
Abstract
Watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai] is an economically important vegetable crop grown extensively worldwide. To facilitate the identification of agronomically important traits and provide new information for genetic and genomic research on this species, a high-density genetic linkage map of watermelon was constructed using an F2 population derived from a cross between elite watermelon cultivar K3 and wild watermelon germplasm PI 189225. Based on a sliding window approach, a total of 1,161 bin markers representing 3,465 SNP markers were mapped onto 11 linkage groups corresponding to the chromosome pair number of watermelon. The total length of the genetic map is 1,099.2 cM, with an average distance between bins of 1.0 cM. The number of markers in each chromosome varies from 62 in chromosome 07 to 160 in chromosome 05. The length of individual chromosomes ranged between 61.8 cM for chromosome 07 and 140.2 cM for chromosome 05. A total of 616 SNP bin markers showed significant (P < 0.05) segregation distortion across all 11 chromosomes, and 513 (83.3 %) of these distorted loci showed distortion in favor of the elite watermelon cultivar K3 allele and 103 were skewed toward PI 189225. The number of SNPs and InDels per Mb varied considerably across the segregation distorted regions (SDRs) on each chromosome, and a mixture of dense and sparse SNPs and InDel SDRs coexisted on some chromosomes suggesting that SDRs were randomly distributed throughout the genome. Recombination rates varied greatly among each chromosome, from 2.0 to 4.2 centimorgans per megabase (cM/Mb). An inconsistency was found between the genetic and physical positions on the map for a segment on chromosome 11. The high-density genetic map described in the present study will facilitate fine mapping of quantitative trait loci, the identification of candidate genes, map-based cloning, as well as marker-assisted selection (MAS) in watermelon breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Watermelon is an important vegetable crop worldwide with production of more than 100 million tons accounting for about 7 % of total vegetable production worldwide (FAO Stat; http://www.fao.org/). Watermelon, Citrullus lanatus (Thunb.) Matsum. & Nakai (2n = 2x = 22) is a diploid species belonging to the family Cucurbitaceae with an estimated genome size of 425 Mb (Arumuganathan and Earle 1991). The development of genetic linkage maps can facilitate the identification of biologically and agronomically important trait loci and further deployment in crop improvement. High-quality and high-density genetic maps can also enable further functional genetics and genomics studies in this plant species (Jones et al. 2009), leading to better understanding of the genetic architecture of complex traits.
Recently, much progress has been made in genetic and genomic research for watermelon, such as construction of expressed sequence tag (EST) libraries (Levi et al. 2006a; Wechter et al. 2008; Guo et al. 2011), development of bacterial artificial chromosome libraries (Joobeur et al. 2006), and genome sequencing (Guo et al. 2013). In the last 20 years, several genetic linkage maps have been developed for watermelon using various molecular marker systems including randomly amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), simple sequence repeat (SSR), sequence-tagged sites (STS) and sequence-related amplified polymorphism (SRAP) (Hashizume et al. 1996; Levi et al. 2002a, 2006b; Hashizume et al. 2003). Ren et al. (2012a) published a high resolution genetic map for watermelon that was constructed using 698 simple sequence repeat (SSR), 219 insertion–deletion (InDel) and 36 structure variation (SV) based on the genome sequences of the elite Chinese line 97103 and the wild accession PI 296341-FR. The map spanned a total of 800 cM with an average distance of 0.8 cM between markers. Sandlin et al. (2012) constructed a single-nucleotide polymorphism (SNP)-based genetic map through comparative mapping of three separate populations developed from crosses Klondike Black Seeded (PI 635609)/New Hampshire Midget (PI 635617), Strain II (PI 279461)/Egusi (PI 560023) and ZWRM50 (PI 593359)/Citroides (PI 244019) and the map comprised 378 SNP markers with an average marker spacing of 5.1 cM. To create a more saturated map for the species, Ren et al. (2014) reported an integrated map based on four populations containing 1,339 (698 SSR, 386 SNP, 219 InDel and 36 SV) markers. This integrated map provided improved resolution, spanning on 798 cM and bringing the average distance between markers down to 0.6 cM. However, marker density still remains far from saturation and the number of markers is relatively few to allow marker-assisted selection (MAS) and cloning of genes of interest in watermelon.
