
Abstract
Genomic selection (GS) is poised to revolutionize eucalypt tree improvement by shortening breeding cycles and increasing selection intensities. This could be particularly valuable for alternative, non-mainstream Eucalyptus species that are still in the initial stages of breeding. Eucalyptus benthamii is important for its adaptation to frost-prone subtropical regions. In this work, we compared seven genomic prediction models, six Bayesian and one frequentist GBLUP (Genomic Best Linear Unbiased Prediction) with the conventional pedigree-based ABLUP approach. Models were evaluated for their ability to estimate heritabilities and predict wood quality traits (wood density, extractives, lignin, and carbohydrates content) and volume growth in 77 open-pollinated families of Eucalyptus benthamii. We also evaluated predictive abilities and heritabilities using variable numbers of SNP in the models. Heritabilities ranged from 0.09 (extractives content) using Bayesian Lasso (BL) to 0.55 (wood density) using ABLUP. Predictive abilities (PA) ranged from 0.12 (for volume using ABLUP) to 0.44 (for wood density using three Bayesian models). All seven genomic models performed similarly well and better than the pedigree model for all traits, except extractives content. Subsets of 5000–7000 SNPs yielded heritabilities and PAs nearly as large as using all 15,293 SNPs. However, a low-density SNP panel might not be economically and technically advantageous compared to the current high-density multi-species Eucalyptus EUCHIP60k. Our results support a positive outlook to implement GS to accelerate Eucalyptus benthamii breeding for adaptation to frost-prone regions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species of Eucalyptus are extensively planted worldwide, delivering fast growing, high productivity plantations with great adaptability and multipurpose products for several industries and small farmers (Mora and Garcia 2000; Myburg et al. 2007). Brazil has become a reference in Eucalyptus forestry due to its advances in breeding, silviculture, and management. The total area of Eucalyptus planted in Brazil was 7.47 million hectares in 2020, ranging from equatorial northern regions to southern areas (Ibá 2021). This wide plantation range has been made possible by exploiting the wide availability of species in the genus with variable adaptive profiles and adequate wood properties. Eucalyptus benthamii Maiden et Cambage has become a species of great importance for its adaptation to frost-prone regions (Butnor et al. 2019) common in southern Brazil (Fonseca et al. 2010). E. benthamii displays high tolerance to cold (Hall et al. 2019) and can withstand absolute minimum temperatures ranging from − 6 to − 10 °C, providing better volume growth and better wood quality compared to tropical species (Lin et al. 2003; Costa et al. 2016). The species has received increasing attention by industries and small farmers in these regions, when planted as a pure species or in hybrid combinations (Pirraglia et al. 2012; Costa et al. 2016).
Eucalyptus benthamii naturally occurs along the coast of New South Wales (NSW), southwest of Sydney, Australia, on the plains along the Nepean River and its tributaries (Butcher et al. 2005; Han et al. 2020). It is now considered an endangered species in its natural range, since its genetic variability was drastically reduced to three small populations. These subpopulations are separated by gene flow barriers, due to direct and indirect anthropic actions. These actions include the introduction of other species, agricultural expansion, flooding, and fires (Hall and Brooker 1973; Skinner 2003; Kjaer et al. 2004; Butcher et al. 2005; Han et al. 2020).
Systematic breeding of E. benthamii is quite recent in Brazil. Current breeding strategies focus on selecting clones from intraspecific recurrent selection programs and developing hybrids with other species. Breeding aims to increase yield, cold resistance, and wood quality for pulp production (Ferraz et al. 2020). Both strategies involve lengthy evaluations, using progeny and sequential clonal trials to identify superior individuals. Up to 18 years are required to select elite clones (Resende et al. 2012a, b), and as many as 15 years to advance a recurrent breeding cycle.
The time required to identify superior genotypes is a common challenge to tree breeding programs (Namkoong et al. 1988; Grattapaglia et al. 2018). To increase genetic gain per unit time in plant and animal breeding, Genomic Selection (GS) was proposed 20 years ago (Meuwissen et al. 2001) and in forest trees, it was anticipated ten years after (Grattapaglia and Resende 2011). Since then, studies have shown its potential to revolutionize tree breeding (reviewed in Grattapaglia et al. 2018). GS has the potential to shorten breeding cycles by skipping some stages of conventional breeding, increasing selection intensity by indirectly evaluating a much larger number of individuals at the seedling stage, and increasing selection accuracy (Grattapaglia et al. 2018). Thus, GS could significantly accelerate the progress of forest tree breeding (Resende et al. 2012a, b; Grattapaglia 2014).
The first step in GS is to estimate the combined effect of a genome-wide panel of markers on the phenotype, using a “training population”. These markers are used to build prediction models that are tested and validated using a different set of genetically related individuals, the “validation population” (Grattapaglia et al. 2018). While traditional breeding is based on the expected genetic relationships represented by an additive relationship matrix (A), GS adjusts and models the random Mendelian segregation term using the realized relationships estimated by a genomic relationship matrix (G) (Grattapaglia et al. 2018).
For successful GS prediction, SNP (single nucleotide polymorphism) markers must capture the existing linkage disequilibrium (LD) between markers and quantitative trait loci (QTLs) and the relatedness between the training population and selection candidates. The extent of LD between marker and QTL is influenced by the effective population size (Ne); the larger the Ne, the more markers are needed to achieve a satisfactory predictive ability (Grattapaglia and Resende 2011; Grattapaglia 2014).
To apply GS, genomic prediction models are required, and these may use different prior assumptions. Bayesian approaches are efficient, flexible in their assumptions, and can generate more realistic credibility intervals, but demand more computational time when many markers are used (Kärkkäinen and Sillanpää 2012; Xavier 2019). In Bayesian approaches, the variance explained by a single locus is assumed to follow a prior distribution, therefore, the variance can vary across loci (Tan et al. 2017). GS studies in Eucalyptus have shown, however, that frequentist mixed models that assume all markers as normally distributed with equal variance, perform as well as Bayesian models, especially for growth and wood quality traits (Gao et al. 2013; Isik et al. 2016; Müller et al. 2017; Tan et al. 2017; Resende et al. 2017).
In this study, we evaluated and compared the ability of seven genomic additive prediction models (six Bayesian models, and one frequentist model) and the commonly used pedigree-based model (ABLUP) to predict growth and wood quality traits. Wood density, extractives, lignin, and carbohydrates content were measured at age four and volume growth was measured at age 6 in an open-pollinated family trial of E. benthamii. We also compared the predictive abilities and heritabilities obtained using variable numbers of SNPs fitted in the genomic models.
