective effects. For the analysis, the following priors were assumed:
p(μ)∝ constant; \( a\mid {K}_1,{\sigma}_a^2\sim N\left(0,{K}_1{\sigma}_a^2\right) \)
For the model that considers both the additive and dominance effects (GAD), the additive (G) and dominance (D) genomic matrices were provided as kernel matrices in order to fit the following mixed model:
where y is the phenotypic measure of the evaluated trait; β is the fixed effect vector associated with the block replication; a is the vector of random effects associated with the additive effect; d is the vector of random effects associated with dominance deviation; e is the random residual effect; X, Za, and Zd are the incidence matrices for the respective effects. For the analysis, the following priors were assumed:
p(μ)∝ constant; \( a\mid {K}_1,{\sigma}_a^2\sim N\left(0,{K}_1{\sigma}_a^2\right) \) e \( d\mid {K}_2,{\sigma}_d^2\sim N\left(0,{K}_2{\sigma}_d^2\right) \)
In the assumptions of both GA and GAD models above, K1 and K2 are, respectively, the kernel matrices G and D.
When fitting the single-step GBLUP, or HBLUP model (H), the G matrix was replaced with the H in the GA model. The same priors were also assumed.
In order to obtain the variance components associated with each model, the standard distributions of the BGLR package (Pérez and De Los 2014) were assumed: \( {\sigma}_i^2\sim {\chi}^{-2}\left({\sigma}_i^2|d{f}_i,{S}_i\right) \) with dfi degrees of freedom and scale parameter Si > 0.
After previous analysis on Markov chains using the coda package (Plummer et al. 2006), 120,000 iterations were considered, with the first 2000 excluded as burn-in with a thinning interval of 100.
Heritabilities were estimated at the broad (\( {h}_G^2 \)) and narrow sense (\( {h}_a^2 \)) as:
where \( {h}_G^2 \) is the broad-sense heritability (only estimated for the GAD model); \( {h}_a^2 \) is the narrow-sense heritability; \( {\sigma}_a^2 \) is the additive variance associated with the respective relationship matrix; \( {\sigma}_d^2 \) is the genetic variance associated with the dominance deviation (only accounting for the GAD model); and \( {\sigma}_e^2 \) is the residual variance.
To assess whether the dominance variance, when included in the model, influences the estimation of the additive variance, we calculated a ratio between the additive variance estimated considering the dominance effect (GAD model - \( {\sigma}_{a\left(\mathrm{GAD}\right)}^2 \)) and the additive variance estimated without considering the dominance effect (GA model - \( {\sigma}_{a\left(\mathrm{GA}\right)}^2 \)):
To ascertain the magnitude of the variance associated with the dominance effect in relation to the additive variance estimated with the GAD model, a ratio was calculated between these variances: \( {\sigma}_d^2/{\sigma}_a^2 \).
Predictive ability and prediction bias
The performance of the models was compared using two criteria:
-
(a)
Prediction bias (bias): estimated by a 10-fold cross-validation procedure, using the regression coefficient obtained between values predicted by the model and the phenotypic values, where 1 is the ideal value (i.e., absence of bias).
-
(b)
Prediction ability (PA): estimated by a 10-fold cross-validation procedure. The data were divided into two subsets, one with 90% of the individuals (training population), and another with the remaining 10% (validation population).
Selection efficiency and selection intensity
Genomic selection models were compared to traditional best linear unbiased prediction (BLUP)-based phenotypic selection (PS) using the selection efficiency (Seff) in terms of percent time reduction in breeding cycle length. The selection efficiency was obtained using a ratio between expected genetic gain from genomic (EggGS) and BLUP-based selection (EggPS) as Seff = [(EggGS/EggPS) − 1] × 100, as described earlier (Grattapaglia and Resende 2011).
The expected genetic gains were obtained by the classical breeder’s equation: \( Egg=\frac{ir{\sigma}_a}{L} \) (Falconer 1989), where i is the selection intensity, r is the method accuracy, σa the additive genetic standard deviation, and L is the breeding cycle length. Since σa is equal for both genomic and BLUP-based selection, and aiming to evaluate only the impact of time and accuracy, we initially considered an equal i for both methods, and thus, the equation was simplified as \( Egg=\frac{r}{L} \).
The BLUP-based selection accuracy (rPS) was estimated for both datasets, the one with only genotyped individuals and the one contemplating all individuals as the square root of narrow-sense heritability (\( {r}_{PS}=\sqrt{h_a^2} \)). To compute the genomic selection accuracies (rGS), two paths were considered depending on the main purpose of selection, recurrent selection (RS), or clonal selection (CS). Since the selection purpose dictates the most suitable model, the accuracies of both additive (GA) and single-step (H), which are the most appropriate models for RS, were obtained as a ratio between its predictive ability (PA) and the square root of its narrow-sense heritability (\( {r}_{GS}= PA/\sqrt{h_a^2} \)). For CS, however, the additive-dominant model (GAD) is the most adequate one. The ratio to obtain rGS, consider both the additive and dominant effect, and so, the broad-sense heritabilty was used (\( {r}_{GS}= PA/\sqrt{h_G^2} \)).
For clonal selection, the total breeding cycle length, that is, the traditional BLUP-based selection (PS) cycle, was assumed to be 18 years (Resende et al. 2012, 2017). This cycle length includes 5 years for flowering, mating, and seedling production, 3 years for progeny testing, 5 years for the first initial clonal screening trial of a larger number of clones, and the final 5 years expanded clonal trial of a reduced number of clones. For RS, the PS cycle length was assumed to be 10 years, where the first 5 years are the same as explained for the clonal route above, and the remaining 5 years are for progeny testing. We considered the progeny test of RS to be longer, in order to adequately evaluate wood quality traits, since this strategy is aimed at selecting parents of the subsequent generation. These values (18 years for CS and 10 years for RS) were used as benchmarks to compare the Seff of genomic selection for different percent time reductions in breeding cycle length, ranging from a more conservative 10% reduction (e.g., 9 years for RS; 16.2 years for CS) to a more aggressive 50% reduction (5 years for RS; 9 years for CS).
Since genomic selection opens the possibility to greatly increase selection intensity (i) by indirectly evaluating a much larger number of plants when compared to BLUP-based selection and still select the same number of trees, we aimed to verify its impact on Seff, and, again, used the breeder’s equation to obtain selection efficiencies with the same estimation method as described above. To calculate the expected genetic gains, we also used the benchmark values for total breeding cycle length, depending on the chosen route (10 years for RS; 18 years for CS), as fixed values in the estimate of EggPS. For EggGS, we then adopted a breeding cycle reduction of 45%, such that a RS cycle would take 5.5 years and a CS cycle 10 years. We simulated selection at the truncation point, with the 100 best individuals selected for the trait, thus varying the total number of evaluated plants. We fixed the selection intensity (i) at 5%, i.e., 100 top individuals in 2000 evaluated plants, for EggPS, and varied i from 5 to 0.25%, i.e., 100 top individuals in 2000 up to 40,000 evaluated plants for EggGS. The value of i was obtained by the ratio between z (height of the standardized normal at a given truncation point) and p (percentage of selected individuals) (Falconer and Mackay 1996). To obtain i, the observations must be normally distributed; therefore, normality was verified through the Kolgomorov-Smirnoff (KS) test.
Results
Phenotypic data
Considerable phenotypic variation was observed for the evaluated traits in the progeny trial (Table 1). For basic wood density (BWD), it ranged from 296 to 505 kg m−3, extractives content (EXT) from 0.22 to 5.31%, lignin content (LIG) from 27 to 35%, carbohydrates content (CBO) from 58 to 66%, and from 0.01 to 1.14 m3 for total tree volume ate age six (VOL6).
Growth (VOL6) displayed the highest phenotypic variation, with a coefficient of variation of 55%. The lowest variation was seen for CBO, with a coefficient of variation of 1.9%.
Pedigree and genomic relatedness
The pedigree matrix (Fig. S1 – A), included 97.4% of the pairwise relationships as not genetically related, 1.1% as half-first cousins and 1.5% as half-sibs. Using SNP marker data, the average relatedness was consistent with the expected pedigree-based relatedness. However the G matrix (Fig. S1 – B) was able not only to correct relationships between pairs of individuals mistakenly considered as half-sibs or unrelated by the Agg matrix, but was also able to capture the more granular relatedness existing in the population, such as the presence of full-sibs or even selfed individuals.
Genetic estimates and predictive abilities
Due to the different sample sizes between the dataset that includes only the genotyped individuals (Ag, GA, and GAD models) and the dataset of all individuals in the trial (At and H models), comparisons of heritabilities between genomic and phenotypic methods were performed only for models adjusted for the same sample size.
