
Abstract
In this study, we characterized DArTseq-derived SNPs for species of Psidium, an important Neotropical genus within the family Myrtaceae. Nine Psidium species were used for the identification and characterization of an interspecific set of SNPs. The samples chosen represented a wide range of variation regarding agronomic traits and geographical origin. The samples were genotyped using the DarTseq methodology. This approach, associated with annotation analysis, generated a set of conserved, polymorphic, and possibly functional SNPs. Most of the SNPs in coding regions of Psidium were annotated for the gene ontology classes. We observed high interspecific diversity among species and considerable intraspecific genetic diversity in native species (P. cattleyanum, P. myrtoides, P. acidum, and P. oblongatum). Psidium guajava, a cultivated species, shows low diversity and shares genetic similarities with the tetraploid P. guineense. The group of markers selected in this study will contribute to the development of a panel of interspecific SNPs for Psidium. Our investigation will support further studies regarding genetic variability, diversity, phylogeny, cytogenetics, and other relationships among Psidium species, constituting essential information for the creation, conservation, management, and use of genetic resources in breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Psidium is one of the most important and explored genera within the family Myrtaceae, including about one hundred species distributed from Mexico and the Caribbean to Uruguay and northern Argentina (Thornhill et al., 2015). The genus belongs to the tribe Myrteae (subtribe Pimentinae), which represents almost all Myrtaceae diversity in Neotropical environments (Wilson et al., 2005; Lucas et al., 2007). Approximately 60 taxa occur in Brazil, and the Atlantic Forest is particularly rich in Psidium species, with a total of 38, of which 18 are endemic to this biome (Flora do Brasil, 2020). Guava (Psidium guajava L.) is prominent among the species of this genus owing to its commercial and medicinal value. However, Psidium comprises other promising species (e.g., the araças), with some significant attributes such as early maturation, resistance to certain pests and diseases, and exotic fruit flavor. These attributes may be used in guava breeding programs (Dias et al., 2015; Mendes et al., 2017; Vitti et al., 2020). In addition, natural interspecific hybrids were found to occur between phylogenetically close species, P. guajava and P. guineense (Landrum et al., 1995). Moreover, an artificial interspecific hybrid of these species has recently become available for use as nematode-resistant rootstock in the guava crop (Costa et al., 2016).
Guava and some other araça species (P. guineense, P. cattleyanum, and P. myrtoides) have been characterized for agronomic traits in Brazil (Nimisha et al., 2013). Chemical analyses have also demonstrated the value of substances from these plants in co-treatments of different illnesses, such as diabetes mellitus, cardiovascular diseases, cancer, and parasitic infections (Cerio et al., 2017). Psidium species also contain functional compounds with antioxidant activities (Mallmann et al., 2020), and they are sources of essential oils rich in terpenes, which are of agronomic, industrial, and medicinal interest (Mendes et al., 2017).
The genus Psidium is karyologically variable, with a suggested basic number of chromosomes of x = 11 (Tuler et al., 2019a). Psidium guajava, P. oblongatum, and P. cauliflorum have a diploid nature (2n = 2x = 22) (Souza et al., 2015; Marques et al., 2016; Tuler et al., 2019a), whereas P. guineense and P. friedrichsthalianum are described as tetraploids (2n = 4x = 44), and P. myrtoides and P. longipetiolatum as hexaploids (2n = 6x = 66), confirming a trend towards polyploidy in the genus. In addition, intraspecific polyploidy (i.e., within the same species) also occurs, as found in P. cattleyanum, with chromosome numbers varying between 44 and 88 (Machado et al., 2020; Tuler et al., 2019a; Souza et al., 2015).
Genetic diversity and phylogenetic relationships of Psidium species have been elucidated through the use of different molecular techniques, such as random amplified polymorphic DNA (RAPD), transferability of simple sequence repeats (SSR) and inter-simple sequence repeats (ISSR), and phylogenetic markers (Abdelmigid and Morsi, 2018; Bernardes et al., 2018; Tuler et al., 2019b; Han et al., 2020; Machado et al., 2020). However, the use of genomic approaches, including SNP markers, is still incipient for these species. Advances in the genetic and genomic tools for distinct Psidium species may provide more reliable taxonomic information and support for the development of cultivars with improved agronomic traits (Costa and Santos, 2017), as well as for studies on diversity, conservation, phylogenetics, and evolution of the species.
SNP markers have been increasingly used owing to their high performance, precision, resolution, and reproducibility (Elshire et al., 2011; Kilian et al., 2012; Sansaloni et al., 2020). They are abundant, widely distributed, and in high density in the genome, favoring the detection of multiple haplotypes by the combination of tightly linked SNP alleles. In addition, these markers have been widely used because of their automated genotyping in next-generation sequencing (NGS) platforms (Deschamps et al., 2012; Ren et al., 2015; Al-Beyroutiová et al., 2016; Garavito et al., 2016; Nimmakayala et al., 2016; Egea et al., 2017; Ndjiondjop et al., 2017; Valdisser et al., 2017; Sukumaran et al., 2018).
DArTseq, a high-throughput whole-genome genotyping technology, enables the sequencing and genotyping of hundreds or thousands of SNPs by using methods that reduce the genome complexity that enriches genic regions (Kilian et al., 2012; Fellers, 2008). The so-called STAGs, fragments sequenced in the population that contain the SNPs, also allow the alignment of these sequences with the genome of the target (or close) species, and the annotation (functional and structural) of the marker regions in the genome. This in silico analysis has been used to identify conserved genomic regions among species, to enable the construction of genotyping panels for multiple species, and to detect and characterize genomic regions with signals of selection (Shams et al., 2019; Merot-L’Anthoene et al., 2019; Valdisser et al., 2017).
Eucalyptus grandis, an important representative of Australian Myrtaceae species, is a reference in the family for omics approaches (Grattapaglia et al., 2012; Myburg et al., 2014). Thus, the identification of STAGs in a suite of evolutionarily conserved genes of E. grandis, shared with non-traditional model species of Neotropical Psidium lineages, is important for genome evolutionary studies in Myrtaceae. In addition, a set of SNPs for multiple species of Psidium is also important for the polyploid species of the genus, as the complexity of the genome and the difficulties in developing high-performance genotyping platforms create challenges in the efforts of carrying out breeding and conservation programs, as well as evolutionary studies, for these species.