Material and methods
Genetic material and SNP data
The phenotypic and genotypic data used in this study were previously described by Paludeto et al. (2021). Briefly, phenotypic data were obtained from an open-pollinated family trial of E. benthamii planted in October 2010 at Otacílio Costa, Santa Catarina state, Brazil. The test plantation design was a randomized complete block design with 20 replicates of 81 open-pollinated families in single-tree plots, totaling 1,620 trees in the trial. Seeds used to establish the trial were collected from selected trees in three seedling seed orchards in Telêmaco Borba (Paraná state) established with 32 open pollinated families introduced from Kedumba Valley, NSW, Australia. SNP genotypes were collected using the genome-wide Eucalyptus SNP chip (EUChip60K) containing 60,904 SNPs on an Illumina Infinium platform (Silva-Junior and Grattapaglia 2015). SNP discovery and ascertainment for the EUChip60K were developed from pooled whole-genome sequencing of 240 trees from the 12 most widely planted Eucalyptus species worldwide, including E. benthamii. The deliberate goal of chip development was to provide a flexible, multi-species genotyping chip to accommodate the genotyping needs of eucalypt-based forest enterprises world-wide at a significantly reduced cost (Silva-Junior and Grattapaglia 2015).
The final training population used in this study consisted of 780 individuals selected in 77 out of the 81 families in the trial. Individuals were selected at age three for stem volume, using a mixed model REML/BLUP procedure:
where y is the vector of measured phenotypic values for volume growth, r is the vector of replicate effects (assumed to be fixed), a is the vector of individual additive genetic effects (assumed to be random), e is the vector of random errors or residues, being \(e\sim \,N(0,I{\sigma }_{e}^{2})\), and \({\sigma }_{e}^{2}\) is the error variance. X and Z are the incidence matrices for their respective effects.
Wood quality was measured at age four on the training population using two increment cores extracted at breast-height. One core was used to determine the wood density, and another core was used for chemical characterization. The cores for wood chemical characterization were ground to sawdust with a Thomas Model 4 Wiley® Mill. These samples were chemically analyzed for lignin content (%) (Goldschmid 1971; Gomide and Demuner 1986), extractives content (%) (TAPPI T280 PM 99 2000), and carbohydrate content (%), i.e. the sum of the polymers that compose cellulose, glycans and hemicelluloses, i.e. xylans, arabinans, galactans and mannans (Wallis et al. 1996). Wood density was determined from the second wood core, using the hydrostatic weighting method, according to the NBR 11941 (ABNT 2003). The wood density was obtained using the ratio between oven dry weight in stove (105 °C ± 1 °C) and the water-saturated volume of the sample, according to the equation: WD = Ms/Vu; where WD is the wood density (g/cm3), Ms is the core dry mass (g), and Vu is the water-saturated volume of the core (cm3). All individual trees were measured at age six for diameter at breast height, and height and wood stem volume was estimated (Volume) using a taper factor equal to 0.45.
Linkage disequilibrium and effective population size
The effective population size (\(N_{e}\)) was estimated using the linkage disequilibrium method (\(LDN_{e}\)) (Waples and Do 2008) implemented by the NeEstimator v2.1 software (Do et al. 2014). This was done using SNPs with MAF (minor allele frequency) ≥ 0.01, since very rare alleles can lead to biased \(N_{e}\) estimates (Waples and Do 2010). We used a random mating model and produced confidence intervals for the estimate of \(N_{e}\) (95% probability) using a jackknife procedure (Waples and Do 2008). Pairwise estimates of LD were estimated using PLINK (Purcell et al. 2007) by the classical measure of the squared correlation of allele frequencies at diallelic loci (r2) without any correction for population structure or relatedness. The decay of r2 versus the pairwise distance between SNPs was modeled using the expression of Hill and Weir (1988), implemented using an R script adapted from Marroni et al. (2011):
where n is the sample size and C is the parameter to be estimated, which representes the product of the population recombination parameter (\(\rho = 4N_{e} r\)) and the distance between SNPs in base pairs. In the recombination parameter r is the recombination rate per base pair and Ne is the effective population size. In this analysis, we considered only SNPs with MAF ≥ 0.01, since very rare alleles can generate unrealistic results (F. Marroni Univ. of Udine, personal communication).
Statistical models and analyses
The ABLUP and GBLUP were fitted using Eq. (1). The only difference is the vector of phenotypic values (y) varied with each analyzed trait. The main difference between the ABLUP and the GBLUP methods is that ABLUP uses the expected genetic relationship between individuals (A matrix) as the variance–covariance matrix to estimate the additive variance and breeding values, where \(a\sim N(0,\mathbf{A}{\sigma }_{a}^{2})\). The GBLUP method, on the other hand, uses the genomic relationship matrix, known as GRM or G matrix (VanRaden 2008), estimated from SNP data, where \(a\sim N(0,\mathbf{G}{\sigma }_{a}^{2})\). ABLUP and GBLUP analyses were carried out using the R package rrBLUP, (Endelman 2011).
Five Bayesian additive genetic models were tested for genomic predictions: Bayes A, Bayes B, Bayes Cπ, Bayesian Lasso (BL) and Bayesian Ridge Regression (BRR). All methods were applied using the following model:
where y is the vector of phenotypic values; X and Z are the incidence matrices for the vectors b (fixed block effects and general mean) and m (random marker effects); \(e\) is the random residual effect being \(e\sim N(0,I{\sigma }_{e}^{2})\); and \({\sigma }_{e}^{2}\) is the error variance.
For these models, the Z matrix was coded numerically, with markers genotypes AA, Aa and aa corresponding to 2, 1 and 0, respectively. In all evaluated Bayesian approaches, the following statistical assumptions were made:
where \({S}_{e}\) is a scale parameter and \({\nu }_{e}\) is the degrees of freedom.
Although all tested Bayesian models have the common assumptions listed above, each model uses different priors for the m vector:
-
Bayesian Ridge Regression (BRR): Assumes that all markers contribute equally to the genetic variance and, therefore, have the same variance (Meuwissen et al. 2001):
$$\begin{aligned}&{m}_{i}|{\sigma }_{m}^{2}\sim N(0,{\sigma }_{m}^{2})\\ & {\sigma }_{m}^{2}|{S}_{m},{\nu }_{m}\sim {\chi }^{-2}({S}_{m},{\nu }_{m}=-2)\end{aligned}$$where \({S}_{m}\) is the scale parameter and \({\nu }_{m}\) is the degrees of freedom for marker effects.