Narrow-sense heritabilities (\( {h}_a^2 \)) estimated using genomic models (H, GA, and GAD) were lower than the estimates using the pedigree matrix (\( {h}_a^2 \)) (Ag and At models) for all evaluated traits, with exception of VOL6, for which the Ag model showed the lowest value (Table 2). The largest differences between the heritabilities estimated using genomic and pedigree methods using only genotyped individuals were found for VOL6 with the additive model (GA) and for LIG the additive-dominant model (GAD). The estimate of heritability for EXT obtained with the GA model (0.30) was 66.7% higher than the estimate of the heritability obtained with the Ag model (0.18), while for LIG it was 45% lower with the GAD model (0.22) when compared to the Ag model (0.40). The smallest differences were seen for BWD and VOL6, where the genomic estimates of the GA model corresponded respectively to 90.9 and 116.7% of the estimates obtained with the Ag model. For the dataset containing all individuals, the largest differences between the single-step (H) and pedigree-based (At) genomic estimates were for LIG and CBO, corresponding to 54.9% and 55.7% of pedigree-based heritability, respectively. The smallest differences were seen for VOL6, where the estimates were practically the same (94.6%) and for BWD (76.1%).
The magnitude of the ratio between dominance and additive variances (\( {\sigma}_d^2/{\sigma}_a^2 \)) indicated the presence of dominance effect for all traits, varying from 0.46 (BWD) to 1.38 (VOL6). For EXT and VOL6, it exceeded the unit, that is, these traits involve a dominance effect larger than the additive effect. The ratio of the additive variance estimated with the additive effect model (GA) and the additive and dominant effect model (GAD), \( {\sigma}_{a\left(\mathrm{GA}\mathrm{D}\right)}^2/{\sigma}_{a\left(\mathrm{GA}\right)}^2 \), was calculated to evaluate the presence of orthogonality between the additive and dominance effects. The ratio varied from 0.68 (EXT) to 0.86 (BWD) indicating that the GAD model contains 68 to 86% of the additive variance estimated by the GA model for EXT and BWD, respectively.
Estimates of prediction bias (bias) show whether the evaluated models underestimate (values >1) or overestimate (values <1) the predicted values (Table 2). For BWD, LIG, and CBO, none of the tested models showed large deviations from the unit, meaning that for these traits, all models are largely free from biased prediction. For EXT, an overestimation was seen with the genomic models considering only genotyped individuals (0.82 - GA; 0.83 - GAD), and an underestimation with the pedigree-based model (1.26 - Ag). However, for the dataset that contains all trees in the trial, no bias was seen in the pedigree (At) and single-step hybrid (H) models, indicating that the inclusion of the additional phenotypic information, together with the increase in sample size, resulted in predicting more accurate values for this trait. For VOL6, known to involve a significant contribution of dominance variance, the inclusion of the D matrix corrected the overestimation of the values predicted by the GA model (0.87), as well as the underestimation of the Ag model (1.18), resulting in an unbiased estimation (1.01 - GAD). Additionally, the H model produced a slight underestimation of the predicted values for VOL6 showing that there is some level of bias associated (1.15).
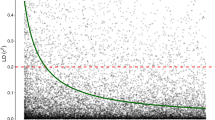
Predictive abilities (Fig. 1) ranged from 0.12 (VOL6 - Model Ag) to 0.44 (BWD - Models H, GA, and GAD). For all evaluated traits, the genomic models (GA and GAD) showed improved predictive abilities when compared to the Ag model. The same was seen for all traits when comparing the single-step (H) and the At models, with the exception of EXT, where both models performed equally well.

Predictive abilities (rgy) with standard error bars of the different models (see text for details) for basic wood density (BWD), extractives (EXT), lignin (LIG), and carbohydrates (CBO) at 4 years of age and volume at 6 years of age (VOL6) for an open-pollinated progeny trial of E. benthamii
BLUP-based vs. genomic selection efficiency with reduced breeding cycle length and increased selection intensity
Following the same approach used for genetic parameters estimation, the estimates of selection efficiency (Seff) were obtained respecting the sample size difference of datasets. The intersection between a model Seff curve and the zero value on the Y-axis corresponds to the point where GS becomes more efficient than PS at a given percent time reduction of breeding cycle length (Fig. 2).

Increase in selection efficiency (Seff) of genomic selection (GS) in comparison to traditional BLUP-based selection (PS) with a progressive reduction in the time necessary to complete a breeding cycle for growth and wood traits in an open-pollinated progeny trial of E. benthamii
For all traits, except volume at age 6 years (VOL6), the GA model presented the highest selection efficiencies. For density (BWD), Seff started near zero for the GA model and became positive for GAD and H model at around 15% of breeding cycle reduction (8.5 years by a GAD model for CS; 15.3 by H model for RS), reaching an increase in efficiency of 233% for GA and 200% for GAD and H models with a 50% reduction in breeding cycle length. Extractives (EXT), carbohydrates (CBO), and lignin (LIG) showed a similar behavior for the GA model, starting at Seff = 8% (EXT) to 19% (CBO) with a 10% reduction of breeding cycle length, up to 248% (EXT) to 286% (CBO) with a 50% reduction. Although both the GAD and H models start with a negative or near zero Seff for these three traits, the H model, for LIG displayed the second highest Seff (235%), while for EXT and CBO, it showed a lower Seff (137% for EXT; 184% for CBO), than the GAD model (183% for EXT; 224% for CBO).
Growth (VOL6) was the only trait for which the GA model did not stand out. The GAD model reached the highest increase in selection efficiency, 356%, with a 50% reduction in breeding cycle length, followed by the GA (299%) and the H models (249%) respectively.
The impact of increasing the selection intensity (i) by adopting genomic selection on the overall Seff, was modeled considering the same final number of selected individuals, set to 100, for increasing numbers of evaluated plants assuming a fixed reduction of 45% in breeding cycle length. The fixed reduction in breeding cycle length explains why the modeled curves do not start at zero but rather at a positive gain in selection efficiency. The proportion selected varied from 5%, i.e.,100 in 2000 plants corresponding to a typical selection intensity in progeny trial by BLUP-based selection, down to 0.25% assuming a very high selection intensity of the same 100 individuals in a genomic selection trial with 40,000 plants (Fig. 3).

Increase in selection efficiency (Seff) of genomic selection (GS) with increasing selection intensity (evaluated plants) for a fixed reduction of 45% in breeding cycle for growth and wood traits in an open-pollinated progeny trial of E. benthamii
When the same selection intensity was used (5%, i.e., 100 selections in 2000 evaluated plants), GS already showed an increased efficiency due to the reduced breeding cycle length. Once again, for all traits, except VOL6, the GA model was superior. A rapid increase in selection efficiency was observed as the selection intensity increased from the evaluation of 2000 to 10,000 plants and less so but still considerably when up to 40,000 showing a logarithmic behavior of the curves. For VOL6 the GAD model, again presented the highest increase in Seff among the models tested, starting, at 105% when 2000 plants are evaluated, up to 210% when 40,000 plants are evaluated.
Discussion
Variation in wood quality traits in Eucalyptus benthamii
Considerable variation was observed for all evaluated wood quality traits in the E. benthamii progeny trial (Table 1), in line with previous estimates in other species of the genus. Comparative assessments of wood quality traits across eucalypts, although potentially useful to make predictions, are difficult to properly make as they depend on a number of factors, including species, the genetic origin of the material, planting site conditions, measurement methods, type of wood sample used, and age of the tree (Rencoret et al. 2011). It is important to note that our measurements were taken at a relatively young tree age. BWD, for example, is known to increase with the age of the eucalypt trees (Santana et al. 2012; Resquin et al. 2019). Extractives have been reported to generally increase with age (Miranda and Pereira 2002; Raymond 2002; Mohammadi et al. 2011; Morais and Pereira 2012). The same can be stated for LIG, with reports showing increased (Miranda and Pereira 2002; Mohammadi et al. 2011; Rencoret et al. 2011), decreased (Healey et al. 2016), or not influenced values with the age of the tree (Santana et al. 2012; Lachowicz et al. 2019). Same for cellulose content, with reports showing increasing (Raymond 2002; Mohammadi et al. 2011; Rencoret et al. 2011) as well as decreasing values with age (Miranda and Pereira 2002). For hemicelluloses, existing reports show decreasing values over the years (Miranda and Pereira 2002; Mohammadi et al. 2011; Lachowicz et al. 2019). Overall, no single trend seems to exist. Nevertheless, our study provides additional data for a Eucalyptus species for which very little data exists and emphasizes the need for further studies to better understand the expected patterns of variation of wood chemical traits with age.