Recently, through the use of next-generation sequencing (NGS) technologies, a rapid SNP discovery method, known as DArTseq™, was developed utilizing DArT marker platform in combination with next-generation sequencing platforms (Sansaloni et al. 2011; Kilian et al. 2012; Courtois et al. 2013; Cruz et al. 2013; Raman et al. 2014). DArTseq™ represents a new implementation of sequencing of complexity-reduced representations (Altshuler et al. 2000) and more recent applications of this concept on the next-generation sequencing platforms (Baird et al. 2008; Elshire et al. 2011). DArTseq™ has been successfully applied in genetic diversity assessment studies (Cruz et al. 2013).
Thus, to increase marker density and expand marker resources for watermelon, we demonstrate high-throughput SNP discovery using DArT-Seq™ method resulting in a high-density genetic linkage map for watermelon based on corresponding SNP markers.
Materials and methods
Plant material and DNA isolation
An F2 population of 144 plants derived from a cross between elite watermelon cultivar K3 (female) and wild watermelon germplasm PI 189225 (male) was used to generate the linkage map. Genomic DNA was extracted from freeze-dried leaves of each F2 plant and parent using the CTAB method as modified by Ren et al. (2012b). DNA quality and concentration were determined by electrophoresis in 0.8 % agarose gel with a λDNA standard and ND-1000 spectrophotometry (NanoDrop, Thermo scientific, Wilmington, DE, USA) and were normalized to 30–50 ng/μl.
High-throughput genotyping using the DArT-Seq™ method
A high-throughput genotyping method using the DArT-Seq™ technology (Sansaloni et al. 2011) was employed to genotype the F2 population. Restriction enzymes PstI and MseI were selected as enzyme combination for genomic complexity reduction. DNA samples are processed in digestion/ligation reactions principally as per Kilian et al. (2012), but replacing a single PstI-compatible adapter with two different adapters corresponding to two different restriction enzyme (RE) overhangs. The PstI-compatible adapter was designed to include Illumina flowcell attachment sequence, sequencing primer sequence and “staggered”, varying length barcode region, similar to the sequence reported by Elshire et al. (2011). The reverse adapter contained the flowcell attachment region and MseI-compatible overhang sequence. Only “mixed fragments” (PstI–HpaII) were effectively amplified in 30 rounds of PCR using the following reaction conditions: 1 min at 94 °C for initial denaturation; 30 cycles each consisting of 20 s at 94 °C for denaturation, 30 s at 58 °C for annealing and 45 s at 72 °C for extension; and finally a 7 min extension step at 72 °C. After PCR, equimolar amounts of amplification products from each sample of the 96-well microtiter plate were bulked and applied to c-Bot (Illumina) bridge PCR followed by sequencing on Illumina Hiseq 2500. The sequencing (single read) was run for 77 cycles. Sequences generated from each lane were processed using proprietary DArT analytical pipelines. In the primary pipeline, the FASTQ files were first processed to filter poor-quality sequences, applying more stringent selection criteria to the barcode region compared to the rest of the sequence. Thus, the assignments of the sequences to specific samples carried in the “barcode split” step are more consistent. Approximately 2,500,000 (±7 %) sequences per barcode/sample are used in marker calling. Finally, identical sequences are collapsed into “fastqcall files”. These files are used in the secondary pipeline for DArT PL’s proprietary SNP calling algorithms (DArTsoft14). To get the physical positions of the corresponding SNP markers, the sequences of the corresponding DNA fragments were BLASTed against a local database containing the watermelon reference genome sequence of 97103, with an expected value (E) < e −10 and minimum base identity >70 % as blast criteria. Sequences for the genomes 97103 were obtained through http://www.icugi.org/cgi-bin/ICuGI/genome/index.cgi?organism=watermelon.
Bin map construction
Prior to the bin map construction, the SNP data that were not homozygous in both parents as well as proportion of missing data greater than 70 % were discarded. Due to potential sources of sequence errors and restrictions on the maximum number of markers that can be analyzed in JoinMap software, the SNP data were analyzed for genotype calling recombinations using a sliding window approach (Huang et al. 2009). Briefly, the consecutive genotypic SNP data was scanned with a window size of 15 SNPs and a step size of 1. Windows with 15 or more SNPs from either parent were considered to be homozygous for an individual, while those with less were classified as heterozygous. Adjacent windows with the same genotypes were combined into blocks based on the recombinant breakpoints and the physical position of each SNP marker and a bin map was generated and presented by using Perl scripts.