The identification of SNPs for multiple Psidium species can promote advances in the characterization of the structure and function of common genomic regions for these species. In addition, a set of SNPs with high discriminatory potential between species, as well as between genotypes, is useful for routine characterization of accessions in germplasm banks and in breeding programs. To support the construction of a multispecies panel of SNPs, the chosen markers must be conserved among species, preferably selected within the gene regions, and should be polymorphic and useful in the study of intra- and interspecific variation. Therefore, the aim of the present study was to identify and characterize a set of SNP markers that may be useful for breeding, phylogenetics, and further approaches regarding the genome of Psidium species.
Materials and methods
Plant material
A total of 94 samples from nine species of Psidium were evaluated in the present study. P. guajava (Voucher: Tuler.A.C 563) samples were the most numerous, totaling 63. Of these, 19 were cloned cultivated genotypes, including 11 cultivars registered in the Brazilian Ministry of Agriculture (Ministério da Agricultura, Pecuária e Abastecimento - MAPA) (http://sistemas.agricultura.gov.br/) (Paluma, Pedro Sato, Século XXI, Cortibel LG, Cortibel LM, Cortibel Branca LG, Cortibel RM, Cortibel Branca RM, Cortibel RM2, Cortibel RG and Cortibel SLG) and 8 cultivated materials (Chinesa Branca, Indiana, Kuse, Maçã, Roxa, Tailandesa, Tailandesa Branca, and Petri). Thirty-two (32) were Cortibel® (C) genotypes obtained by selection from the orchard of seminal origin in the state of Espírito Santo (Frucafé) (VII, XI, XIII, XVIII, XIX, XX, XXII, XXIII, XXIV, XXV, XXVI, XXVII, XXVIII, XXIX, XXX, XXXI, XXXII, XXXIII, XXXIV, XXXV, XXXVI, XXXVII, XXXVIII, XXXIX, XL, XLI, XLII, XLIII, XLV, XLVI, XLVII, and XLVIII). All these P. guajava cultivars and selected genotypes were collected from adult plants in the Frucafé Nursery, certified by the Brazilian Ministry of Agriculture. In addition, 12 samples of naturally occurring guava trees were also used from different states of Brazil: Alagoas (1), Bahia (2), Ceará (1), Espírito Santo (2), Pernambuco (1), Paraíba (1), Sergipe (1), Paraná (2), and São Paulo (1). We also included an individual with morphological traits of a natural hybrid (P. guajava × P. guineense) sampled in the state of Sergipe.
Eight (8) more Psidium species with different numbers of samples were added (number of samples/voucher): P. gaudichaudianum (01/Tuler.A.C 637), P. acidum (01/Tuler.A.C 524), P. cattleyanum (10/Tuler.A.C 553), P. friedrichsthalianum (01/UFES collection), P. guineense (05/Tuler.A.C 548), P. myrtoides (07/Tuler.A.C 510), P. oblongatum (04/Tuler.A.C 567), and one unidentified sample, P. sp1. The samples were collected from adult plants in the municipality of Alegre, ES, most of which are found in the germplasm collection of the Universidade Federal do Espírito Santo (UFES), which maintains naturally occurring plants collected from different states of Brazil. The samples chosen represent a wide range of variation based on agronomic traits and different geographical origins (Table 1).
Genotyping by sequencing on the DArTseq platform
The DNA of each sample was extracted from leaves macerated with liquid nitrogen according to the CTAB method (Doyle and Doyle, 1990). After DNA extraction, all quality parameters were checked according to recommendations of the Diversity Arrays Technology Pty. Ltd. company (Canberra, Australia) (https://www.diversityarrays.com/ faq /). The quantity and quality of the DNA samples were verified using NanoDrop and visualized in 0.8% agarose gel. Only intact DNA samples were used. A volume of 60 μL of each sample, containing 60 ng/μL of DNA, was pipetted into PCR plates and sent to the Diversity Arrays Technology Pty. Ltd. company, according to recommendations (https://www.diversityarrays.com / faq /), for production of the library, genotyping by sequencing, and marker identification (Kilian et al., 2012). The genome representation was obtained from a genomic library attained from digestion with the restriction enzymes PstI and MseId. The ends of the cleaved fragments were connected to a barcode adapter and to a common adapter. The mixed fragments were amplified with two primers with sequences complementary to the attached adapters and to the oligonucleotides of the Illumina sequencing platform. All the successful amplifications were clustered and applied to a flow cell for amplification (Kilian et al., 2012). The clusters were sequenced on the Illumina HiSeq2500 sequencing platform, and the sequences were processed using proprietary DArT analytical pipelines (Sansaloni et al., 2020). The sequences per barcode/sample were identified and used in marker calling. Poor-quality sequences were filtered out, and identical ones were collapsed into fastqcall files. These files were used in a pipeline for DArT PL’s proprietary SNP-calling algorithms (DArTsoft-seq14), as described by Sansaloni et al. (2020). The DArTseq quality markers were determined by the reproducibility parameter (the proportion of technical replicate assay pairs for which the marker score is consistent) of at least 0.95 and call rate parameter (the proportion of samples with genotypic score, i.e., not recorded as missing data) with a threshold of 0.7, which constitute the main parameters for marker selection. In addition, the statistics of the filtered DArTseq markers included the expected heterozygosity (He), the minor allele frequency (MAF), and the polymorphic information content (PIC).
Data analysis
SNP markers from common and specific species were identified for all of the species. For P. guajava, P. cattleyanum, P. guineense, P. myrtoides, and P. oblongatum, the DArTseq quality markers were determined by call rate = 1 and reproducibility > 0.95 (Wenzl et al., 2004), number of polymorphic SNP markers, monomorphic SNPs, and total SNPs per species. The non-amplified markers (from the total amplified in all species) were also calculated.