Basic wood density (BWD) ranging from 414 to 539 kg m−3 has been reported for Eucalyptus species at 5 to 10 years of age (Chen et al. 2018; Lima et al. 2019; Resquin et al. 2019; Carrillo-Varela et al. 2019). Our average BWD (380 kg m−3) was on the low end of these estimates and even lower than previous reports for E. benthamii (Resquin et al. 2019; Carrillo-Varela et al. 2019). For total lignin content (LIG), average content in the trial (31.7%) was higher than previous reports for other Eucalyptus species, which ranged from 23.2 to 31.5% at 5 to 7.5 years of age (Pereira et al. 2013; Samistraro et al. 2015; Lima et al. 2019; Carrillo-Varela et al. 2019). In contrast to LIG, carbohydrate content (CBO) (61.5%) was lower than the literature reports for this trait, usually ranging from 62.2 to 70.8% at 5 to 10 years of age (Longue Júnior et al. 2010; Pereira et al. 2013; Samistraro et al. 2015; Chen et al. 2018; Lima et al. 2019). This contrast is expected, since CBO is a trait composed by the sum of the polymers that compose cellulose and hemicellulose: glucan, xylan, arabinan, galactan, mannan, and acetyl (Wallis et al. 1996); therefore, it is inversely proportional to LIG. Estimates for extractives content (EXT) (2.15%) were found within the reported ranges for E. urophylla × E. grandis (1.27%), E. grandis (1.20%), E. saligna (3.14%), E. dunnii (3.21%), and E. benthamii (3.69%) at age seven (Simetti et al. 2018). Overall, our estimates of LIG and CBO were similar to the ones previously reported for E. benthamii (Samistraro et al. 2015), suggesting that this species has higher LIG and lower CBO values in the spectrum of the most widely planted Eucalyptus species.
Hidden relatedness and pedigree reconstruction
Our results corroborate the ability of genome-wide SNP data to capture and reveal a considerable amount of hidden relatedness among individuals in the trial and possibly correcting pedigree errors although an individualized parentage analysis was outside the scope of this study. This can be seen in the realized relationship matrix (Fig. S1 - B), showing a greater range of relationship values, and thus a more complex relationship structure expected from the numerator pedigree matrix, even revealing a considerable inter-relatedness among families, also neglected by the pedigree matrix. Advancing breeding cycles by an open-pollinated strategy with exclusive maternal control has frequently been adopted by tree breeders for its simplicity and lower cost when compared to full pedigree tracking through controlled pollination, especially in hermaphrodite species with small seed yield per fruit like Eucalyptus that require intensive labor to complete a full mating design (Gamal El-Dien et al. 2016; Klápště et al. 2018). However, with the introduction of marker data in tree breeding, this strategy was shown to frequently carry considerable pedigree errors (Kumar and Richardson 2005; Bush et al. 2011; Gamal El-Dien et al. 2016; Müller et al. 2017; Lima et al. 2019). This hidden relatedness can affect the accuracy of genetic parameter estimation, genotypic values, and predictive ability and inflate the additive variance (Squillace 1974; Namkoong et al. 1988; Tambarussi et al. 2018; Klápště et al. 2018). With the current accessibility to inexpensive large-scale breeder-friendly SNP genotyping data, we envisage its increased routine adoption to improve the accuracy of genetic parameter estimates and predicted genetic values, minimize pedigree errors, and precisely manage inbreeding as generations of mating and selection advance. For E. benthamii, a species still in the initial stages of breeding and typically advanced by incomplete pedigree control, the use of marker data should prove particularly valuable.
Heritability, orthogonality of effects, and dominance variance
Wood chemical traits and BWD showed a moderate to high levels of genetic control, considerably higher than volume growth, corroborating the higher complexity of the latter, irrespective of the model used, in agreement with studies in other Eucalyptus species (Resende et al. 2017; Tan et al. 2017, 2018; Cappa et al. 2019; Lima et al. 2019). Notwithstanding the difference in estimates obtained with pedigree and genomic data, the magnitudes of the narrow-sense genomic heritabilities (\( {h}_a^2 \)) obtained for volume growth (ranging from 0.21 to 0.35 for the GAD and H models respectively) agree with the only estimates reported for E. benthamii (Müller et al. 2017) between 0.10 and 0.28 with different pedigree and genomic models. Our estimates for BWD (0.43 - GAD, up to 0.51 - H) are also consistent with previous studies in other Eucalyptus species reporting values between 0.30 and 0.67 (Resende et al. 2017; Tan et al. 2018; Lima et al. 2019; Suontama et al. 2019). For LIG, our estimates (0.22 - GAD to 0.28 - H / GA) match those reported earlier for E. grandis × E. urophylla hybrids ranging from 0.23 to 0.24 (De Moraes et al. 2018) but are considerably lower than those reported for these same hybrids in a recent study where values varied between 0.58 and 0.68 (Lima et al. 2019). Estimates for the sum of cellulose and hemicellulose content, here referred to as carbohydrate content (CBO), were very similar to those reported separately for these two components for E. grandis × E. urophylla hybrids (Lima et al. 2019)
The substitution of the pedigree matrix for a GRM and the inclusion of the non-additive component allowed disentangling the non-additive from the additive variance providing, as a result, more realistic estimates of variance components (Table S1) and narrow-sense heritability (Table 2). Narrow-sense heritabilities estimated using all three genomic models used (H, GA, and GAD) were lower than the estimates obtained from the pedigree matrix (Ag and At models) exception made for volume growth at age six. These results are generally in line with a number of recent studies in forest trees reporting higher pedigree-based heritabilities than genomic-based estimates for a range of traits in Picea (Beaulieu et al. 2014; Gamal El-Dien et al. 2015), E. grandis × E urophylla hybrids (Resende et al. 2017; Lima et al. 2019), and E. benthamii (Müller et al. 2017). Despite a general trend showing an overestimated pedigree-based heritability, this does not appear to be a rule. In Eucalyptus nitens, for example, these two estimates were found to be relatively similar in magnitude, with small deviations, both for growth and wood quality traits (Suontama et al. 2019).
The main reason behind the higher heritability estimated with both A models (Ag and At) when compared to the genomic heritability is the assumed nature of the A matrix itself. Expected pedigrees are prone to pedigree mislabeling and unaccounted relatedness in the population (Fig. S1), leading to overestimated additive genetic variance (Table S1) (Squillace 1974; Namkoong et al. 1988; Goddard et al. 2011; Ratcliffe et al. 2017; Tambarussi et al. 2018; Klápště et al. 2018), a problem that tends to be more pronounced in mixed mating species, such as E. benthamii for which an outcrossing rate of tm= 0.62 has been estimated, therefore allowing a significant amount of selfing and biparental inbreeding at least in natural populations (Butcher et al. 2005; Tambarussi et al. 2018). Although inbreeding might contribute to the drawback of matrix A not reflecting the correct relatedness, this does not seem to be the case in this population. By taking the average mean of the genomic relationship matrix (G) diagonal, we estimated an F statistic called FGRM (Ghoreishifar et al. 2020) equal to one. In a non-inbred population, this value is in fact expected to be equal to one (Isik et al. 2017).
Narrow-sense heritabilities obtained with the GAD model, that is, including the dominance effect, were lower than those estimated with the GA model. When the GA model is used, the dominance effect is not modeled and, therefore, becomes part of the residual error (Duenk et al. 2017). The GA model thus provides less accurate estimates of the additive variance, as it assumes that the residuals are identically distributed, which is known not to be the case, since the dominance deviations vary from genotype to genotype. In addition, reports in the literature have shown a lack of independence between additive and dominant effects, which are often mistakenly treated as orthogonal (Tan et al. 2018; Xiang et al. 2018). The non-orthogonality between additive and non-additive effects has been reported in Eucalyptus (Tan et al. 2018) comparing additive with additive-dominant models for growth and wood quality traits in Eucalyptus hybrids. That study found that, by including the dominance effect, part of the additive variance was removed, reducing its magnitude and consequently decreasing the genomic estimate of narrow-sense heritability. The same effect was seen in our study when looking at the variance components (Table S1) and the \( {\sigma}_{a\left(\mathrm{GA}\mathrm{D}\right)}^2/{\sigma}_{a\left(\mathrm{GA}\right)}^2 \) ratio (Table 2). The closer to unity this ratio is, the more orthogonal are the effects for that given trait. Apparently, a trend was seen in the magnitude of the dominance variance, indicated by the \( {\sigma}_d^2/{\sigma}_a^2 \) ratio, and the non-orthogonality between the effects. VOL6 and EXT, which displayed greater dominance variance, appeared to be less orthogonal in terms of the associated effects. This non-orthogonality most likely occurs because only under ideal conditions of the epistatic model described by Cockerham (1954), where allele frequencies are assumed to be independent between loci (linkage equilibrium), there is a true orthogonality between the additive and non-additive effects. However, as these conditions are not easily met in breeding populations, these effects can be entangled (Muñoz et al. 2014). This phenomenon has also been reported in other studies (Su et al. 2012; Muñoz et al. 2014; Bouvet et al. 2016; Vitezica et al. 2017).
Typically, full-sib families are necessary to estimate dominance variance using pedigree data. In our data set, there were few known full-sib relationships based exclusively on pedigree information. However, genome-wide markers revealed those additional unknown full-sib relationships allowing one to estimate dominance (Vitezica et al. 2013).