Genetic linkage map construction
Resulting bins on each chromosome were treated as genetic markers and analyzed for linkage using JoinMap version 4.1 (Van Ooijen 2011). Linkage group (LG) were established and the marker order within each linkage group were determined using the maximum likelihood (ML) mapping algorithm (Jansen et al. 2001). The Kosambi mapping function was used to convert the recombination frequency into cM values (Kosambi 1943). The LG maps of each chromosome were drawn and aligned using MapChart v2.2 (Voorrips 2002). Chi square (χ 2) test analysis of all the bin makers was also performed to test for deviations from the expected 1:2:1 ratio of K3 and PI 189225 alleles for the current F2 population using R statistical analysis package (Team 2012).
Relationship between genetic position and physical position
In addition to the genetic map position, the physical positions of the bin markers were also determined based on the alignment with the watermelon reference genome sequence of 97103. Colinearity between genetic and physical positions was determined by plotting genetic marker positions (in centimorgans) against their physical positions (in megabases) using R statistical analysis package (Team 2012). We have also calculated the average recombination rate for each chromosome through the plot of the cM distance of each marker versus its physical distance.
Genomic variance analysis in segregation distortion regions
To investigate the causal relationships between segregation distortion regions and genomic variance, InDels and SNPs data from the watermelon genome of 97103 and PI 296341-FR in Cucurbit Genomics Database (http://www.icugi.org/) were studied. The physical locations of SNPs and InDels between97103 and PI 296341-FR were aligned to the equivalent segregation distorted regions (SDRs) in the present study and the number of SNPs and InDels per megabase (Mb) were compared between the segregation distortion regions (SDRs) and the non-segregation distorted regions.
Results
Characterization of DArTseq-based SNP marker and SNP bin marker
A total of 4,808 SNPs were identified from the DArTseq-based genotyping systems. The average call rate of the markers was 91.3 % and their PIC values ranged from 0.022 to 0.5, with an average of 0.454. The physical position along the chromosome as determined for most of the SNPs based on the alignment to the reference genome of 97103, using the selecting criteria of minimum base identity, was >90 % and E-value <10−5.
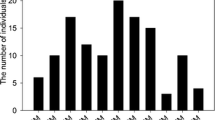
Based on markers with proportion of missing genotype greater than 70 % as well as the markers whose parental genotypes were not homozygous or could not be determined on their chromosome location, 1,343 were removed from the total markers and the remaining 3,465 (72.1 %) SNP markers were assembled in a total of 1,161 bin signatures representing the corresponding recombination events according to their physical positions and their genotypes (Table 1). Each bin contained different numbers of SNP markers and the two bins with the largest number of SNP markers were 70 on bin 50 of chromosome 07 and 43 on bin 1 of chromosome 06 (Fig. 1, Table S1). In total, 87.9 % of the bin markers were less than 0.5 Mb in length (Table S2). There were 54 bins larger than 1.0 Mb in size and 2 big bins of more than 5.0 Mb dispersed on chromosomes 08 (Bin6) and 09 (Bin89) (Table S2).

Distribution of DArTseq-based SNP markers within each bin on different watermelon chromosomes. The x axis shows the number of bins. The y axis shows the number of SNP markers within each bin. The number of SNPs mapping to a single bin is shown by the height of the bars
Genetic map construction
The final genetic linkage map consisted of 1,161 SNP bin markers, giving a total map length of 1,099.2 cM, with individual linkage groups ranging from 61.8 cM for chromosome 07 to 140.2 cM for chromosome 5 (Table 1; Fig. 2; Table S3). The number of markers per chromosome ranged from 62 for chromosome 7 to 160 for chromosome 5, with an average number of 105.5 bin markers. The average distance between neighboring bin markers on the maps was almost similar for all the 11 chromosomes with ether 0.9 cM/marker (chromosome 02, 05, 08, 09 and 11) or 1.0 cM/marker (chromosome 01, 06, 07 and 10) or 1.1 cM/marker (chromosome 03 and 04) and the whole genome average density was 1.0 cM/marker. In general, the SNP bin markers were well distributed across the genome and most of the spaces between two bin markers for all the 11 chromosomes were smaller than 5.0 cM with some exceptions where we observed gaps of 5.5 and 9.9 cM on chromosome 01, 6.2 and 6.9 cM on chromosome 03, 5.4 cM on chromosome 10 and 10.4 cM chromosome 11(Table 1; Fig. 2; Table S3). The total physical length of the map was 330.6 Mb and the average physical length for the whole genome was 30.0 Mb, ranging from 24.3 Mb for chromosome 04 to 35.0 Mb for chromosome 09 (Table 1; Fig. 2; Table S3).