DArTseq-derived sequences of 40–69 base pairs (bp) containing SNPs (STAGs) (Shams et al., 2019) obtained from 94 individuals of Psidium were aligned against the reference genome of Eucalyptus grandis, a pivotal species for genomic studies in Myrtaceae (Grattapaglia et al., 2012; Myburg et al., 2014). This analysis was performed and provided by the DArT Pty. Ltd. company.
Identification and annotation of STAGs present in coding regions
We performed an in silico functional annotation of each of the STAGs, which were classified regarding their presence in coding and non-coding regions. E. grandis annotation and genomic sequence data were used to obtain the functional annotation. To identify the most conserved regions among Myrtaceae species, the unique STAGs were classified as being present in the E. grandis genome (anchored) or not found in the genome (non-anchored). The localization of each anchored STAG was obtained by a search with the Basic Local Alignment Search Tool (BLAST) on E. grandis gene sequences, available at the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html). For the STAGs non-anchored in Eucalyptus, a BLAST was performed against P. guajava gene sequences provided by the research group (http://guava.ufes.br/). The BLAST was carried out using the BLASTn software (NCBI n.d.), with default parameters and output format changed to csv (comma-separated values) and e-value 1e-7.
The functional annotations of the anchored STAGs present in coding regions were obtained from the annotation of E. grandis, available in the Phytozome Database. For the non-anchored STAGs, it was necessary to obtain the amino acid sequence translated by each available gene in the data from the structural annotation of P. guajava (from the research group database), then perform BLASTp in the NR database and obtain a predicted gene association in P. guajava X gene identifier (gi). The functional annotation of each gi was obtained with the Uniprot database support (www.uniprot.org/uploadlists/).
Files from the processing were analyzed to generate abundance graphs for GOs (Gene Ontology) in each sequence group. With this data, the STAGs were classified as being present in coding regions, when derived from polymorphic sites inside coding regions, or as present in non-coding regions, when found in polymorphic and random sites of the genome (Andersen and Lubberstedt, 2003).
Marker selection
A descriptive analysis was performed for the SNPs of each species using the DArTR package in the R software (R Core Team, 2020). To select SNP markers for multiple species of Psidium, we filtered the SNPs with call rate = 1.00 and MAF > 0.01 and PIC parameters. The excess of heterozygosity was verified for each SNP to identify possible highly similar sequences that occupy different chromosomal locations (multicopy loci), as described by Willis et al. (2017).
The analysis within and between species was performed using Euclidean distance for principal coordinate analysis (PCoA) with the R software (R Core Team, 2020). In addition, this set of markers was applied for analysis of UPGMA clustering of the genotypes used (Brazilian cultivars, selected genotypes, and guava trees of natural occurrence) for application in Brazilian guava breeding.
Phylogenetic reconstruction
We analyzed the SNP data with the SNPhylo software (Lee et al., 2014). A vcf file was used as data input and analyzed with the script snphylo.sh -v VCF_file A -b -B 1500, where -A performs multiple alignments by MUSCLE, -b performs the bootstrap analysis and generates a tree, and -B is the number of bootstrap samples. After that, the Newick file (tree format file) was generated using the iTol software (Letunic and Bork, 2019). Finally, the modified file was edited using the Affinity Designer® program.
Results
A total of 124,069 loci were obtained with SNPs, representing 64,672 unique sequences. Of these, 22,050 STAGs were anchored on the eleven chromosomes and scaffolds of the reference genome E. grandis (Myburg et al., 2014). Considering only the anchored SNPs, the averages of PIC and the frequency of homozygote and heterozygote loci per chromosome were similar, whereas the average of heterozygosity of the SNPs in the chromosomes was low (0.026) (Supplementary File S1). The SNPs anchored to chromosomes 10 and 11 had an average PIC above 0.2, revealing variability. SNPs with high PIC values (0.5) were detected in chromosomes 06, 08, 10, and 11, demonstrating the possibility of conserving selection, but selection of polymorphic loci markers. The SNPs exclusive to Psidium species (102,019) had an average PIC of 0.2 (Supplementary File S1).
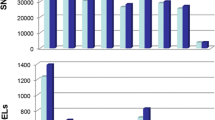
Psidium guajava and P. guineense had similar numbers of SNPs and STAGs anchored to the E. grandis genome, whereas P. oblongatum and P. myrtoides had lower numbers of markers anchored in this manner (Fig. 1A).

(A) Distribution of 124,069 SNP markers identified in nine species into the classes: Coding and non-coding regions; anchored in E. grandis genome and coding anchors; non-anchored and non-anchored in coding regions. Distribution of SNP markers: (B) total, (C) anchored in Eucalyptus genome, and (D) non-anchored. In each class, the SNPs were subdivided by species and recorded as monomorphic, polymorphic, polymorphic in coding regions, polymorphic in non-coding regions, and absence of the marker (NA)
Sixty percent of the total SNPs (75,074 STAGs) were recorded in coding regions, indicating the effectiveness of the DArTseq methodology in selecting the fraction of the genome that corresponds to coding regions. For the SNPs anchored to the E. grandis genome, 82% were recorded in coding regions, while for non-anchored markers this percentage was 55% (Fig. 1A).
A similar pattern was found for the parameters evaluated in P. acidum and P. friedrichsthalianum. Psidium oblongatum, exceptionally, had a similar number of loci in coding and non-coding regions, while the other species had a higher number of loci in coding regions (Fig. 1A). Loci monomorphic within species and polymorphic among species were verified. A large number of monomorphic loci per species indicate genomic regions shared between species, but do not indicate intraspecific polymorphism (Fig. 1B, C, D). The number of monomorphic loci was higher in P. guajava, P. guineense, and P. cattleyanum. These markers can be investigated for interspecific differences.
Psidium guajava, P. guineense, and P. cattleyanum had a larger number of polymorphic loci than P. myrtoides and P. oblongatum did. These species also had the smallest number of loci in shared coding regions and the highest marker absence. The large number of loci not shared with the other species showed the divergence of P. oblongatum and P. myrtoides. The number of non-shared regions for P. cattleyanum was greater than the number observed in P. guajava and P. guineense (Fig. 1B, C, and D). For indication of a multispecies set of SNP markers, those conserved in the species (with intra- and interspecific polymorphisms) were prioritized.