For Eucalyptus, the contribution of dominance variance tends to vary greatly between traits, species, and hybrids in the genus (Tan et al. 2018; Lima et al. 2019). Still, in general, a substantial effect of non-additive genetic control has been reported for growth traits. Our estimate of the \( {\sigma}_d^2/{\sigma}_a^2 \) ratio for VOL6 (1.38) is in agreement with previous reports for growth traits in the genus (Bison et al. 2006; Volker et al. 2008; Resende et al. 2017; Tan et al. 2018; Chen et al. 2018; Lima et al. 2019; Mora et al. 2019) although on the lower end. Estimates have been reported in Eucalyptus where the dominance variance was considerably higher than the additive for a range of growth traits (Resende et al. 2017; Tan et al. 2018; Lima et al. 2019). It is important to note, however, that these high values were all reported for E. grandis x E. urophylla hybrids where the genetic architecture of this trait might be the result of different non-additive sources of variation non-accounted for by the models emerging from their unique interspecific nature. In pure species of the genus, the few studies available have not shown a significant contribution of dominance effects to the variation in growth (Costa e Silva et al. 2009; Klápště et al. 2017) with the exception of a recent study in E. globulus (Mora et al. 2019).
The estimated ratios of \( {\sigma}_d^2/{\sigma}_a^2 \) for CBO (0.87) and LIG (0.80) indicated an almost equal contribution of dominance and additivity to trait variation. The few reports available to date in Eucalyptus were published for hybrids and generally indicated a small contribution of dominance to wood chemical trait variation (Chen et al. 2018; Lima et al. 2019) with values ranging from 0 to 0.08 for cellulose, although for hemicellulose values were 0.08 to 3.5. For lignin, values ranging from 0.19 to 1.15 were found by Chen et al. (2018) and 0 to 0.19 by Lima et al. (2019). Overall, it appears that dominance variation for wood chemical traits is considerably less important than for growth, in line with another recent study (Resende et al. 2017) and anecdotal statements based on unpublished studies where lignin and hemicellulose content are considered predominantly inherited in a additive fashion (Assis 2000). Finally, dominance was also important for extractives content (EXT) in E. benthamii (1.28). However, only one study with Picea abies to date has studied this trait showing unimportant dominance (Hannrup et al. 2004).
Predictive ability
Overall, the use of genomic data resulted in considerably more accurate predictions of trait values when compared to pedigree-only-based models in E. benthamii for the majority of traits, exception made for EXT where the single-step (H) and the At models performed equally well. This result corroborates previous reports in Eucalyptus species and hybrids as well as mainstream conifer species, showing the outstanding value of the convergence between quantitative genetics and genomics in providing more reliable selective breeding potential (Grattapaglia et al. 2018). The HBLUP method, or single-step GBLUP (model H in this study), combining the benefit of an estimate with a larger sample size with the robustness of relationships estimated from genotype data further illustrates the power of this convergence. In our study, HBLUP displayed performance equivalent to the other genomic models for BWD (0.44 – H, GA, and GAD), LIG (0.32 – H and GA; 0.31 – GAD), and CBO (0.32 – H; 0.31 – GA and GAD), but superior performances for EXT and VOL6. Similar results have been reported showing that HBLUP is superior or at least comparable to models based on pedigree (A) and additive GBLUP (GA) in terms of predictive ability (Beaulieu et al. 2014; Legarra et al. 2014; Ashraf et al. 2016; Cappa et al. 2017, 2019; Pérez-Rodríguez et al. 2017; Ratcliffe et al. 2017; Morais Júnior et al. 2018). The superiority of the H model over traditional genomic methods (GA) was more apparent for growth with a PA 35.5% higher than that obtained with model GA for VOL6 consistent with a previous study in Eucalyptus (Cappa et al. 2019). For this trait, the H model was only inferior to the GAD model, both in PA (Fig. 1), as well as bias (Table 2), likely due to the larger importance of dominance for volume growth. Although the H model was not the best for VOL6, its predictive ability (0.31) was close to that obtained with the GAD model (0.34), as well as higher than the other tested models (0.12 - Ag; 0, 22 - At; 0.23 - GA). For the other traits, although model H did not stand out as the best, it was as good as the best models (Fig. 1). Different from the other traits, for extractive content (EXT), the increased sample size used in the At and H models resulted in higher predictive abilities (0.32 for both models) when compared to the other models, which ranged from 0.16 (Ag) to 0.22 (GAD). This possibly occurred due to a larger training population generating a more reliable prediction, with less predictive bias (Table 2) and greater PA (Fig. 1) (Daetwyler et al. 2010; Grattapaglia and Resende 2011; Tan et al. 2017).
It is important to note that by accounting for all trees in the trial, the HBLUP model allows improved selection of any individual tree in the trial, not just the genotyped portion, with an equivalent or even superior predictive ability of other genomic models that only predict for the genotyped individuals. We therefore foresee an increased use of this modeling approach especially in forest tree breeding programs where typically large interconnected progeny trials are established across sites and only a portion of trees can actually be genotyped due to budget constraints. With the HBLUP model, while the genomic data provides information unavailable in the expected pedigrees, the non-genotyped trees not only provide useful additional phenotypic data but they also become selection candidates with equivalent accuracy.
When dominance effects constitute a considerable portion of the phenotypic variance, the inclusion of this effect in modeling has been shown to increase the accuracy of prediction (Nishio and Satoh 2014; De Almeida Filho et al. 2016; Duenk et al. 2017). In our study, this effect was detected even with a small variance associated with the dominance effect for BWD (0.46), LIG (0.80), and CBO (0.87) (Table 2). For these traits, the GAD model reached a similar predictive ability to the other genomic models (H and GA) (Fig. 1). Similar observations were reported for BWD and pulp yield (Resende et al. 2017; Tan et al. 2018), where, despite a relevant dominance variance found, it did not influence the predictive ability.
For volume (VOL6), an increase of the predictive ability was seen when modeling the D matrix. Although dominance variance was detected for all traits in the study, for EXT and VOL6 dominance was preponderant, exceeding the magnitude of the additive variance (\( {\sigma}_d^2/{\sigma}_a^2 \) > 1) (Table 2). As a result, a much higher PA for VOL6 was obtained with the GAD model (0.34) when compared to the GA model (0.23) (Fig. 1). However, for EXT, the GAD model did not show a higher performance suggesting that for this trait additional sources of genetic variation were not adequately captured by the models. Increased predictive abilities between 5 and 14% for growth traits by including a genomic-based dominance component was previously reported in interspecific E. grandis × E. urophylla hybrids (Resende et al. 2017; Tan et al. 2018). Besides the effect of dominance variance, this much larger increase in predictive ability may also be due to the estimation method used. The non-parametric RKHS method used in our study was shown to provide greater predictive abilities for growth traits, especially in the presence of non-additive effects (Tan et al. 2017; Momen et al. 2018; De Almeida Filho et al. 2019). From the breeding standpoint, the predictive abilities estimated for VOL6, exceed the only existing estimate for volume growth in E. benthamii (0.14) (Müller et al. 2017), and are within the same range of estimates for this trait in other species of the genus, such as E. pellita (0.42) (Müller et al. 2017), E. globulus (0.04 to 0.42) (Ballesta et al. 2018), and E. urophylla x E, grandis (0.27 to 0.30) (Tan et al. 2017).
Selection efficiency increases with reduction of breeding cycle length and increased selection intensity
Since its original conception, genomic selection (GS) was proposed as a powerful method to reduce the time associated with breeding cycles (Meuwissen et al. 2001). Devising ways to accelerate breeding cycles while keeping the same or closer selection accuracies has always been a mantra in forest tree breeding, where breeding cycles are usually significantly longer than almost all other plant species (Grattapaglia and Resende 2011; Ratcliffe et al. 2015; Grattapaglia et al. 2018). The main anticipated advantage of GS over BLUP-based selection (PS) arises when the selection of yet unknown phenotypes is made with a time reduction in traditional breeding cycle length as predicted by simulations and empirical results (Grattapaglia and Resende 2011; Resende et al. 2012; Grattapaglia et al. 2018)
We evaluated the reduction of breeding cycle length and increased selection intensity for two main breeding routes where genomic selection could be implemented in Eucalyptus breeding, reminding that both routes would be implemented in parallel for the two complementary objectives as illustrated earlier (Grattapaglia et al. 2018). For clonal selection (CS), a total breeding cycle length of 18 years was assumed for PS and an additive-dominant model (GAD) used in order to capture both additive and dominance effects. For recurrent selection (RS), where typically only the additive variance is accounted for, additive models were used (GA and H) and a total breeding cycle length of 10 years was assumed. These breeding cycle lengths were chosen to reflect the current operational practice of tropical Eucalyptus breeding (Resende et al. 2012, 2017). Overall, in all scenarios of time reduction of the length of the breeding cycle, a considerable increase of selection efficiency was observed (Fig. 2). In general, conservative reductions of 30% already resulted in gains of selection efficiency above 50% or more depending on the trait and model used.
For the recurrent selection breeding route, an increase in selection efficiency (Seff) up to 137% for basic wood density (BWD), using an H model, to 299% for volume at age six (VOL6), with the GA model, would be expected with a reduction of 50% (Fig. 2). Although a 50% reduction in the breeding cycle may seem aggressive, it would essentially correspond to precluding the progeny trial, and carrying out selections at the seedlings stage followed by successfully completing flower induction and mating in 5 years, which is currently considered a feasible option from the operational standpoint. Reductions below 50% would correspond to situations where although progeny trial is precluded, the breeder would take a bit longer to complete genomic selection and mating due to delays in genotyping, flowering or mating.