-
Bayes A: Assumes that marker effects have heterogeneous variances (Meuwissen et al. 2001; Pérez and De Los Campos 2014):
$$\begin{aligned}& {m}_{i}|{\sigma }_{{m}_{i}}^{2}\sim N(0,{\sigma }_{{m}_{i}}^{2})\\ & {\sigma }_{{m}_{i}}^{2}|{S}_{m},{\nu }_{m}\sim {\chi }^{-2}({S}_{m},{\nu }_{m})\end{aligned}$$ -
Bayes B: Is similar to Bayes A for the marker variance assumptions, but also assumes that a proportion of markers have null effects (parameter π) (Meuwissen et al. 2001; Pérez and De Los Campos 2014):
$$\begin{aligned}&{m}_{i}|{\sigma }_{m}^{2}\left\{\begin{array}{ll} \sim N(0,{\sigma }_{m}^{2}) &\text{with probability equal}1-\pi\\=0 &\text{with probability equal}\, \pi \end{array}\right.\\ & {\sigma }_{{m}_{i}}^{2}|{S}_{m},{\nu }_{m}\sim {\chi }^{-2}({S}_{m},{\nu }_{m})\end{aligned}$$ -
Bayes Cπ: This model assumes that all markers have the same variance, and, similar to BayesB, assumes that a proportion of markers have null effects (parameter π) (Habier et al. 2011):
$$\begin{aligned}&{m}_{i}|{\sigma }_{m}^{2}\left\{\begin{array}{ll}\sim N(0,{\sigma }_{m}^{2}) &\text{with probability equal}1-\pi\\ =0 &\text{with probability equal}\, \pi \end{array}\right.\\ & {\sigma }_{{m}_{i}}^{2}|{S}_{m},{\nu }_{m}\sim {\chi }^{-2}({S}_{m},{\nu }_{m})\\ & \pi \sim Beta({p}_{0},{\pi }_{0})\end{aligned}$$where \({p}_{0}\) and \({\pi }_{0}\) are hyperparameters of the beta distribution.
-
Bayesian Lasso (BL): Similar to Bayes A and Bayes B, this approach assumes that all markers have effects, and the variance of marker effects obeys the double exponential distribution. This approach leads to a strong shrinkage of marker effects for a large number of markers (Park and Casella 2008; De Los Campos et al. 2009):
$$\begin{aligned} & {m}_{i}|{\sigma }_{e}^{2},{\tau }_{i}^{2}\sim N(0,{\sigma }_{e}^{2}\times {\tau }_{i}^{2})\\ & {\tau }_{i}^{2}|\lambda \sim Exp(0.5{\lambda }^{2})\\ & \lambda |r,s\sim G(r,s) \end{aligned}$$where \(\lambda\) is a regularization parameter, \({\tau }_{i}^{2}\) is the extra variance component associated with each marker locus, and \(r\) and \(s\) are hyperparameters of the gamma distribution.
We also tested the RKHS model (Reproducing Kernel Hilbert Space) (Gianola et al. 2006, 2010), a Bayesian semi-parametric method, that has the same assumptions as ABLUP and GBLUP for the \(a\) vector, where \(a\sim N(0,\mathbf{K}{\sigma }_{a}^{2})\). In this model \(\mathbf{K}\) is a Gaussian kernel matrix given by \(\mathrm{exp}(-h{d}_{ij})\), with \(h\) as the bandwidth parameter that controls how fast the prior covariance among individuals declines with increasing Euclidean distance (\({d}_{ij}\)). The \({d}_{ij}\) between two individuals was computed as \({d}_{ij}={\sum }_{k=1}^{p}{({m}_{ik}-{m}_{jk})}^{2}\) at a normalized range from 0 to 1, where \({m}_{ik}\) and \({m}_{jk}\) are the \(k\)th SNP markers for the \(i\)th and \(j\)th individuals.
All Bayesian models were implemented using the R package BGLR (Pérez and De Los Campos 2014) using 200,000 iterations of the Markov Chain Monte Carlo method (MCMC), with 50,000 cycles of burn-in. The convergence of the Markov chains was checked with a Geweke diagnostic (Geweke 1992).
We estimated predictive abilities (PA) as the correlations (rgy) between the GEBVs (genomic estimated breeding values) and the measured, unadjusted, phenotypic values. PAs were estimated using a tenfold cross validation method where the whole dataset was equally partitioned into k folds (k = 10). The GEBVs for the 10% individuals in each fold were predicted based on a model trained with the remaining 90% of individuals, and the obtained GEBV correlated with phenotypic values.
Narrow-sense heritabilities (\(h_{a}^{2}\)) for all traits and all models (ABLUP, GBLUP, and Bayesian) were estimated by the same equation: \(h_{a}^{2} = \sigma_{a}^{2} /(\sigma_{a}^{2} + \sigma_{e}^{2} )\), where \(\sigma_{a}^{2}\) and \(\sigma_{e}^{2}\) are the additive and residual variance estimates obtained from each model. Standard deviations of the heritability estimates were obtained using the same cross validation procedure described above for PA.
The impacts of progressive reductions in the number of SNPs fitted in the model on the estimates of heritabilities and PA were evaluated using the BRR model. The BRR model was fitted using different subsets of SNPs between 15,000 and 1000 SNPs in 1000-SNP reductions. This was followed by of 100-SNP drops. For each number of SNPs tested, ten replicates were randomly sampled and the average heritability and PA were calculated.
Results
Data for 15,293 high-quality SNP markers with call rates > 95% were obtained for 671 individuals from 77 families. To include all rare alleles in the analyses no MAF cutoff was applied (i.e., MAF ≥ 0.000745 given that 671 individuals were sampled). The genomic relationship matrix (GRM) identified 53 putatively selfed individuals (pairwise relatedness values above 0.75). The diagonal elements of the GRM for these putatively selfed individuals ranged from 0.91 to 3.68, with a mean of 1.33. Thus, the FGRM, which is the diagonal elements minus 1, ranged from − 0.09 to 2.68, with mean 0.33. For the entire population, the GRM diagonal elements ranged from 0.82 to 3.68, with mean 1.00. This means that the FGRM ranged from − 0.18 to 2.68, with mean zero.
Linkage disequilibrium and effective population size
Pairwise estimates of LD were obtained using SNPs with MAF ≥ 0.01 and no correction for population structure or relatedness (Fig. 1). The modeled LD dropped below 0.2 within 12.3 Kb. Considering SNPs with MAF ≥ 0.01, the estimated effective population size (Ne) was 80.1 with CI95% = [75.3–87.7].

Trend line of the average decay of the pairwise LD (r2) with physical distance, estimated using SNPs with MAF > 0.01 in 77 open-pollinated families of E. benthamii. The dashed line corresponds to the commonly used threshold of r2 = 0.2 at which LD stops to exist and equilibrium is reached
Heritabilities
Narrow-sense heritabilities estimated using the pedigree-based model (ABLUP) were generally higher than those obtained using all genomic models, except for Volume (Table 1). Bayesian estimates of heritability were similar across models and ranged from a low 0.09 for extractives content (BL model) to a high 0.50 for wood density (BayesA and BRR models). Compared to the other Bayesian models the BL model yielded lower estimates for extractives content and Volume. Bayesian estimates were also similar to those from GBLUP, which ranged from 0.20 (extractives content) to 0.49 (wood density).