We verified the potential gene ontologies (GO) related to STAGs present in coding regions of anchored (18,211 STAGs) and non-anchored (56,836 STAGs) through functional annotation. A coding region can be present in one or more of three main ontological structure categories: biological process, cellular component, and molecular function. From the 18,211 STAGs anchored in coding regions of the Eucalyptus genome, 6325 were aligned in coding regions with functional annotation, represented by 959 GOs. From the remaining non-anchored STAGs in coding regions (56,836), 11,539 annotations were found, which identified 1384 different GOs.
The groups of genes with STAGs of Psidium species are represented in Supplementary File S2. The 20 most abundant gene classes within the biological processes ontology (Supplementary File S2-A) showed 4810 GOs associated with STAGs anchored to the Eucalyptus genome, and 2499 were specific to the Psidium species (non-anchored). For the cellular component graph (Supplementary File S2-B), specific regions of STAGs for Psidium (non-anchored) had a larger number of GO terms (6343), demonstrating the diversification of these regions in the Psidium genomes. Both anchored and non-anchored STAGs showed the integral components of the membrane class as the most abundant. The STAGs in regions of genes with molecular function are listed in Supplementary File S2-C. In this group, the class of genes related to ATP binding was the most abundant, with 1518 GOs in anchored and 2276 in non-anchored regions. Molecular function was the ontological class most represented by the annotation of STAGs in this study, exhibiting 48% of GOs, followed by cellular components (28.5%) and biological processes (23.5%). When differentiated into anchored STAGs and non-anchored STAGs, only the cellular components showed higher values for the non-anchored STAGs (72%).
The genetic relationship within a species and between species was evidenced by the results of principal coordinate analysis (PCoA), performed with three data groups: all SNPs, anchored SNPs, and non-anchored SNPs (Fig. 2). The results provided a spatial representation of the relative genetic distances between individuals, with little difference in the proportion of the variation explained in the analysis with all of the SNPs (59.6%) and with non-anchored ones (58.1%). The greater representativeness of the data was observed in the analysis with the anchored SNPs (explained variation = 66.1%), justifying the selection of these markers as a priority for interspecific evaluations.

Principal coordinate analysis (PCoA) based on SNP markers for 94 Psidium samples. Each species is represented by a different color for all markers, anchored markers, and non-anchored markers
The PCoA distribution resulted in three distinct groups. Psidium guajava and P. guineense were grouped on the first axis, showing less intraspecific variation compared to the other wild and polyploid species. Psidium myrtoides and P. oblongatum were close on one of the PCoA axes. Less intraspecific variation was observed in individuals of P. oblongatum than in individuals of P. myrtoides.
Considerable variability was found for P. acidum, P. cattleyanum, P. friedrichsthalianum, P. gaudichaudianum, and Psidium sp1, which consist of tree species with large fruit, many of these tree species grown due to their socioeconomic importance. The ten samples of P. cattleyanum represented a subdivision. Six samples were close to P. acutangulum, P. friedrichsthalianum, and P. gaudichaudianum, which share many SNPs. The other four samples of P. cattleyanum were clustered with a sample harvested in Espírito Santo, identified as Psidium sp.1 (not yet identified).
The 5951 SNPs whose call rate was 1 were considered the most conserved of the species. Of these only 14 showed heterozygotes above the expected 50% for bi-allelic SNPs (0.5–0.69) (Willis et al., 2017). For the 5951 SNPs, a new analysis was performed for the anchored and non-anchored SNPs (Supplementary File S1; see Supplemental Data with this article). Similar values between groups of SNPs were observed, and the average PIC increased from 0.20 (Supplementary File S1-A) to 0.24 (Supplementary File S1-B), suggesting loci diversity. Of these SNPs, 78% showed functional annotation and 37% were anchored to the E. grandis genome (Fig. 3A, B, and C). The percentage of anchored SNPs in coding regions was 93% (Fig. 3D). Of the 5951 sequences, 4846 are present in the assembly of the P. guajava genome. A total of 6459 alignments were obtained; of 4258 STAGs aligned against 5719 genes, 2734 exhibit GO annotation.

List of the 5951 SNP markers selected with their gene ontology (GO) for genes related to (A) biological processes (blp), (B) cellular components (cc), and (C) molecular function (mf). (D) Distribution of selected markers in coding and non-coding regions, considering all markers, markers anchored in E. grandis genome, and markers exclusive to Psidium species (non-anchored). Principal coordinate analysis (PCoA) based on the SNP markers selected (E) for 94 samples of Psidium and (F) for P. guajava samples only.
The biological process and cellular component ontological classes were represented with 39.28% and 39.32% of the GOs found, respectively (Fig. 3A and B). Among the biological processes with most abundant GO terms, responses to stress, responses to external stimuli, and reproduction were most prominent. In turn, the class of molecular function, widely represented previously, corresponded to 21.4% of the results in this analysis (Fig. 3C).
The PCoA analysis performed with the SNPs from the STAGs of all samples reveals the potential of this tool for genomic approaches of interspecific (Fig. 3E) and also intraspecific discrimination, as shown by the guava genotypes used (Fig. 3F). These SNPs increased the power of discrimination between species (only one component explained 84%). As in the first cluster, here we also observed the formation of three distinct groups: (1) P. guajava and P. guineense, very close species; (2) P. myrtoides and P. oblongatum; and (3) P. acidum, P. cattleyanum, P. friedrichsthalianum, P. gaudichaudianum, and Psidium sp1. These SNPs also showed ability to discriminate genotypes within species (with genotypes distributed on four axes) (Fig. 3F). The low variability of P. guajava in comparison with the other native species was also evident, indicating high diversity, as well as inter- and intraspecific variability (Fig. 3E).
A phylogeny was also provided (Fig. 4). The reconstructed tree was formed by five well-supported clades (ML bootstrap = 100), showing that the markers used had sufficient resolution to separate five species of Psidium sp.: P. guajava, P. guineense, P. oblongatum, P. myrtoides, and P. cattleyanum. Three genotypes, previously classified as P. gaudichaudianum (x92), P. acidum (x93), and P. friedrichsthalianum (x94), were allocated to the larger clade of P. cattleyanum. The distance of P. guajava genotypes to those of other species is shown by the size of the central branch, which demonstrates an evolutionary process distinct from the others.