When considering the recurrent selection route, even though the H model provided better predictive abilities than the GA model for EXT and VOL6 (Fig. 1), the GA model was superior in terms of increasing Seff when compared the H model for all traits (Fig. 2). This may be explained by the impact of sample size on BLUP-based accuracy. Since the GA model is fitted with only the genotyped individuals, it is directly compared with the BLUP-based accuracy obtained from the genotyped dataset only (given by \( \sqrt{h_a^2} \)). The H model, on the other hand, is fitted with both genotyped and non-genotyped individuals, and must be compared with BLUP-based accuracies obtained for the full dataset. These BLUP-based accuracies, although not shown in our study, were heavily influenced by sample size, showing higher values for the full dataset. Increasing sample size can increase PA values for some traits, but an increase in BLUP-based accuracy is much more relevant to increase the final selection efficiency Seff of PS when compared to GS. In order to better assess the relative value of GS, studies must go beyond just reporting PAs. For example, in our study, although the highest PA estimated by a genomic model was for BWD (Fig. 1), this was not the trait where GS would have the largest impact on selection efficiency.
For the clonal selection route, GS would be implemented under a GAD model, either more conservatively, i.e., testing selected individuals as clones in two stages, initial and expanded clonal trials or more aggressively, with just a single expanded clonal trial, but in both approaches, the progeny trial would be precluded. With the aggressive approach, considering a 40 to 50%-time reduction, Seff gains ranged between 150% (EXT) to 300% (VOL6). On the conservative path, with breeding cycle reduction around 20%, gains in efficiency would be considerably more modest down to a point that GS would not be attractive (Fig. 2). Such an exponential nature of the rapid gain in selection efficiency with breeding cycle reduction corroborates earlier reports based on deterministic simulations of predictive ability and heritabilities (Grattapaglia and Resende 2011; Resende et al. 2012). In our study, we now used actual estimates of these parameters from experimental data and adopted a 50% ceiling for time reduction in the breeding cycle length which is realistic for tropical Eucalyptus breeding. Greater reductions in time as described in those simulations would apply to temperate species with longer breeding cycles such as conifers.
Besides accelerating breeding cycles, an additional foreseen advantage of GS is the possibility of increasing selection intensity in the progeny trial phase, provided that a valuable cost/benefit relationship between genotyping costs and selection gains is obtained (Grattapaglia 2014; Heslot et al. 2015; Grattapaglia et al. 2018; Muleta et al. 2019). Overall, increased selection intensities had a positive impact on gains in selection efficiency and models that performed best for each trait for breeding cycle reduction were also the best when increasing selection intensity (Fig. 3). However, the modeled curves had a contrasting logarithmic behavior when compared to the exponential growth curves seen when shortening breeding cycles, with a distinct rate of diminishing returns when greatly increasing the number of evaluated individuals. These simulations suggest that generally a two-fold gain in selection efficiency happens when going from 2000 up to 10,000 plants evaluated, with modest additional gains afterwards. This result is in line with what could be realistically implemented in practice, considering that it is unlikely that the cost of genotyping more than 10,000 seedlings would currently be economically feasible in most eucalyptus breeding programs. However, a number of lower density genotyping methods based on targeted next-generation sequencing of up to ~3000 SNPs have become available in recent times, that allow significantly reduced costs per sample as long as much larger samples sizes are multiplex genotyped (Campbell et al. 2015; Ruff et al. 2020).
Conclusion
To summarize, the genetic analysis carried out for E. benthamii adds further experimental data advocating the positive forecasts for the routine adoption of genomic data to deliver the power of realized genomic relationships in eucalypt breeding programs. A number of recent studies recently reviewed (Grattapaglia et al. 2018) have shown that the integration of genomic data in breeding practice provides a powerful approach to genetic parameters estimation by tracking the variation due to the random Mendelian segregation in pedigrees. In our study, this effect was crystalline for volume growth, indisputably still the central trait in most industrially oriented Eucalyptus breeding programs, regardless of the final use of wood. This major contribution of dominance variance to growth in Eucalyptus has consistently been shown in a number of studies that used genomic data to model this effect when compared to studies that relied exclusively on expected pedigrees. The positive impact of dominance variance for growth, on the estimates of heritability and predictive ability, has a critical consequence on the prospects of genetic gain from selective breeding, especially in Eucalyptus where clonal deployment of elite genotypes exploits all sources of variation, additive and non-additive. Nevertheless, an overestimation of the dominance variance cannot be ruled out, an issue reported to be associated with the RKHS method for the GAD model used in our work (Azevedo et al. 2015; Santos et al. 2017; De Almeida Filho et al. 2019).
Our experimental results also provide additional pieces of evidence that the single-step method (HBLUP) improves genomic prediction of growth and wood traits when phenotypes of the non-genotyped trees in the trial are used. This has been shown in Eucalyptus and other forest trees (Forni et al. 2011; Christensen 2012; Beaulieu et al. 2014; Cappa et al. 2017, 2019; Ratcliffe et al. 2017; Velazco et al. 2019) and is rapidly becoming the preferred analytical approach when considering the use of genomic data in forest tree breeding. From the practical standpoint, the use of the hybrid matrix (H) can be seen as an approximate pedigree reconstruction of the entire trial based on the genotypes of only a portion of family-related individuals. The benefit of genetic relationship is leveraged two ways: while on one hand the genotyped trees provide the pedigree correction with a limited genotyping cost, the non-genotyped trees increase overall prediction accuracies by increasing the samples size. Because forest tree pedigrees frequently contain unknown proportions of mislabeling and undesired inbreeding, or are deliberately advanced by exclusive maternal control, SNP data not only provide an overall correction of parentage errors but also allow advancing populations as though they were derived from labor-intensive fully controlled mating designs. Most importantly, however, the HBLUP method extends the genomic enabled predictive ability to all genetically connected trees across trials, a particularly relevant advantage for large networks of field trials commonly deployed in forest tree breeding.
Our modeling of the expected gain in selection efficiency based on the actual estimates of predictive ability and heritabilities obtained for the E. benthamii trial corroborated previous deterministic simulations (Grattapaglia and Resende 2011) and also forecasted exponential gains in efficiency by implementing genomic prediction even with realistic time reductions in breeding cycle length between 30 and 50% in a recurrent selection Eucalyptus breeding program by precluding progeny testing. In a clonal selection route, the elimination of progeny testing alone results in only modest gain in efficiency, depending on the trait, requiring the additional exclusion of the initial clonal testing trial to accrue a meaningful gain in efficiency. Additional gains in efficiency when compared to standard BLUP-based selection were also seen when an increased selection intensity is applied using genomic data. However, in this scenario, the increase in selection efficiency was quick up to a 5-fold increase in the number of evaluated progeny individuals (2000 to 10,000) and slow thereafter, therefore requiring a careful cost/benefit analysis about increasing selection intensity beyond this point. Finally, an additional projected advantage of GS not evaluated in our study is the possibility to perform an early indirect selection for all traits of interest simultaneously using index selection methods, especially with the inclusion of wood chemical properties that are late expressing and require expensive and time-consuming assays. In operational Eucalyptus breeding, such traits are usually only evaluated on a limited number of trees in the final stages of clonal trials, after truncation selection for volume growth is applied. The possibility of increasing selection intensity by GS provided that genotyping costs are kept within budget, would be a game changer in exploiting the entire spectrum of genetic variation for all traits of interest in a tree breeding program, not only for eucalypts but especially for species that have much longer breeding cycles.