Empirical prediction ability from cross-validation
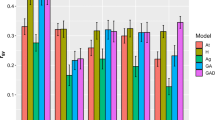
Predictive abilities obtained using the GBLUP model ranged from 0.18 for extractives content to 0.43 for wood density. These were generally higher than those estimated using the pedigree-based model (ABLUP), which ranged from 0.13 for Volume to 0.27 for wood density (Fig. 2). It is worth reminding that the two predictions are completely different. While ABLUP predictions are based on the midparent value, GBLUP predictions are based on the individual itself. The same was observed for the other Bayesian models employed, with PAs ranging from 0.16 for extractives content using the RKHS model, to 0.44 for wood density, using BayesCπ, BRR, and RKHS (Fig. 2). Overall, the PAs using the genomic models were higher than those obtained using the pedigree-based model, with the exception of extractives content. The PAs were essentially equivalent between the pedigree and the genomic models for this last trait (Fig. 2). In general, all Bayesian genomic models provided PAs similar to those using the standard GBLUP approach. However, for Volume, the RKHS method yielded a better prediction. Bayesian Lasso (BL) was inferior to the other genomic methods for wood density.

Predictive abilities (rgy) and standard error bars estimated using different genomic prediction models and a pedigree-based model (ABLUP) for wood properties and volume growth in 77 open-pollinated families of E. benthamii
Genomic heritabilities and predictions with variable numbers of SNPs
To evaluate the impact of varying the number of SNPs on the estimates of heritability and PA, we used the BRR model, since it closely resembles the traditional and most commonly used GBLUP method in its prior assumptions. Heritabilities increased rapidly and reached a plateau around 10,000 SNPs for all traits (Fig. 3a). PAs also increased rapidly as more SNPs were added to the model, plateauing at around 5000–7000 SNPs. (Fig. 3b).

a Genomic narrow-sense heritability (\(h_{a}^{2}\)) estimated using increasing numbers of SNPs and the Bayesian Ridge Regression (BRR) genomic model for wood properties and volume growth in 77 open-pollinated families of E. benthamii. b Predictive abilities (rgy) estimated using a Bayesian Ridge Regression (BRR) genomic model with increasing numbers of SNPs for wood properties and volume growth in 77 open-pollinated families of E. benthamii
Discussion
Inbreeding, linkage disequilibrium, and effective population size
The value of SNP data for more precisely estimating genetic parameters in forest trees has been well documented (reviewed in Grattapaglia et al. 2018). A genomic relationship matrix (GRM) built with marker data provides the realized genetic relatedness among individuals, instead of the expected relatedness, which may be based on inaccurate pedigree information. Additionally, the diagonal of the GRM can be used to detect inbreeding in the population, since the average of the diagonal elements corresponds to the average inbreeding coefficient for a given population (Isik et al. 2017). If we subtract one from the diagonal of a GRM, an F statistic called FGRM is obtained (Zhang et al. 2015; Ghoreishifar et al. 2020). In a non-inbred population, the average of the diagonal elements of the GRM is expected to be 1 (Isik et al. 2017), which means an average zero for the FGRM. This is the average estimate we obtained in our E. benthamii breeding population. For the 53 putatively selfed individuals, the average of the GRM diagonal elements was greater than one (1.33), indicating that the average FGRM for these individuals is 0.33. This is explained by the typically mixed mating system of E. benthamii with outcrossing rate (tm) varying from 0.45 to 0.68. This suggests there is considerable amount of self-fertilization or biparental inbreeding in this species (Butcher et al. 2005).
Genome-wide linkage disequilibrium was estimated to decay below 0.2 within 12.3 Kb (Fig. 1). This estimate is close to a previous estimate of 15.6 kb for another E. benthamii population studied by Müller et al. (2017). While that population was mostly composed of selected seed seed sources from wild Australian populations, our population had substantial family structure and had experienced one round of selection. Nonetheless, the estimates of LD decay were similar, suggesting that our population still resembles a natural population in terms of effective population size.
Earlier LD estimates for natural populations of Eucalyptus, which were based on short range sequence data in candidate genes, suggested that LD decayed within 1 kb (Grattapaglia and Kirst 2008; Thavamanikumar et al. 2011; Denis et al. 2013). However, genome-wide estimates in a natural population of E. grandis showed that LD decayed over a larger distance (4–6 kb), with considerable variation from absence to complete LD up to 50 kb (Silva-Junior and Grattapaglia 2015). Our LD values also indicate that LD extends over several kb, which facilitates GS and the durability of genomic prediction models across generations.
Our estimate of \(N_{e}\) (80.1) was obtained using SNP markers with MAF ≥ 0.01 to provide a good balance between maximizing precision and reducing bias (Waples and Do 2010). This \(N_{e}\) should provide abundant variability for sustained long term genetic gains (Namkoong et al. 1988; White et al. 2007). The effective population size also affects the success of genomic selection. The smaller the \(N_{e}\) the smaller the number of independently segregating chromosome segments and the fewer SNPs are needed to capture all the QTLs co-segregating with those segments. Conversely, the larger the \(N_{e}\) the lower will be the average genetic relatedness with the selection candidates in the next generation. Thus, the larger the \(N_{e}\), the more markers are needed to capture the LD between markers and quantitative trait loci (QTLs). In other words, more markers are needed to ensure adequate levels of genetic relatedness to drive accurate predictions across generations (Grattapaglia and Resende 2011; Grattapaglia 2014; Lee et al. 2017). Deterministic simulations showed that ~ 15 markers/cM will provide a PA of 0.6–0.7 for a population with \(N_{e}\) = 60–100 and a trait with a heritability of 0.2 (Grattapaglia and Resende 2011). With a Eucalyptus recombining genome around 1000–1200 cM (Silva-Junior and Grattapaglia 2015) the 15,293 SNPs provided by the EUChip60K should be enough to obtain accurate genomic predictions in our E. benthamii population.
Heritabilities and predictive abilities
As in other Eucalyptus species, our E. benthamii population displayed a high heritability for wood density, moderate heritabilities for extractives, lignin, and carbohydrate content, and a low heritability for volume growth (Resende et al. 2017; De Moraes et al. 2018; Tan et al. 2018; Lima et al. 2019; Suontama et al. 2019; Mphahlele et al. 2020). All genomic models provided similar heritability estimates, with only slight differences (Table 1) attributable to the variable prior assumptions of the models. GBLUP and BRR had very similar heritabilities, as expected, given their same assumptions regarding homogeneity of marker effects. Heritabilities estimated using ABLUP were somewhat higher than those obtained with all genomic models, except for volume growth. Regarding the trend observed for the other traits, it has been shown that ABLUP may overestimate the additive variance and, thus, heritability. This is because estimates of additive variance may contain non-additive variance following the lack of orthogonality of additive and non-additive effects in breeding (non-idealized) populations (Muñoz et al. 2014). Experimental evidences have shown that the inclusion of dominance effects in genomic models decreased the magnitude of the estimated additive genetic variance and heritability (Müller et al. 2017; Resende et al. 2017; Tan et al. 2017; De Almeida Filho et al. 2019; Paludeto et al. 2021). However, the ABLUP estimate of heritability for volume (0.18) was smaller when compared to the GBLUP estimate (0.24) which is different from previous reports and therefore possibly underestimated.