Genetic relationships among Psidium species. Phylogram based on the maximum likelihood inference of 5951 SNPs of Psidium sp. The bootstrap probability is indicated near the branch nodes, and only values greater than 75 are shown. An asterisk denotes genotypes previously classified as P. gaudichaudianum (x92), P. acidum (x93), and P. friedrichsthalianum (x94)
In addition, the application of this set of markers for breeding of P. guajava was demonstrated by analysis of the UPGMA clustering of the genotypes used, and seven clusters were shown (Fig. 5).

Clustering analysis of Brazilian germplasm by the UPGMA method, including cultivars, selections, and individuals of natural occurrence
The STAGs shared with E. grandis represent a genomic tool for the family. The distribution of these markers in chromosomes revealed enrichment of Psidium-conserved coding regions in chromosomes 2, 6, and 11 of E. grandis. The STAGs exclusive to the Psidium species represent specific evolution in the genus. All information about these SNP markers is available (Supplementary File S3).
Discussion
The DArTseq technology provided a large number of SNPs, which can be anchored to reference genomes (Sansaloni et al., 2011). However, in most studies, the STAGs that contain the SNPs remain unused. These sequences provide important information for identification of regions evolutionarily conserved across related species (Shams et al., 2019). Here, a set of STAGs, conserved for Psidium species and associated with protein-coding regions and random sequences, revealed an important genomic tool for Psidium species, which can help increase knowledge regarding the genetics and evolution of these species, as well as assist in their breeding.
The SNPs provided robust information about the genetic relationships among species, corroborating morphological, cytogenetic, and genetic patterns previously described for these species (Tuler et al., 2015, 2019a, 2019b; Marques et al., 2016). In addition, they exposed the wide divergence among individuals of native species, particularly P. cattleyanum, for which intraspecific polyploidization has been reported (Machado et al., 2020). Moreover, the set of SNPs allowed intraspecific discrimination of the species evaluated, which supplements their morphological evaluation and allows the development of tools related to selection and genetic breeding, which is especially relevant for breeding of guava.
A higher number of loci shared by Psidium species and E. grandis, which represent genera belonging to distant tribes of Myrtaceae (Vasconcelos et al., 2017), increases the applications of this study due to the possible transferability of these markers to other genera of the family. Previous studies showed that markers conserved among species of these genera could also be used in many other genera of the family (Bernardes et al., 2018). In addition, the polymorphic STAGs shared by Psidium and Eucalyptus, the majority of which were present in coding regions, suggest ancient variants that arose before the division of these taxa and that persisted in separate strains (Silva-Junior et al., 2015). The fact that they are mostly located in coding regions suggests the functionality of these markers. The relatively small number of SNPs anchored to Eucalyptus in relation to the total indicates the phylogenetic distance between Psidium and Eucalyptus, also previously described by SSR and phylogenetic markers (Bernardes et al., 2018; Vasconcelos et al., 2017). Therefore, the set of markers presented here can be valuable for the analysis of evolution, phylogeny, synteny, and breeding programs for many species of Myrtaceae.
The data also showed a high percentage (60.5%) of markers in coding regions. This increases the possibility of developing functional markers, which are considered superior in several applications. Their complete link to functional motifs allows their use in populations with no available genetic maps, due to more efficient allele fixation and better representation of genetic variation in natural or selected populations (Andersen and Lubberstedt, 2003).
The proximity observed between P. guajava and P. guineense is corroborated by studies of morphological attributes (Landrum et al., 1995), microsatellite markers (Bernardes et al., 2018), phylogeny (Tuler et al., 2019b), and evidence of evolutionary proximity via cytogenetic analyses (Marques et al., 2016). However, the low intraspecific variability of P. guajava and P. guineense in relation to the other species studied is a new observation and has implications for breeding, since they are cultivated species. The low diversity observed in P. guajava, a diploid species, is suggested by its extensive breeding, which increases the homozygosity of the populations even though the samples were harvested in different locations. Hence, the results shown here can be expected for the cultivars and clonal selections from the same orchard. However, they reveal low diversity of P. guajava in relation to the other species when comparing the samples of naturally occurring trees.
However, the application of this set of markers for breeding of P. guajava was also demonstrated by analysis of the UPGMA clustering of the genotypes used, revealing the following: two divergent groups of cultivated genotypes in Brazil; the power of the set of markers in molecular discrimination and determination of genetic relationships among genotypes selected from the same population; and the importance of genetic resources for this species of natural occurrence, given the divergence shown by this type of individual in the present study in relation to selected and cultivated genotypes in Brazil. These results confirm, refine, and expand previous reports from our research group for a common set of genotypes and species studied using SSR markers (Coser et al., 2012).
The narrow diversity of P. guineense may be related to recent divergence of the species, suggesting an event of self-polyploidy in relation to P. guajava, also reported by Marques et al. (2016) and Tuler et al. (2019b). The wide diversity observed for the other species is probably due to their multiple ploidies (Marques et al., 2016; Tuler et al., 2019a). Furthermore, the geographical dispersion of the species may also contribute, as the genetic characteristics of the populations depend on the interaction between genetic drift, gene flow, and natural selection, processes that can be strongly affected by the spatial distribution of the populations (Eckert et al., 2008).
Psidium oblongatum and P. myrtoides were in the same group and were the species that most differed from the others. Moreover, they shared the fewest genomic regions with Eucalyptus and the other Psidium species, which shows their genetic distance. The genetic relationship between these two species was reported in a previous study of cluster analysis based on microsatellite amplification patterns performed by Tuler et al. (2015), in which they clustered with P. sartorianum and P. brownianum.