References
Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ (2010) Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J Dairy Sci 93:743–752. https://doi.org/10.3168/jds.2009-2730
Aliloo H, Pryce JE, González-Recio O, Cocks BG, Hayes BJ (2016) Accounting for dominance to improve genomic evaluations of dairy cows for fertility and milk production traits. Genet Sel Evol 48:8. https://doi.org/10.1186/s12711-016-0186-0
Arnold R, Li B, Luo J, Bai F, Baker T (2015) Selection of cold-tolerant Eucalyptus species and provenances for inland frost-susceptible, humid subtropical regions of southern China. Aust For 78:180–193. https://doi.org/10.1080/00049158.2015.1063471
Ashraf B, Edriss V, Akdemir D, Autrique E, Bonnett D, Crossa J, Janss L, Singh R, Jannink JL (2016) Genomic prediction using phenotypes from pedigreed lines with no marker data. Crop Sci 56:957–964. https://doi.org/10.2135/cropsci2015.02.0111
Assis T (2000) Production and use of Eucalypts hybrids for industrial purposes. In: Dungey H, Dieters M, Nikles D (eds) QFRI/CRC-SPF Symposium: Hybrid Breeding and Genetics of Forest Trees, Queensland. Australia, Department of Primary Industries, pp 63–74
Azevedo CF, Resende MDV, FF ES et al (2015) Ridge, Lasso and Bayesian additive-dominance genomic models. BMC Genet 16:105. https://doi.org/10.1186/s12863-015-0264-2
Ballesta P, Serra N, Guerra FP, Hasbún R, Mora F (2018) Genomic prediction of growth and stem quality traits in Eucalyptus globulus Labill. at its southernmost distribution limit in Chile. Forests 9:1–18. https://doi.org/10.3390/f9120779
Beaulieu J, Doerksen T, Clément S, MacKay J, Bousquet J (2014) Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity (Edinb) 113:343–352. https://doi.org/10.1038/hdy.2014.36
Bison O, Ramalho MAP, Rezende GDSP, Aguiar AM, Resende MDVD (2006) Comparison between open pollinated progenies and hybrids performance in Eucalyptus grandis and Eucalyptus urophylla. Silvae Genet 55:192–196. https://doi.org/10.1515/sg-2006-0026
Bouvet JM, Makouanzi G, Cros D, Vigneron P (2016) Modeling additive and non-additive effects in a hybrid population using genome-wide genotyping: prediction accuracy implications. Heredity (Edinb) 116:146–157. https://doi.org/10.1038/hdy.2015.78
Brondani GE, Dutra LF, Wendling I, Grossi F, Hansel FA, Araujo MA (2011) Micropropagação de um híbrido de Eucalyptus (Eucalyptus benthamii x Eucalyptus dunnii). Acta Sci - Agron 33:655–663. https://doi.org/10.4025/actasciagron.v33i4.8317
Bush D, Kain D, Matheson C, Kanowski P (2011) Marker-based adjustment of the additive relationship matrix for estimation of genetic parameters-an example using Eucalyptus cladocalyx. Tree Genet Genomes 7:23–35. https://doi.org/10.1007/s11295-010-0312-z
Butcher PA, Skinner AK, Gardiner CA (2005) Increased inbreeding and inter-species gene flow in remnant populations of the rare Eucalyptus benthamii. Conserv Genet 6:213–226. https://doi.org/10.1007/s10592-004-7830-x
Campbell NR, Harmon SA, Narum SR (2015) Genotyping-in-Thousands by sequencing (GT-seq): a cost effective SNP genotyping method based on custom amplicon sequencing. Mol Ecol Resour 15:855–867. https://doi.org/10.1111/1755-0998.12357
Cappa EP, El-Kassaby YA, Muñoz F et al (2017) Improving accuracy of breeding values by incorporating genomic information in spatial-competition mixed models. Mol Breed 37. https://doi.org/10.1007/s11032-017-0725-6
Cappa EP, de Lima BM, da Silva-Junior OB, Garcia CC, Mansfield SD, Grattapaglia D (2019) Improving genomic prediction of growth and wood traits in Eucalyptus using phenotypes from non-genotyped trees by single-step GBLUP. Plant Sci 284:9–15. https://doi.org/10.1016/j.plantsci.2019.03.017
Carrillo-Varela I, Retamal R, Pereira M, Mendonça RT (2019) Structure and reactivity of cellulose from bleached kraft pulps of different Eucalyptus species upgraded to dissolving pulp. Cellulose 26:5731–5744. https://doi.org/10.1007/s10570-019-02491-0
Chen S, Weng Q, Li F, Li M, Zhou C, Gan S (2018) Genetic parameters for growth and wood chemical properties in Eucalyptus urophylla × E. tereticornis hybrids. Ann For Sci 75:1–11. https://doi.org/10.1007/s13595-018-0694-x
Christensen OF (2012) Compatibility of pedigree-based and marker-based relationship matrices for single-step genetic evaluation. Genet Sel Evol 44:37. https://doi.org/10.1186/1297-9686-44-37
Christensen OF, Lund MS (2010) Genomic prediction when some animals are not genotyped. Genet Sel Evol 42. https://doi.org/10.1186/1297-9686-42-2
Cockerham CC (1954) An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 39:859–882
Costa e Silva J, NMG B, Araújo JA et al (2009) Genetic parameters for growth, wood density and pulp yield in Eucalyptus globulus. Tree Genet Genomes 5:291–305. https://doi.org/10.1007/s11295-008-0174-9
Crossa J, De Los CG, Pérez P et al (2010) Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 186:713–724. https://doi.org/10.1534/genetics.110.118521
Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA (2010) The impact of genetic architecture on genome-wide evaluation methods. Genetics 185:1021–1031. https://doi.org/10.1534/genetics.110.116855
De Almeida Filho JE, Guimarães JFR, Fonseca e Silva F et al (2016) The contribution of dominance to phenotype prediction in a pine breeding and simulated population. Heredity (Edinb) 117:33–41. https://doi.org/10.1038/hdy.2016.23
De Almeida Filho JE, Guimarães JFR, Fonsceca E, Silva F et al (2019) Genomic prediction of additive and non-additive effects using genetic markers and pedigrees. G3 Genes. Genomes, Genet 9:2739–2748. https://doi.org/10.1534/g3.119.201004
De Gonçalves PS, Bortoletto N, Fonseca FDS et al (1998) Early selection for growth vigor in rubber tree genotypes in northwestern Sao Paulo State (Brazil). Genet Mol Biol 21:515–521. https://doi.org/10.1590/s1415-47571998000400018
De Moraes BFX, dos Santos RF, de Lima BM et al (2018) Genomic selection prediction models comparing sequence capture and SNP array genotyping methods. Mol Breed 38. https://doi.org/10.1007/s11032-018-0865-3
Denis M, Bouvet J-M (2011) Genomic selection in tree breeding: testing accuracy of prediction models including dominance effect. BMC Proc 5:1–2. https://doi.org/10.1186/1753-6561-5-s7-o13
Duenk P, Calus MPL, Wientjes YCJ, Bijma P (2017) Benefits of dominance over additive models for the estimation of average effects in the presence of dominance. G3 Genes. Genomes, Genet 7:3405–3414. https://doi.org/10.1534/g3.117.300113
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R Package rrBLUP. Plant Genome 4:250–255. https://doi.org/10.3835/plantgenome2011.08.0024
Falconer DS (1989) Introduction to quantitative genetics. Longman Scientific and Technical, Essex
Falconer DS, Mackay TF (1996) Introduction to quantitative genetics, 4th edn. Longman, Essex, UK
Fisher RA (1918) The correlation between relatives on the suposition of mendelian inheritance. Trans R Soc Edinburgh 53:399–433
Fonseca SM, Resende MDV, Alfenas AC et al (2010) Manual Prático de Melhoramento Genético do Eucalipto, 1st edn. Universidade Federal de Viçosa, Viçosa
Forni S, Aguilar I, Misztal I (2011) Different genomic relationship matrices for single-step analysis using phenotypic, pedigree and genomic information. Genet Sel Evol 43:1–7. https://doi.org/10.1186/1297-9686-43-1
Gamal El-Dien O, Ratcliffe B, Klápště J et al (2015) Prediction accuracies for growth and wood attributes of interior spruce in space using genotyping-by-sequencing. BMC Genomics 16:1–16. https://doi.org/10.1186/s12864-015-1597-y
Gamal El-Dien O, Ratcliffe B, Klápště J et al (2016) Implementation of the realized genomic relationship matrix to open-pollinated white spruce family testing for disentangling additive from nonadditive genetic effects. G3 Genes. Genomes, Genet 6:743–753. https://doi.org/10.1534/g3.115.025957
Gamal El-Dien O, Ratcliffe B, Klápště J et al (2018) Multienvironment genomic variance decomposition analysis of open-pollinated Interior spruce (Picea glauca x engelmannii). Mol Breed 38:26. https://doi.org/10.1007/s11032-018-0784-3
Ghoreishifar SM, Moradi-Shahrbabak H, Fallahi MH, Jalil Sarghale A, Moradi-Shahrbabak M, Abdollahi-Arpanahi R, Khansefid M (2020) Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo. Bubalus bubalis BMC Genet 21:16. https://doi.org/10.1186/s12863-020-0824-y
Goddard ME, Hayes BJ, Meuwissen THE (2011) Using the genomic relationship matrix to predict the accuracy of genomic selection. J Anim Breed Genet 128:409–421. https://doi.org/10.1111/j.1439-0388.2011.00964.x
Goldschmid O (1971) Ultraviolet spectra. In: Sarknanen K, Ludwig C (eds) Lignin: Occurrence, formation, structure and reactions. Wiley-Interscience, New York, pp 241–266