It is important to point out that genomic models are expected to provide more realistic estimates of variance components and heritabilities compared to pedigree-based models. This is especially true for species with mixed-mating systems which are common in forest trees that result in unknown levels of inbreeding, incomplete or imprecise pedigrees (Klápště et al. 2017; Paludeto et al. 2021; Ratcliffe et al. 2017; Tambarussi et al. 2018). Pedigree based methods not only ignore relationships beyond those included in the known pedigree, but also operate with expected rather than actual relationships. Moreover, pedigree relationships are exact only under an infinitesimal model. Under a more realistic finite-locus model with linkage, the actual relationships will be distributed around the expectation, with variable relationships among half and full-sibs. Furthermore, because pedigree-based analyses assume homogeneous relationships among the same type of relatives, the genetic (co)variance components are estimated based on between-family variation only. This is because the Mendelian sampling deviations cannot be separated from residual non-genetic sources of error (Ødegård and Meuwissen 2012). Finally, the use of a GRM has a major impact when major pedigree errors exist, allowing the rescue of field trials that suffer from erroneous identification of trees and, and would otherwise be completely useless for genetic parameter estimation (Müller et al. 2017; Tan et al. 2018).
Except for extractives content, predictive abilities based on genomic approaches were generally higher than those based on the pedigree-based approach, ABLUP (Fig. 2). Similar results were reported for Eucalyptus hybrids (Tan et al. 2017), Eucalyptus pellita (Müller et al. 2017), Eucalyptus polybractea (Kainer et al. 2018), Picea abies (Lenz et al. 2020), and Pinus taeda (De Almeida Filho et al. 2019). In general, PAs using GBLUP and the Bayesian models were similar for wood quality and volume growth, in agreement with results reported previously for Eucalyptus (Müller et al. 2017; Tan et al. 2018) and other forest tree species (Resende et al. 2012a, b; Beaulieu et al. 2014a; Ratcliffe et al. 2015; Isik et al. 2016). Interestingly, the machine-learning RKHS model yielded a considerably greater PA for volume when compared to all other models (Fig. 1). This is consistent with previous results for growth traits in Eucalyptus sp. and Pinus taeda (Müller et al. 2017; Tan et al. 2017; De Almeida Filho et al. 2019). Two factors may explain these results. First, RKHS can account for non-additive variation, and dominance variation is known to be important for growth traits in Eucalyptus (Resende et al. 2017; Tan et al. 2018; Lima et al. 2019). Second, because RKHS is a semi-parametric method, it is less affected by the choice of priors. Given its focus on prediction, the RKHS method is better for dealing with noisy, redundant or inconsistent information (Gianola and van Kaam 2008; González-Recio et al. 2009). Nevertheless, RKHS performed slightly worse than the other models for all traits, except wood density.
Overall, only slight differences in PAs were found using different Bayesian models, despite the differences in priors. BayesA, BayesB, and BL, do not assume a normal distribution of marker effects, but instead assume that some major QTL loci are present. BayesB, nevertheless, performed slightly better than all other Bayesian models, and almost as well as RKHS for Volume. BayesB is frequently suggested as the best approach for traits controlled by major effect QTLs (Daetwyler et al. 2010; Wang et al. 2019), although this would not be expected for volume growth in Eucalyptus. A possible explanation for the better performance of BayesB might be its ability to capture non-additive effects. Among all models Bayesian Lasso had the poorest performance. From the practical standpoint, it is relevant to highlight the equivalent efficiency of the frequentist GBLUP and Bayesian models to predict growth and wood quality traits in Eucalyptus. These results further support the hypothesis that these traits have a multifactorial genetic architecture with no major effect loci, thus adequately fitting the infinitesimal model.
Impact of SNP number on genomic predictions
We simulated the impact of SNP number on the estimates of heritability and PA. Results showed that smaller numbers of SNPs, in the range of a few thousand, can result in similar prediction abilities and heritabilities when compared to using all 15,000 SNPs, even in our population with relatively large effective size (\(N_{e}\) = 80.1). Similar results have been reported in Picea glauca (Beaulieu et al. 2014b), Picea mariana (Lenz et al. 2017) and Picea abies (Chen et al. 2018). These results suggest that relatedness rather than LD, is the main driver of PA (Müller et al. 2017; Chen et al. 2018).
Despite the declining costs of SNP chip genotyping technologies, high-density chips are still expensive for tree breeding programs run on tight budgets. Thus, low-density SNP chips could be an attractive alternative, provided that the final cost per genotyped sample decreases linearly with the number of SNPs (Gorjanc et al. 2015; Bhandari et al. 2019), which is not currently the case. However, there are two hurdles to using low-density SNP chips in the case of Eucalyptus. First, this would entail selecting SNPs optimized for polymorphism for a particular breeding population, rather than using the more flexible multi-species EUChip60K. Currently, the cost of manufacturing and using a SNP chip mostly depends on the prospective number of samples to be genotyped, rather than on the number of SNPs. In other words, the cost reduction from a 60 K SNP chip to a 10 K SNP chip is not linear. Thus, unless tens of thousands of samples can be contracted upfront, a custom chip will be considerably more expensive than using the existing multi-species EUChip60K. This “community” chip has been used by a large number of breeding programs that jointly contract and process thousands of samples per year. The second hurdle involves changes in SNP allele frequencies and LD structure from one generation to the next. The number of informative SNPs in the initial generation may decline as allele frequencies change or even become fixed after a few generations as result of selection, drift and recombination. This might be a particular concern in aggressive eucalypt genomic selection programs, that involve small effective population sizes, short breeding cycles with early flower induction, combined with high selection intensities (Grattapaglia and Resende 2011; Grattapaglia et al. 2018; Paludeto et al. 2021). Furthermore, simulations have shown that higher marker densities can keep genomic selection effective for more generations and yield larger genetic gains when compared to using lower marker density, as the accuracy and the effectiveness of selection drop very quickly in the latter case (Long et al. 2011; DoVale et al. 2021). Furthermore, even in the absence of selection, recombination under low marker density more rapidly dissipates descent relationships when compared to higher marker densities, negatively impacting genomic predictions (Long et al. 2011). Therefore, it is valuable or even necessary to have an excess of informative SNPs on the chip in the best interest of ensuring some level of redundancy while avoiding the risk of over-optimization of the system. Evidently, this can be achieved in a high-density SNP chip and not in a customized low-density SNP chip. The original concept behind the design of the multi-species EuCHIP60K was to provide a common SNP platform to encompass all main planted Eucalyptus species and provide the necessary flexibility, redundancy and low cost for the implementation of long-term genomic selection by many breeding programs worldwide (Silva-Junior et al. 2015).