Psidium myrtoides is a polyploid species known in the Atlantic Forest, Cerrado, and Caatinga in the Southeast, West-Central, and Northeast of Brazil. This wide distribution exposes individuals from this species to different environmental conditions, which may explain the separation of two groups of individuals within this same species, but the two groups were not maintained in the PCoA analysis only with the conserved markers. It is noteworthy that P. oblongatum also showed wide genetic diversity, yet it consists of a diploid species endemic to the Atlantic Forest (Tuler et al., 2019a).
Psidium cattleyanum and P. friedrichsthalianum also formed a cluster. The proximity between these species has already been identified in other studies by different molecular markers (Costa and Santos, 2013). P. cattleyanum has an intraspecific ploidy variation, which explains the wide genetic variability detected in the samples analyzed (Machado et al., 2020; Tuler et al., 2019a; Souza et al., 2015).
Costa and Santos (2017) also evaluated SNPs in Psidium species, using EUChip60Kchip, developed by Silva-Junior et al. (2015) for Eucalyptus species, with transfer of 3523 SNPs (around 5%) to Psidium samples. The authors concluded that the transfer of SNPs between genera was very reliable, in agreement with previous genetic divergence studies using microsatellites. In the present study, we found around 17.7% of markers of Psidium species common to Eucalyptus, thus increasing study possibilities.
In this study, we selected SNP markers of greater informativeness for nine Psidium species. These markers showed efficiency in detecting intra- and interspecific variations, with 37% shared with E. grandis and 78% aligned in sites with annotation in coding regions. These marker regions were delimited in the genome of P. guajava and provide data for the development of a large-scale and flexible panel of SNPs for genotyping different species of Psidium.
The evolution of Psidium gave rise to several endemic species differing in ploidy level, distribution range, and habitat preferences. In the present study, polyploidy contributed to the high diversity observed in the Psidium species studied. Furthermore, we verified wide genetic diversity in little-known native species, such as P. gaudichaudianum, P. acidum, and P. oblongatum. Investigating genetic variability and the relationships between Psidium species is important for the creation, conservation, management, and use of these genetic resources in breeding programs. The information found here may be used as an aid for these purposes.
The lack of phylogenetic resolution for some species reported above may be due to the small number of individuals, or even related to the markers used. It is believed that very close species need more markers with higher resolving power in order to be grouped in different clades (Parks et al., 2009; Swenson, 2009). Phylogenetic diversity can be directly affected by phylogenetic resolution when working with large datasets, and cryptic species are formed (Swenson, 2009; Santos et al., 2020).
The distance of P. guajava genotypes from the other species may be due to the fact that this species has long been domesticated. Moreover, the species may have been separated according to the type of evolution to which each gene was subjected, which can be more or less conserved.
Here, DArTseq-derived SNPs were used for the genetic analysis of diploid and polyploid species of Psidium. The present study also made advances in the detection and characterization of intra- and interspecific polymorphic genomic regions. In addition, a set of SNPs with high discriminatory potential among species, as well as among individuals, was proposed for routine characterization of accessions in gene banks and in breeding programs. The STAGs exclusive to the Psidium species represent specific evolution in the genus. The associated GO terms reveal classes of genes conserved in evolution of the family. Furthermore, the distribution of a subset of markers in E. grandis chromosomes has importance for guava breeding, comparative genomics, and genome evolution studies.
Conclusions
The DArTseq approach, associated with annotation analysis, generated a large set of conserved, polymorphic, and possibly functional SNPs for Psidium species. A large number of markers conserved between Psidium and Eucalyptus contributes to Psidium research, which can benefit from studies on Eucalyptus, a pivotal genus in Myrtaceae. Species-specific markers were identified, and most of the functional polymorphisms in Psidium were annotated for the GO classes: biological processes, cellular components, and molecular functions. These SNPs generated clusters that showed intra- and interspecific relationships. These clusters revealed that P. guajava shows substantial genetic similarity to the tetraploid P. guineense and low variability in relation to the other species. A group of SNPs was selected and annotated, allowing the development of a panel of interspecific SNPs for Psidium. The association of STAGs with a suite of evolutionarily conserved genes reveals the conserved nature of orthologs along the genome of E. grandis, which are shared by the Psidium lineages. This result is important for guava breeding and evolutionary genomics studies in the Myrtaceae family, and highlights how STAGs can be used for comparative genomics and genome evolution studies in these non-traditional model species of Psidium for which whole-genome sequences are unavailable.
References
Abdelmigid HM, Morsi MM (2018) Genetic fidelity testing of soybean plants under allelopathic stress of Eucalyptus ground leaves through RAPD and ISSR markers. Int J Bot 14(1):14–23
Al-Beyroutiová M, Sabo M, Sleziak P, Dušinský R, Birčák E, Hauptvogel P, Kilian A et al (2016) Evolutionary relationships in the genus Secale revealed by DArTseq DNA polymorphism. Plant Syst Evol 302(8):1083–1091
Andersen JR, Lubberstedt T (2003) Functional markers in plants. Trends in Plant Science 8(11):554–560
Bernardes CO, Tuler AC, Ferreira, Carvalho AMS, Nogueira AM, Ferreira MFS (2018) Transferability of Psidium microsatellite loci in Myrteae (Myrtaceae): a phylogenetic signal. Euphytica. 214. https://doi.org/10.1007/s10681-018-2195-2
Cerio EDC, Verardo V, Caravaca AMG, Gutiérrez AF, Carretero AS (2017) Health effects of Psidium guajava L. leaves: an overview of the last decade. Int J Mol Sci 18:897. https://doi.org/10.3390/ijms18040897
Costa SR, Santos CAF (2013) Allelic database and divergence among Psidium accessions by using microsatellite markers. Genet Mol Res 12(4):6802–6812
Costa SR, Santos CAF, Castro JMCE (2016) Inheritance of resistance to Meloidogyne enterolobii in Psidium guajava x P. guineense hybrid. Eur J Plant Pathol:1001–1008. https://doi.org/10.1007/s10658-016-1098-0
Costa SR, Santos CAF (2017) Genetic divergence among Psidium accessions based on single nucleotide polymorphisms developed for Eucalyptus. Genet Mol Res 16(2):gmr16029566. https://doi.org/10.4238/gmr16029566
Deschamps S, Llaca V, May GD (2012) Genotyping-by-sequencing in plants. Biology 1(3):460–483. https://doi.org/10.3390/biology1030460
Dias CN, Alves LP, Rodrigues KA, Brito MC, dos Rosa C, dos Amaral FM, Monteiro OS, Andrade EH, Maia JG, Moraes DF (2015, 2015) Chemical composition and larvicidal activity of essential oils extracted from Brazilian legal Amazon plants against Aedes aegypti L. (Diptera: Culicidae). J Evid-Based Integr Med. https://doi.org/10.1155/2015/490765
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus:1213–1215
Egea LA, Mérida-García R, Kilian A, Hernandez P, Dorado G (2017) Assessment of genetic diversity and structure of large garlic (Allium sativum) germplasm bank, by diversity arrays technology “genotyping-by-sequencing” platform (DArTseq). Front Genet 8:1–9. https://doi.org/10.3389/fgene.2017.00098
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. Plos One 6(5):1–10. https://doi.org/10.1371/journal.pone.0019379
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol Ecol 17:1170–1188. https://doi.org/10.1111/j.1365-294X.2007.03659.x
Fellers JP (2008) Genome filtering using methylation-sensitive restriction enzymes with six base pair recognition sites. The Plant Genome 1(2):146–152
Flora do Brasil (2020). Website under construction. Jardim Botânico do Rio de Janeiro. Available at: http://floradobrasil.jbrj.gov.br/. [].