Gomide J, Demuner B (1986) Determination of lignin in woody material: modified Klason method. O Pap 47:36–38
Grattapaglia D (2014) Breeding forest trees by genomic selection: current progress and theway forward. In: Genomics of Plant Genetic Resources: Volume 1. Managing, Sequencing and Mining Genetic Resources. Springer Netherlands, pp 651–682
Grattapaglia D, Resende MDV (2011) Genomic selection in forest tree breeding. Tree Genet Genomes 7:241–255. https://doi.org/10.1007/s11295-010-0328-4
Grattapaglia D, Silva-Junior OB, Resende RT, Cappa EP, Müller BSF, Tan B, Isik F, Ratcliffe B, el-Kassaby YA (2018) Quantitative genetics and genomics converge to accelerate forest tree breeding. Front Plant Sci 871:1–10. https://doi.org/10.3389/fpls.2018.01693
Hall N, Brooker MIH (1973) Camden white gum, Eucalyptus benthamii Maiden et Cambage. Australian Government Pub, Service
Han L, Love K, Peace B, Broadhurst L, England N, Li L, Bush D (2020) Origin of planted Eucalyptus benthamii trees in Camden NSW: checking the effectiveness of circa situm conservation measures using molecular markers. Biodivers Conserv 29:1301–1322. https://doi.org/10.1007/s10531-020-01936-4
Hannrup B, Cahalan C, Chantre G, Grabner M, Karlsson B, Bayon IL, Jones GL, Müller U, Pereira H, Rodrigues JC, Rosner S, Rozenberg P, Wilhelmsson L, Wimmer R (2004) Genetic parameters of growth and wood quality traits in Picea abies. Scand J For Res 19:14–29. https://doi.org/10.1080/02827580310019536
Harwood C (2011) New introductions—doing it right. In: Developing a Eucalypt Resource for New Zealand. Blenheim, New Zealand, p 10
Healey AL, Lupoi JS, Lee DJ, Sykes RW, Guenther JM, Tran K, Decker SR, Singh S, Simmons BA, Henry RJ (2016) Effect of aging on lignin content, composition and enzymatic saccharification in Corymbia hybrids and parental taxa between years 9 and 12. Biomass Bioenergy 93:50–59. https://doi.org/10.1016/j.biombioe.2016.06.016
Heslot N, Jannink JL, Sorrells ME (2015) Perspectives for genomic selection applications and research in plants. Crop Sci 55:1–12. https://doi.org/10.2135/cropsci2014.03.0249
Inglis PW, de CR PM, Resende LV, Grattapaglia D (2018) Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLoS One 13:e0206085. https://doi.org/10.1371/journal.pone.0206085
Isik F, Holland J, Maltecca C (2017) Genetic data analysis for plant and animal breeding. Springer International Publishing, Cham
Kageyama PY, Vencovsky R (1983) Variação genética em progênies de uma população de Eucalyptus grandis (Hill) Maiden. IPEF 24:9–26
Kjaer E, Amaral W, Yanchuk A, Graudal L (2004) Strategies for conservation of forest genetic resources. Conservation of Eucalyptus benthamii: an endangered eucalypt species from eastern Australia. In: Forest genetic resources conservation and management: overview, concepts and some systematic approaches, 1st edn. Interntional Plant Genetic Resources Institute, Rome, pp 5–24
Klápště J, Suontama M, Telfer E, Graham N, Low C, Stovold T, McKinley R, Dungey H (2017) Exploration of genetic architecture through sib-ship reconstruction in advanced breeding population of Eucalyptus nitens. PLoS One 12:e0185137. https://doi.org/10.1371/journal.pone.0185137
Klápště J, Suontama M, Dungey HS, Telfer EJ, Graham NJ, Low CB, Stovold GT (2018) Effect of hidden relatedness on single-step genetic evaluation in an advanced open-pollinated breeding program. J Hered 109:802–810. https://doi.org/10.1093/jhered/esy051
Kumar S, Richardson TE (2005) Inferring relatedness and heritability using molecular markers in radiata pine. Mol Breed 15:55–64. https://doi.org/10.1007/s11032-004-2059-4
Lachowicz H, Wróblewska H, Wojtan R, Sajdak M (2019) The effect of tree age on the chemical composition of the wood of silver birch (Betula pendula Roth.) in Poland. Sci Technol 53:1135–1155 https://doi.org/10.1007/s00226-019-01121-z
Legarra A, Aguilar I, Misztal I (2009) A relationship matrix including full pedigree and genomic information. J Dairy Sci 92:4656–4663. https://doi.org/10.3168/jds.2009-2061
Legarra A, Christensen OF, Aguilar I, Misztal I (2014) Single Step, a general approach for genomic selection. Livest Sci 166:54–65. https://doi.org/10.1016/j.livsci.2014.04.029
Lima BM, Cappa EP, Silva-Junior OB et al (2019) Quantitative genetic parameters for growth and wood properties in Eucalyptus “urograndis” hybrid using near-infrared phenotyping and genome-wide SNP-based relationships. PLoS One 14:1–24. https://doi.org/10.1371/journal.pone.0218747
Lin M, Arnold RJ, Li B, Yang M (2003) Selection of cold-tolerant eucalypts for Hunan Province. In: Turnbull JW (ed) Proceedings of Eucalypts in Asia - A Symposium Held in Zhanjiang, People’s Republic of China, 7-11 April 2003. ACIAR Proceedings. Australian Centre for International Agricultural Research, Zhanjiang, Guangdong, People’s Republic of China. Canberra, pp 107–116
Longue Júnior D, Colodette JL, Gomes VJ (2010) Extraction of wood hemicelluloses through NaOH leaching. Cerne 16:423–429
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Miranda I, Pereira H (2002) The variation of chemical composition and pulping yield with age and growth factors in young Eucalyptus globulus. Wood Fiber Sci 34:140–145
Misztal I, Legarra A, Aguilar I (2009) Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. J Dairy Sci 92:4648–4655. https://doi.org/10.3168/jds.2009-2064
Mohammadi NS, Nemai M, Samariha A et al (2011) Studying the efect of the age of a tree on chemical composition and degree of polymerization cellulose. Indian J Sci Technol 4:1679–1680
Momen M, Mehrgardi AA, Sheikhi A, Kranis A, Tusell L, Morota G, Rosa GJM, Gianola D (2018) Predictive ability of genome-assisted statistical models under various forms of gene action. Sci Rep 8:1–11. https://doi.org/10.1038/s41598-018-30089-2
Mora F, Ballesta P, Serra N (2019) Bayesian analysis of growth, stem straightness and branching quality in full-sib families of Eucalyptus globulus. Bragantia 78:328–336. https://doi.org/10.1590/1678-4499.20180317
Morais Júnior OP, Duarte JB, Breseghello F, Coelho ASG, Morais OP, Magalhães Júnior AM (2018) Single-step reaction norm models for genomic prediction in multienvironment recurrent selection trials. Crop Sci 58:592–607. https://doi.org/10.2135/cropsci2017.06.0366
Morais MC, Pereira H (2012) Variation of extractives content in heartwood and sapwood of Eucalyptus globulus trees. Wood Sci Technol 46:709–719. https://doi.org/10.1007/s00226-011-0438-7
Muleta KT, Pressoir G, Morris GP (2019) Optimizing genomic selection for a sorghum breeding program in Haiti: a simulation study. G3 Genes. Genomes, Genet 9:391–401. https://doi.org/10.1534/g3.118.200932
Müller BSF, Neves LG, de Almeida Filho JE, Resende MFR Jr, Muñoz PR, dos Santos PET, Filho EP, Kirst M, Grattapaglia D (2017) Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genomics 18:1–17. https://doi.org/10.1186/s12864-017-3920-2
Muñoz PR, Resende MFR, Gezan SA et al (2014) Unraveling additive from nonadditive effects using genomic relationship matrices. Genetics 198:1759–1768. https://doi.org/10.1534/genetics.114.171322
Namkoong G, Kang HC, Brouard JS (1988) Tree breeding: principles and strategies. Springer New York
Nishio M, Satoh M (2014) Including dominance effects in the genomic BLUP method for genomic evaluation. PLoS One 9:e85792. https://doi.org/10.1371/journal.pone.0085792
Paludzyszyn Filho E, Telles PE, Carlos S, Ferreira A (2006) Eucaliptos Indicados para Plantio no Estado do Paraná, 1st edn. Embrapa Florestas, Colombo
Pereira AB, Marques Júnior OG, Ramalho MAP, Althoft P (1997) Eficiência da seleção precoce em famílias de meios-irmãos de Eucalyptus camaldulensis Dehnh., avaliadas na região noroeste do Estado de Minas Gerais. Rev Cern 3:67–81
Pereira BLC, Carneiro ACO, Carvalho AMM et al (2013) Influence of chemical composition of Eucalyptus wood on gravimetric yield and charcoal properties. BioResources 8:4574–4592
Pérez P, De Los CG (2014) Genome-wide regression and prediction with the BGLR statistical package. Genetics 198:483–495. https://doi.org/10.1534/genetics.114.164442
Pérez-Rodríguez P, Crossa J, Rutkoski J, Poland J, Singh R, Legarra A, Autrique E, Campos G, Burgueño J, Dreisigacker S (2017) Single-step genomic and pedigree genotype × environment interaction models for predicting wheat lines in international environments. Plant Genome 10:plantgenome2016.09.0089. https://doi.org/10.3835/plantgenome2016.09.0089
Plummer M, Best N, Cowles K, Vines K (2006) CODA: convergence diagnosis and output analysis for MCMC. R News 6:7–11
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Ratcliffe B, Gamal El-Dien O, Klápště J et al (2015) A comparison of genomic selection models across time in interior spruce (Picea engelmannii × glauca) using unordered SNP imputation methods. Heredity (Edinb) 115:547–555. https://doi.org/10.1038/hdy.2015.57