Conclusions
From the applied breeding standpoint our study has some limitations that will require further investigation before genomic selection is routinely adopted in E. benthamii. Estimates of prediction ability were based on contemporary training and validation sets grown in the same environment. Thus, these estimates do not necessarily match what to expect on the performance of GS in a future generation after genetic recombination. Moreover, heritability and PAs estimates apply to this particular population test and tree age. Given our single experimental trial, no assessment was made of the impact of genotype by environment interaction on the accuracy of predictions and how these will perform with exposure to the evolving environment. Nevertheless, our results corroborate a large number of reports in forest trees showing that SNP markers can be used to improve estimates of genetic parameters and predictions of wood properties and growth (Grattapaglia et al. 2018). Benefits result from the ability to capture realized genetic relationships among individuals, particularly for open pollinated breeding populations. This in turn provides more realistic estimates of heritabilities to inform more accurate expectations of genetic gains in a breeding program.
Furthermore, the implementation of genomic selection in E. benthamii can shorten breeding cycles by early selection of genetically superior material at the seedling stage. By eliminating the need for field progeny trials, the breeding cycle could be reduced by four to nine years depending on the duration of the follow up clonal trials of the genomically selected seedlings (Resende et al. 2017). For wood quality traits, the GBLUP model provided similar PAs to all Bayesian methods tested. However, for volume growth, RKHS and BayesB were better and are recommended, given their ability to capture non-additive genetic variance. Finally, satisfactory estimates of predictive ability in this population were obtained with as few as ~ 5000 SNPs. However, the use of a custom low-density SNP chip should only be considered aware of the cost and long-term technical advantages of using the more flexible and higher density SNP chip currently available for Eucalyptus species.
Data Archiving Statement
Genotypic (10.6084/m9.figshare.14806449) and phenotypic (10.6084/m9.figshare.14807916) data used in this study are available in the Figshare Digital Repository.
References
ABNT (2003) Wood—Determination of basic density. NBR 11941:2003. ABNT—Assoc Bras Normas Técnicas 6
Beaulieu J, Doerksen T, Clément S et al (2014a) Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity (edinb) 113:343–352. https://doi.org/10.1038/hdy.2014.36
Beaulieu J, Doerksen TK, MacKay J, Rainville A, Bousquet J (2014b) Genomic selection accuracies within and between environments and small breeding groups in white spruce. BMC Genom 15:1048
Bhandari A, Bartholomé J, Cao-Hamadoun TV et al (2019) Selection of trait-specific markers and multi-environment models improve genomic predictive ability in rice. PLoS ONE 14:e0208871. https://doi.org/10.1371/JOURNAL.PONE.0208871
Butcher PA, Skinner AK, Gardiner CA (2005) Increased inbreeding and inter-species gene flow in remnant populations of the rare Eucalyptus benthamii. Conserv Genet 6:213–226. https://doi.org/10.1007/s10592-004-7830-x
Butnor JR, Johnsen KH, Anderson PH et al (2019) Growth, photosynthesis, and cold tolerance of Eucalyptus benthamii planted in the Piedmont of North Carolina. For Sci 65:59–67. https://doi.org/10.1093/forsci/fxy030
Chen Z-Q, Baison J, Pan J et al (2018) Accuracy of genomic selection for growth and wood quality traits in two control-pollinated progeny trials using exome capture as the genotyping platform in Norway spruce. BMC Genom 19:946. https://doi.org/10.1186/s12864-018-5256-y
da Costa RML, Estopa RA, Biernaski FA, Mori ES (2016) Predição de ganhos genéticos em progênies de Eucalyptus benthamii Maiden et Cambage por diferentes métodos de seleção. Sci for. https://doi.org/10.18671/scifor.v44n109.10
Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA (2010) The impact of genetic architecture on genome-wide evaluation methods. Genetics 185:1021–1031. https://doi.org/10.1534/genetics.110.116855
De Almeida Filho JE, Guimarães JFR, Fonsceca E, Silva F et al (2019) Genomic prediction of additive and non-additive effects using genetic markers and pedigrees. G3 Genes Genomes Genet 9:2739–2748. https://doi.org/10.1534/g3.119.201004
De Los Campos G, Naya H, Gianola D et al (2009) Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 182:375–385. https://doi.org/10.1534/genetics.109.101501
De Moraes BFX, dos Santos RF, de Lima BM et al (2018) Genomic selection prediction models comparing sequence capture and SNP array genotyping methods. Mol Breed. https://doi.org/10.1007/s11032-018-0865-3
Denis M, Favreau B, Ueno S et al (2013) Genetic variation of wood chemical traits and association with underlying genes in Eucalyptus urophylla. Tree Genet Genomes 9:927–942. https://doi.org/10.1007/s11295-013-0606-z
Do C, Waples RS, Peel D et al (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214. https://doi.org/10.1111/1755-0998.12157
DoVale JC, Carvalho HF, Sabadin F, Fritsche-Neto R (2021) Reduction of genotyping marker density for genomic selection is not an affordable approach to long-term breeding in cross-pollinated crops. bioRxiv 2021.03.05.434084. https://doi.org/10.1101/2021.03.05.434084
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4:250–255. https://doi.org/10.3835/plantgenome2011.08.0024
Ferraz AG, Cruz CD, dos Santos GA et al (2020) Potential of a population of Eucalyptus benthamii based on growth and technological characteristics of wood. Euphytica 216:1–15. https://doi.org/10.1007/s10681-020-02628-4
Fonseca SM, Resende MDV, Alfenas AC et al (2010) Manual Prático de Melhoramento Genético do Eucalipto, 1st edn. Universidade Federal de Viçosa, Viçosa
Gao H, Su G, Janss L et al (2013) Model comparison on genomic predictions using high-density markers for different groups of bulls in the Nordic Holstein population. J Dairy Sci 96:4678–4687. https://doi.org/10.3168/jds.2012-6406
Geweke J (1992) Evaluating the accuracy of sampling-based approaches to calculating posterior moments. Bayesian Stat 4:641
Ghoreishifar SM, Moradi-Shahrbabak H, Fallahi MH et al (2020) Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo, Bubalus bubalis. BMC Genet. https://doi.org/10.1186/s12863-020-0824-y
Gianola D, van Kaam JBCHM (2008) Reproducing kernel hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics 178:2289–2303. https://doi.org/10.1534/genetics.107.084285