Garavito A, Montagnon C, Guyot R, Bertrand B (2016) Identification by the DArTseq method of the genetic origin of the Coffea canephora cultivated in Vietnam and Mexico. BMC Plant Biol 16(1):1–12
Grattapaglia D, Vaillancourt RE, Shepherd M, Thumma BR, Foley W, Külheim C, Potts BM, Myburg AA (2012) Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet Genomes 8:463–508. https://doi.org/10.1007/s11295-012-0491-x
Han L, Love K, Peace B, Broadhurst L, England N, Li L, Bush D (2020) Origin of planted Eucalyptus benthamii trees in Camden NSW: checking the effectiveness of circa situm conservation measures using molecular markers. Biodivers Conserv 29:1301–1322
Kilian A, Wenzl P, Huttner E et al (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89
Landrum LR, Denis WC, Sharp WP, Brendecke J (1995) Hybridization between Psidium guajava and P. guineense (Myrtaceae). Econ Bot 49(2):153–161
Lee T-H, Guo H, Wang X, Kim C, Paterson AH (2014) SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15:162
Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. https://doi.org/10.1093/nar/gkz239
Lucas EJ, Harris AS, Mazine FF, Belsham SR, Nic Lughadha EM, Telford A, Chase MW (2007) Suprageneric phylogenetics of Myrteae, the generically richest tribe in Myrtaceae (Myrtales). Taxon 56:1105–1128
Machado RM, Oliveira FA, Alves FM, Souza AP, Forni-Martins ER (2020) Population genetics of polyploid complex Psidium cattleyanum Sabine (Myrtaceae): preliminary analyses based on new species-specific microsatellite loci and extension to other species of the genus. Biochem Genet. 59:219–234. https://doi.org/10.1007/s10528-020-10002-1
Mallmann, L. P., Tischer, B., Vizzotto, M., Rodrigues, E. and Manfroia, V. (2020). Comprehensive identification and quantification of unexploited phenolic compounds from red and yellow araçá (Psidium cattleianum Sabine) by LC-DAD-ESI-MS/MS. Food Res Int 131. doi: 10.1016/j.foodres .2020.108978
Marques AM, Tuler AC, Carvalho CR, Carrijo TT, Ferreira MFS, Clarindo WR (2016) Refinement of the karyological aspects of Psidium guineense (Swartz, 1788): a comparison with Psidium guajava (Linnaeus, 1753). Comparative Cytogenetics 10(1):117–128
Mendes LA, Martins GF, Valbon WR, de Souza TS, Menini L, Ferreira A, Ferreira, M. F. da S. (2017) Larvicidal effect of essential oils from Brazilian cultivars of guava on Aedes aegypti L. Ind Crop Prod 108:684–689
Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Jenkins J, Lindquist E, Tice H, Bauer D, Goodstein DM, Dubchak I, Poliakov A, Mizrachi E, Kullan ARK, Hussey SG, Pinard D, van der Merwe K, Singh P, van Jaarsveld I, Silva-Junior OB, Togawa RC, Pappas MR, Faria DA, Sansaloni CP, Petroli CD, Yang X, Ranjan P, Tschaplinski TJ, Ye CY, Li T, Sterck L, Vanneste K, Murat F, Soler M, Clemente HS, Saidi N, Cassan-Wang H, Dunand C, Hefer CA, Bornberg-Bauer E, Kersting AR, Vining K, Amarasinghe V, Ranik M, Naithani S, Elser J, Boyd AE, Liston A, Spatafora JW, Dharmwardhana P, Raja R, Sullivan C, Romanel E, Alves-Ferreira M, Külheim C, Foley W, Carocha V, Paiva J, Kudrna D, Brommonschenkel SH, Pasquali G, Byrne M, Rigault P, Tibbits J, Spokevicius A, Jones RC, Steane DA, Vaillancourt RE, Potts BM, Joubert F, Barry K, Pappas GJ, Strauss SH, Jaiswal P, Grima-Pettenati J, Salse J, van de Peer Y, Rokhsar DS, Schmutz J (2014) The genome of Eucalyptus grandis. Nature 510:356–362
NCBI (n.d.). NCBI BLAST. Website https://www.ncbi.nlm.nih.gov/ [].