Ratcliffe B, El-Dien OG, Cappa EP et al (2017) Single-step BLUP with varying genotyping effort in open-pollinated Picea glauca. G3 Genes. Genomes, Genet 7:935–942. https://doi.org/10.1534/g3.116.037895
Raymond CA (2002) Genetics of Eucalyptus wood properties. Ann For Sci 59:525–531. https://doi.org/10.1051/forest:2002037
Rencoret J, Gutiérrez A, Nieto L, Jiménez-Barbero J, Faulds CB, Kim H, Ralph J, Martínez ÁT, del Río JC (2011) Lignin composition and structure in young versus adult Eucalyptus globulus plants. Plant Physiol 155:667–682. https://doi.org/10.1104/pp.110.167254
Resende MDV, Assis T (2008) Seleção Recorrente Recíproca entre Populações Sintéticas Multi-Espécies (SRR-PSME) de Eucalipto. Pesqui Florest Bras 57:57–60
Resende MDV, Resende MFR, Sansaloni CP et al (2012) Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol 194:116–128 https://doi.org/10.1111/j.1469-8137.2011.04038.x
Resende RT, Resende MDV, Silva FF, Azevedo CF, Takahashi EK, Silva-Junior OB, Grattapaglia D (2017) Assessing the expected response to genomic selection of individuals and families in Eucalyptus breeding with an additive-dominant model. Heredity (Edinb) 119:245–255. https://doi.org/10.1038/hdy.2017.37
Resquin F, Navarro-Cerrillo RM, Carrasco-Letelier L, Casnati CR (2019) Influence of contrasting stocking densities on the dynamics of above-ground biomass and wood density of Eucalyptus benthamii, Eucalyptus dunnii, and Eucalyptus grandis for bioenergy in Uruguay. For Ecol Manag 438:63–74. https://doi.org/10.1016/j.foreco.2019.02.007
Ruff TM, Marston EJ, Eagle JD, Sthapit SR, Hooker MA, Skinner DZ, See DR (2020) Genotyping by multiplexed sequencing (GMS): a customizable platform for genomic selection. PLoS One 15:e0229207. https://doi.org/10.1371/journal.pone.0229207
Samistraro G, Hart PW, Colodette JL, Ricardo P (2015) Chemical, anatomical, and technology aspects of Eucalyptus benthamii and Eucalyptus dunii for use in an integrated pulp and paper mill. TAPPI J 14:73–81. https://doi.org/10.32964/TJ14.2.73
Santana WMS, Calegario N, Arantes MDC, Trugilho PF (2012) Efeito da idade e da classe diamétrica nas propriedades da madeira de Eucalyptus clonal. Cerne 18:1–8. https://doi.org/10.1590/S0104-77602012000100001
Santos VS, Martins Filho S, Resende MDV, Fonseca Silva FE (2017) Proposta de BLUP genômico com efeitos aditivos e de dominãncia em ambiente R. Rev Bras Biom:361–375
Silva-Junior OB, Faria DA, Grattapaglia D (2015) A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol 206:1527–1540. https://doi.org/10.1111/nph.13322
Simetti R, Bonduelle GM, Da Silva DA (2018) Wood quality of five Eucalyptus species planted in Rio Grande do Sul, Brazil for charcoal production. J Trop For Sci 30:175–181. https://doi.org/10.26525/jtfs2018.30.2.175181
Squillace AE (1974) Average genetic correlations among offspring from open-pollinated forest trees. Silvae Genet 23:149–156
Su G, Christensen OF, Ostersen T et al (2012) Estimating additive and non-additive genetic variances and predicting genetic merits using genome-wide dense single nucleotide polymorphism markers. PLoS One 7:e45293. https://doi.org/10.1371/journal.pone.0045293
Suontama M, Klápště J, Telfer E, Graham N, Stovold T, Low C, McKinley R, Dungey H (2019) Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity (Edinb) 122:370–379. https://doi.org/10.1038/s41437-018-0119-5
Swain TL (1996) A status report on the trials in the ICFR cold tolerant breeding program. 100
Tambarussi EV, Pereira FB, da Silva PHM, Lee D, Bush D (2018) Are tree breeders properly predicting genetic gain? A case study involving Corymbia species. Euphytica 214:1–11. https://doi.org/10.1007/s10681-018-2229-9
Tan B, Grattapaglia D, Martins GS, Ferreira KZ, Sundberg B, Ingvarsson PK (2017) Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol 17:110. https://doi.org/10.1186/s12870-017-1059-6
Tan B, Grattapaglia D, Wu HX, Ingvarsson PK (2018) Genomic relationships reveal significant dominance effects for growth in hybrid Eucalyptus. Plant Sci 267:84–93. https://doi.org/10.1016/j.plantsci.2017.11.011
TAPPI TA of the P and P (2000) Tappi T280 pm-99 standard – acetone extractives of wood and pulp. TAPPI Press
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423. https://doi.org/10.3168/jds.2007-0980
Velazco JG, Malosetti M, Hunt CH, Mace ES, Jordan DR, van Eeuwijk FA (2019) Combining pedigree and genomic information to improve prediction quality: an example in sorghum. Theor Appl Genet 132:2055–2067. https://doi.org/10.1007/s00122-019-03337-w
Vitezica ZG, Varona L, Legarra A (2013) On the additive and dominant variance and covariance of individuals within the genomic selection scope. Genetics 195:1223–1230. https://doi.org/10.1534/genetics.113.155176
Vitezica ZG, Legarra A, Toro MA, Varona L (2017) Orthogonal estimates of variances for additive, dominance, and epistatic effects in populations. Genetics 206:1297–1307. https://doi.org/10.1534/genetics.116.199406
Volker PW, Potts BM, Borralho NMG (2008) Genetic parameters of intra- and inter-specific hybrids of Eucalyptus globulus and E. nitens. Tree Genet Genomes 4:445–460. https://doi.org/10.1007/s11295-007-0122-0
Wallis AFA, Wearne RH, Wright PJ (1996) Analytical characteristics of plantation eucalypt woods relating to kraft pulp yields. Appita J 49:427–432
Wellmann R, Bennewitz J (2012) Bayesian models with dominance effects for genomic evaluation of quantitative traits. Genet Res (Camb) 94:21–37. https://doi.org/10.1017/S0016672312000018
Xiang T, Christensen OF, Vitezica ZG, Legarra A (2016) Genomic evaluation by including dominance effects and inbreeding depression for purebred and crossbred performance with an application in pigs. Genet Sel Evol 48:1–14. https://doi.org/10.1186/s12711-016-0271-4
Xiang T, Christensen OF, Vitezica ZG, Legarra A (2018) Genomic model with correlation between additive and dominance effects. Genetics 209:711–723. https://doi.org/10.1534/genetics.118.301015
Yu A, Gallagher T (2015) Analysis on the growth rhythm and cold tolerance of five-year old Eucalyptus benthamii plantation for bioenergy. Open J For 05:585–592. https://doi.org/10.4236/ojf.2015.56052
Zeng J, Toosi A, Fernando RL, Dekkers JCM, Garrick DJ (2013) Genomic selection of purebred animals for crossbred performance in the presence of dominant gene action. Genet Sel Evol 45:11. https://doi.org/10.1186/1297-9686-45-11
Acknowledgements
The authors are very grateful to Klabin for providing the data. João Gabriel Zanon Paludeto received scholarships from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). Evandro V. Tambarussi and Dario Grattapaglia were supported by research productivity fellowships granted by CNPq.
Code availability
Not applicable.
Data archiving statement
Genotype (https://doi.org/10.6084/m9.figshare.14806449) and phenotype data (https://doi.org/10.6084/m9.figshare.14807916) have been submitted to the Figshare Digital Repository.
Funding
This work was partially supported by PRONEX-FAP-DF grant 2009/00106-8 ‘NEXTREE’, CNPq grant 400663/2012-0, and EMBRAPA grant 03.11.01.007.00.00 to DG that allowed the development and validation of the multi-species EuCHIP60K used in this work. CNPq productivity fellowship number 04899/2019-4 and 304899/2019-4.
Author information
Authors and Affiliations
Contributions
JGZP conceptualization, methodology, software, formal analysis, writing—original draft. DG conceptualization, resources, investigation, writing—review & editing. RAE conceptualization, resources, visualization. EVT writing—review & editing, visualization, supervision, project administration.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by F.P. Guerra
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Fig. S1
Pedigree and genomic based relatedness. Heatmaps and barplots of relationship frequencies contained in the (A) expected relationship matrix and (B) genomic realized relationship (G) for the open-pollinated progeny trial of E. benthamiii. There is values above 1 in the diagonal of the G matrix (B), however, the upper bound of the color key was set to 1 to facilitate comparisons with the A matrix (A) (PDF 265 kb)
ESM 2
(DOCX 48.6 kb)
Rights and permissions
About this article

Cite this article
Paludeto, J.G.Z., Grattapaglia, D., Estopa, R.A. et al. Genomic relationship–based genetic parameters and prospects of genomic selection for growth and wood quality traits in Eucalyptus benthamii. Tree Genetics & Genomes 17, 38 (2021). https://doi.org/10.1007/s11295-021-01516-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-021-01516-9