Gianola D, Fernando RL, Stella A (2006) Genomic-assisted prediction of genetic value with semiparametric procedures. Genetics 173:1761–1776. https://doi.org/10.1534/genetics.105.049510
Gianola D, De Los Campos G, González-Recio O et al (2010) Statistical learning methods for genome-based analysis of quantitative traits. In: 9th world congress of genetics applied to livestock production, Leipzig, Germany, pp 1–6
Goldschmid O (1971) Ultraviolet spectra. In: Sarknanen K, Ludwig C (eds) Lignin: occurrence, formation, structure and reactions. Wiley, New York, pp 241–266
Gomide J, Demuner B (1986) Determination of lignin in woody material: modified Klason method. O Pap 47:36–38
González-Recio O, Gianola D, Rosa GJ et al (2009) Genome-assisted prediction of a quantitative trait measured in parents and progeny: application to food conversion rate in chickens. Genet Sel Evol 41:3. https://doi.org/10.1186/1297-9686-41-3
Gorjanc G, Cleveland MA, Houston RD, Hickey JM (2015) Potential of genotyping-by-sequencing for genomic selection in livestock populations. Genet Sel Evol 47:12. https://doi.org/10.1186/s12711-015-0102-z
Grattapaglia D, Kirst M (2008) Eucalyptus applied genomics: from gene sequences to breeding tools. New Phytol 179:911–929
Grattapaglia D, Resende MDV (2011) Genomic selection in forest tree breeding. Tree Genet Genomes 7:241–255. https://doi.org/10.1007/s11295-010-0328-4
Grattapaglia D, Silva-Junior OB, Resende RT et al (2018) Quantitative genetics and genomics converge to accelerate forest tree breeding. Front Plant Sci 871:1–10. https://doi.org/10.3389/fpls.2018.01693
Grattapaglia D (2014) Breeding forest trees by genomic selection: current progress and theway forward. In: Genomics of plant genetic resources: volume 1. Managing, sequencing and mining genetic resources. Springer, Netherlands, pp 651–682
Habier D, Fernando RL, Kizilkaya K, Garrick DJ (2011) Extension of the Bayesian alphabet for genomic selection. BMC Bioinform 12:186. https://doi.org/10.1186/1471-2105-12-186
Hall KB, Stape J, Bullock BP et al (2019) A growth and yield model for Eucalyptus benthamii in the southeastern United States. For Sci. https://doi.org/10.1093/forsci/fxz061
Hall N, Brooker MIH (1973) Camden white gum, Eucalyptus benthamii Maiden et Cambage. Australian Government Pub. Service
Han L, Love K, Peace B et al (2020) Origin of planted Eucalyptus benthamii trees in Camden NSW: checking the effectiveness of circa situm conservation measures using molecular markers. Biodivers Conserv 29:1301–1322. https://doi.org/10.1007/s10531-020-01936-4
Hill WG, Weir BS (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33:54–78. https://doi.org/10.1016/0040-5809(88)90004-4
Ibá (2021) 2021—Ibá Annual Report, São Paulo
Isik F, Bartholomé J, Farjat A et al (2016) Genomic selection in maritime pine. Plant Sci 242:108–119. https://doi.org/10.1016/j.plantsci.2015.08.006
Isik F, Holland J, Maltecca C et al (2017) Genomic relationships and GBLUP. In: Genetic data analysis for plant and animal breeding. Springer, pp 311–354
Kainer D, Stone EA, Padovan A et al (2018) Accuracy of genomic prediction for foliar terpene traits in Eucalyptus polybractea. G3 Genes Genomes Genet 8:2573–2583. https://doi.org/10.1534/g3.118.200443
Kärkkäinen HP, Sillanpää MJ (2012) Back to basics for Bayesian model building in genomic selection. Genetics 191:969–987. https://doi.org/10.1534/genetics.112.139014
Kjaer E, Amaral W, Yanchuk A, Graudal L (2004) Strategies for conservation of forest genetic resources. Conservation of Eucalyptus benthamii: an endangered eucalypt species from eastern Australia. In: Forest genetic resources conservation and management: overview, concepts and some systematic approaches, 1st edn. Interntional Plant Genetic Resources Institute, Rome, pp 5–24
Klápště J, Suontama M, Telfer E et al (2017) Exploration of genetic architecture through sib-ship reconstruction in advanced breeding population of Eucalyptus nitens. PLoS ONE 12:e0185137. https://doi.org/10.1371/journal.pone.0185137
Lee SH, Clark S, Van Der Werf JHJ (2017) Estimation of genomic prediction accuracy from reference populations with varying degrees of relationship. PLoS ONE. https://doi.org/10.1371/journal.pone.0189775
Lenz PRN, Beaulieu J, Mansfield SD et al (2017) Factors affecting the accuracy of genomic selection for growth and wood quality traits in an advanced-breeding population of black spruce (Picea mariana). BMC Genom 18:335. https://doi.org/10.1186/s12864-017-3715-5
Lenz PRN, Nadeau S, Mottet M et al (2020) Multi-trait genomic selection for weevil resistance, growth, and wood quality in Norway spruce. Evol Appl 13:76–94. https://doi.org/10.1111/eva.12823
Lima BM, Cappa EP, Silva-Junior OB et al (2019) Quantitative genetic parameters for growth and wood properties in Eucalyptus “urograndis” hybrid using near-infrared phenotyping and genome-wide SNP-based relationships. PLoS ONE 14:1–24. https://doi.org/10.1371/journal.pone.0218747
Lin M, Arnold RJ, Li B, Yang M (2003) Selection of cold-tolerant eucalypts for Hunan Province. In: Turnbull JW (ed) Proceedings of Eucalypts in Asia—a symposium held in Zhanjiang, People’s Republic of China, 7–11 April 2003. ACIAR Proceedings. Australian Centre for International Agricultural Research, Zhanjiang, Guangdong, People’s Republic of China. Canberra, pp 107–116
Long N, Gianola D, Rosa GJM, Weigel KA (2011) Long-term impacts of genome-enabled selection. J Appl Genet 52:467–480. https://doi.org/10.1007/s13353-011-0053-1
Marroni F, Pinosio S, Zaina G et al (2011) Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genet Genomes 7:1011–1023. https://doi.org/10.1007/s11295-011-0391-5
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Mora AL, Garcia CH (2000) Eucalypt cultivation in Brazil. Sociedade Brasileira de Silvicultura, São Paulo
Mphahlele MM, Isik F, Mostert-O’Neill MM et al (2020) Expected benefits of genomic selection for growth and wood quality traits in Eucalyptus grandis. Tree Genet Genomes 16:49. https://doi.org/10.1007/s11295-020-01443-1
Müller BSF, Neves LG, de Almeida Filho JE et al (2017) Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genom 18:1–17. https://doi.org/10.1186/s12864-017-3920-2
Muñoz PR, Resende MFR, Gezan SA et al (2014) Unraveling additive from nonadditive effects using genomic relationship matrices. Genetics 198:1759–1768. https://doi.org/10.1534/genetics.114.171322
Myburg AA, Potts BM, Marques CM et al (2007) Eucalypts. In: Kole C (ed) Forest trees, 1st edn. Springer, Berlin, pp 115–160
Namkoong G, Kang HC, Brouard JS (1988) Tree breeding: principles and strategies. Springer, New York