Ndjiondjop M-N, Semagn K, Gouda AC, Kpeki SB, Dro Tia D, Sow M, Goungoulou A, Sie M, Perrier X, Ghesquiere A, Warburton ML (2017) Genetic variation and population structure of Oryza glaberrima and development of a mini-core collection using DArTseq. Front Plant Sci 8:1–19. https://doi.org/10.3389/fpls.2017.01748
Nimisha S, Kherwar D, Ajay KM, Singh B, Usha K (2013) Molecular breeding to improve guava (Psidium guajava L.): current status and future prospective. Sci Hortic 164:578–588. https://doi.org/10.1016/j.scienta.2013.10.017
Nimmakayala P, Tomason YR, Abburi VL, Alvarado A, Saminathan T, Vajja VG, Salazar G et al (2016) Genome-wide differentiation of various melon horticultural groups for use in GWAS for fruit firmness and construction of a high resolution genetic map. Front Plant Sci 7:1–15
Parks M, Cronn R, Liston A (2009) Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol 7:84. https://doi.org/10.1186/1741-7007-7-84
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing. Website, Vienna, Austria http://www.R-project.org/ [
Ren R, Ray R, Li P, Xu J, Zhang M, Liu G, Yao X, Kilian A, Yang X (2015) Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Gen Genomics 290(4):1457–1470
Sansaloni C, Franco J, Santos B, Percival-Alwyn L, Singh S, Petroli C, Campos J, Dreher K, Payne T, Marshall D, Kilian B, Milne I, Raubach S, Shaw P, Stephen G, Carling J, Pierre CS, Burgueño J, Crosa J, Li HH, Guzman C, Kehel Z, Amri A, Kilian A, Wenzl P, Uauy C, Banziger M, Caccamo M, Pixley K (2020) Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nature Communication 11:4572. https://doi.org/10.1038/s41467-020-18404-w
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D, Kilian A (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5(Suppl 7):54
Santos PHD, Carvalho BM, Aredes FAS, Mussi-Dias V, Pinho DB, Pereira MG, da Silveira SF (2020) Is Lasiodiplodia theobromae the only species that causes leaf blight disease in Brazilian coconut palms? Trop Plant Pathol 45:434–442
Swenson NG (2009) Phylogenetic resolution and quantifying the phylogenetic diversity and dispersion of communities. PLoS One 4:e4390
Silva-Junior OB, Faria DA, Grattapaglia D (2015) A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytologist 206:1527–1540
Souza AG, Resende LV, Lima IP, Martins LSS, Techio VH (2015) Chromosome number and nuclear DNA amount in Psidium spp. resistant and susceptible to Meloidogyne enterolobii and its relation with compatibility between rootstocks and commercial varieties of guava tree. Plant Syst Evol 301:231–237
Sukumaran S, Reynolds MP, Sansaloni C (2018) Genome-wide association analyses identify QTL hotspots for yield and component traits in Durum wheat grown under yield potential, drought, and heat stress environments. Front Plant Sci 9:1–16
Thornhill AH, Ho SYW, Külheim C, Crisp MD (2015) Interpreting the modern distribution of Myrtaceae using a dated molecular phylogeny. Molecular Phylogenetics and Evolution 93:29–43
Tuler AC, Carrijo TT, Nóia LR, Ferreira A, Peixoto AL, Ferreira, M. F. da S. (2015) Short communication – SSR markers: a tool for species identification in Psidium (Myrtaceae). Molecular Biology Reports. 42:1501–1513. https://doi.org/10.1007/s11033-015-3927-1
Tuler AC, Carrijo TT, Peixoto AL, Garbin ML, da Ferreira MFS, Carvalho CR, Spadeto MS, Clarindo WR (2019a) Diversification and geographical distribution of Psidium (Myrtaceae) species with distinct ploidy levels. Trees. https://doi.org/10.1007/s00468-019-01845-2
Tuler A, Carrijo T, Sheu Y, Peixoto AL (2019b) Re-establishment of Psidium macahense (Myrtaceae, Myrteae), an endemic species from the Brazilian Atlantic Forest. Phytotaxa 397(1). https://doi.org/10.11646/phytotaxa.397.1.3
Valdisser PAMR, Pereira WJ, Almeida Filho JE et al (2017) In-depth genome characterization of a Brazilian common bean core collection using DArTseq high-density SNP genotyping. BMC Genomics 18(1):1–19
Vitti KA, Lima LM, Martines Filho JG (2020) Agricultural and economic characterization of guava production in Brazil. Rev Bras Frutic 42(1). https://doi.org/10.1590/0100-29452020447
Wenzl P, Carling J, Kudrna D, Jaccoud D, Huttner E, Kleinhofs A, Kilian A (2004) Diversity arrays technology (DArT) for whole-genome profiling of barley. Proc Natl Acad Sci U S A. 101:9915–9920
Willis SC, Hollenbeck CM, Puritz JB, Gold JR, Portnoy DS (2017) Haplotyping RAD loci: an efficient method to filter paralogs and account for physical linkage. Mol Ecol Resour 17(5):955–965
Wilson PG, O’Brien MM, Heslewood MM, Quinn CJ (2005) Relationships within Myrtaceae sensu lato based on a matK phylogeny. Plant Syst Evol 251:3–19
Acknowledgements
We would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, DF, Brazil), the Fundação de Amparo à Pesquisa do Espírito Santo (FAPES/VALE, Vitória, ES, Brazil), and VALE company for financial support. This study was partly financed by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. We would also like to thank José and Isabel Corti and Erli Röpke for providing the samples of Cortibel genotypes.
Data archiving statement
The sequence data of the 5951 STAGs are available in Supplementary File S5.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J.P. Jaramillo-Correa
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary File 1
Description, per chromosome, of the distribution of 22,050 SNPs present in Psidium species that were anchored in the genome of Eucalyptus grandis (DOCX 19 kb)
Supplementary File 2
Relationship of SNP markers in coding regions with their gene ontology (GO) for genes related to A) Biological Processes (blp), B) Cellular Components (cc), and C) Molecular Function (mf). The information for each graph is divided into markers anchored and non-anchored to the Eucalyptus genome. (DOCX 295 kb)
ESM 3
(PDF 7455 kb)
Supplementary File S3
Sequence of 5951 STAGs conserved in the Psidium species. (XLSX 1020 kb)
Rights and permissions
About this article

Cite this article
Grossi, L.L., Fernandes, M., Silva, M.A. et al. DArTseq-derived SNPs for the genus Psidium reveal the high diversity of native species. Tree Genetics & Genomes 17, 23 (2021). https://doi.org/10.1007/s11295-021-01505-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-021-01505-y