Ødegård J, Meuwissen THE (2012) Estimation of heritability from limited family data using genome-wide identity-by-descent sharing. Genet Sel Evol 44:16. https://doi.org/10.1186/1297-9686-44-16
Paludeto JGZ, Grattapaglia D, Estopa RA, Tambarussi EV (2021) Genomic relationship-based genetic parameters and prospects of genomic selection for growth and wood quality traits in Eucalyptus benthamii. Tree Genet Genomes 17:38. https://doi.org/10.1007/s11295-021-01516-9
Park T, Casella G (2008) The Bayesian Lasso. J Am Stat Assoc 103:681–686. https://doi.org/10.1198/016214508000000337
Pérez P, De Los Campos G (2014) Genome-wide regression and prediction with the BGLR statistical package. Genetics 198:483–495. https://doi.org/10.1534/genetics.114.164442
Pirraglia A, Gonzalez R, Saloni D et al (2012) Fuel properties and suitability of Eucalyptus benthamii and Eucalyptus macarthurii for torrefied wood and pellets. BioResources 7:217–235
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Ratcliffe B, Gamal El-Dien O, Klápště J et al (2015) A comparison of genomic selection models across time in interior spruce (Picea engelmannii × glauca) using unordered SNP imputation methods. Heredity (Edinb) 115:547–555. https://doi.org/10.1038/hdy.2015.57
Ratcliffe B, El-Dien OG, Cappa EP et al (2017) Single-step BLUP with varying genotyping effort in open-pollinated Picea glauca. G3 Genes Genomes Genet 7:935–942. https://doi.org/10.1534/g3.116.037895
Resende MFR Jr, Muñoz P, Resende MDV et al (2012a) Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190:1503–1510. https://doi.org/10.1534/genetics.111.137026
Resende MDV, Resende MFR, Sansaloni CP et al (2012b) Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol 194:116–128. https://doi.org/10.1111/j.1469-8137.2011.04038.x
Resende RT, Resende MDV, Silva FF et al (2017) Assessing the expected response to genomic selection of individuals and families in Eucalyptus breeding with an additive-dominant model. Heredity (Edinb) 119:245–255. https://doi.org/10.1038/hdy.2017.37
Silva-Junior OB, Grattapaglia D (2015) Genome-wide patterns of recombination, linkage disequilibrium and nucleotide diversity from pooled resequencing and single nucleotide polymorphism genotyping unlock the evolutionary history of Eucalyptus grandis. New Phytol 208:830–845. https://doi.org/10.1111/nph.13505
Silva-Junior OB, Faria DA, Grattapaglia D (2015) A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol 206:1527–1540. https://doi.org/10.1111/nph.13322
Skinner A (2003) The effects of tree isolation on the genetic diversity and seed production of Camden White Gum (Eucalyptus benthamii Maiden et Cambage). In: Centre for Plant Biodiversity Research. http://www.cpbr.gov.au/cpbr/summer-scholarship/2002-projects/skinner-alison-report.html. Accessed 12 May 2020
Suontama M, Klápště J, Telfer E et al (2019) Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity (Edinb) 122:370–379. https://doi.org/10.1038/s41437-018-0119-5
Tambarussi EV, Pereira FB, da Silva PHM et al (2018) Are tree breeders properly predicting genetic gain? A case study involving Corymbia species. Euphytica 214:1–11. https://doi.org/10.1007/s10681-018-2229-9
Tan B, Grattapaglia D, Martins GS et al (2017) Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol. https://doi.org/10.1186/s12870-017-1059-6
Tan B, Grattapaglia D, Wu HX, Ingvarsson PK (2018) Genomic relationships reveal significant dominance effects for growth in hybrid Eucalyptus. Plant Sci 267:84–93. https://doi.org/10.1016/j.plantsci.2017.11.011
TAPPI TA of the P and P (2000) Tappi T280 pm-99 standard—acetone extractives of wood and pulp. TAPPI Press
Thavamanikumar S, McManus LJ, Tibbits JFG, Bossinger G (2011) The significance of single nucleotide polymorphisms (SNPs) in Eucalyptus globulus breeding programs. Aust For 74:23–29. https://doi.org/10.1080/00049158.2011.10676342
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423. https://doi.org/10.3168/jds.2007-0980
Wallis AFA, Wearne RH, Wright PJ (1996) Analytical characteristics of plantation eucalypt woods relating to kraft pulp yields. Appita J 49:427–432
Wang X, Miao J, Chang T et al (2019) Evaluation of GBLUP, BayesB and elastic net for genomic prediction in Chinese simmental beef cattle. PLoS ONE. https://doi.org/10.1371/journal.pone.0210442
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756. https://doi.org/10.1111/j.1755-0998.2007.02061.x
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262. https://doi.org/10.1111/j.1752-4571.2009.00104.x
White TL, Adams WT, Neale DB (2007) Forest genetics. CABI, Wallingford
Xavier A (2019) Efficient estimation of marker effects in plant breeding. G3 Genes Genomes Genet 9:3855–3866. https://doi.org/10.1534/g3.119.400728
Zhang Q, Calus MP, Guldbrandtsen B et al (2015) Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. https://doi.org/10.1186/s12863-015-0227-7
Acknowledgements
João Gabriel Zanon Paludeto received scholarships from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). Evandro V. Tambarussi and Dario Grattapaglia were supported by research productivity fellowships granted by CNPq (Brazilian National Council for Scientific and Technological Development).
Funding
This work was partially supported by PRONEX-FAP-DF Grant 2009/00106-8 ‘NEXTREE’, CNPq (Brazilian Council for Scientific and Technological Development) Grant 400663/2012-0 and EMBRAPA Grant 03.11.01.007.00.00 to DG that allowed the development and validation of the multi-species EuCHIP60K used in this work.
Author information
Authors and Affiliations
Contributions
RAE Methodology, Formal analysis, Investigation, Resources, Data curation, Funding acquisition, Writing—Original Draft. JGZP Software, Formal analysis, Investigation, Data curation, Writing—Original Draft. BMSF Methodology, Formal analysis, Software, Investigation, Data curation, Writing—Original Draft. RAO Conceptualization, Methodology. CFA Formal analysis, Data curation, Writing—Review and Editing. MDVR Conceptualization, Methodology, EVT Visualization, Writing—Review and Editing. DG Conceptualization, Methodology, Investigation, Resources, Writing—Final Review and Editing, Supervision, Project administration.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Consent to participate
Not applicable
Consent for publication
Not applicable
Ethical approval
Not applicable
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Estopa, R.A., Paludeto, J.G.Z., Müller, B.S.F. et al. Genomic prediction of growth and wood quality traits in Eucalyptus benthamii using different genomic models and variable SNP genotyping density. New Forests 54, 343–362 (2023). https://doi.org/10.1007/s11056-022-09924-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11056-022-09924-y