The SNP-based genetic linkage map for watermelon using the K3 × PI 189225 population. Bin marker names and map distances are shown on the left and right side of each linkage group, respectively
Comparison of genetic and physical positions
Genetic distance of a map was generated based on the relative recombination frequencies between markers during crossover of homologous chromosomes. With the availability of complete watermelon genome sequences, we also carried out the comparisons between genetic and physical positions of each chromosome. In the present study, the relationship between genetic and physical positions was mostly linear for each chromosome except for a large segment of approximately 21.2 Mb on chromosome 11, which was in opposite orientation between genetic and physical positions (Fig. 3; Table S1; Table S3). The variation in the relationship between the genetic and physical positions for each chromosome can be directly reflected by the different slopes of the curves of each chromosome (Fig. 3; Table S1; Table S3).

Relationship between genetic and physical positions of the bin markers within each chromosome. In each plot, the x axis represents the physical positions in megabases (Mb) based on 97103 reference v2 sequence. The y axis represents the genetic marker positions in centimorgans
Besides the relationship between genetic and physical positions, to compare the genetic and physical distances, we also calculated recombination rates for each chromosome by comparing the genetic distance to the physical distance (Mb). The recombination rate among all the 11 chromosomes varied greatly for each of the chromosome, ranging from 2.0 centimorgans per megabase (cM/Mb) for chromosome 07 to 4.2 cM/Mb for chromosome 05 with a genome-wide average recombination rate is 3.3 cM/Mb (Table 1; Table S2).
Distortion analysis
Chi square testing revealed that 616 of 1,161 (53.1 %) SNP bin markers showed significant (P = 0.05) segregation distortion in the F2 population. Five hundred and thirteen bin markers (44.2 %) showed distortion in favor of the elite watermelon cultivar K3 allele, whereas 103 (8.9 %) showed distortion in favor of the wild watermelon germplasm PI 189225 allele (P = 0.05). The 513 markers that showed distortion in favor of the cultivated allele were distributed among all the chromosomes [01 (49), 02 (52), 03 (60), 04 (49), 05 (151), 07 (15), 08 (50), 09 (60), 10 (13) and 11 (26)] except chromosome 06 (Fig. 4). The 103 markers that showed distortion in favor of the wild allele were distributed among five chromosomes as follows: 04 (2), 06 (11), 07 (16), 10 (39) and 11 (35) (Fig. 4). The 616 distorted segregation markers were clustered in 44 main SDRs on 11 chromosomes as follows: 01 (2), 02 (6), 03 (4), 04 (4), 05 (3), 06 (2), 07 (3), 08 (3), 09 (9), 10 (3) and 11 (5) (Fig. 5; Table 2).

Segregation distortion of SNP bin markers on each chromosome. The x axis shows the segregation ratios for each SNP bin marker on each chromosome. The y axis shows the −logP value of allele ratios of the number of individuals carrying the marker compared to the number of individuals not carrying the marker. The expected distribution of K3 and PI 189225 alleles across the 144 F2 individuals should result in a ratio of 1:2:1 and is depicted as a horizontal line at −logP = 0. SNP markers with allele ratios above the line indicate segregation distortion toward the K3 allele, whereas SNP markers with allele ratios below the line indicate segregation distortion toward the PI 189225 allele. SNP markers with allele ratios between the values of 0 and −log (0.05) indicate no significant segregation distortion from the expected ratio of 1:2:1 between K3 and PI 189225 genotypes. SNP markers with allele ratios beyond the line at the value of −log (0.05) indicate significant (P < 0.05) segregation distortion toward the K3 or PI 189225 allele


The physical map and the segregation distortion regions’ (SDRs’) distribution in the F2 population of K3 × PI 189225. Bin marker names and locations are shown on the left and right side of each chromosome, respectively. The physical location (in Mb) of each bin marker represents the physical location along the chromosomes of reference genome of 97103. Segregation distortion markers in favor of K3 alleles and PI 189225 alleles are shown in red and pink, respectively (color figure online)
Genomic variance in segregation distortion regions
A total of 4,335,338 SNPs and 35,197 InDels were detected between 97103 and PI 296341-FR from the SNPs and InDels data available in the Cucurbit Genomics Database. To examine if the markers with distorted segregation were in genomic regions with major differences in genome sequence between cultivated and wild watermelon, we placed all of the polymorphisms (including both InDels and SNPs) between 97103 and PI 296341-FR into the segregation distortion regions (SDRs) across each chromosome and 2,289,215 SNPs and 18,023 InDels were located in 44 SDRs. Large size SDRs were identified on some chromosomes, including SDR5.3 (27.35 Mb) on chromosome 05, SDR1.1 (20.82 Mb) on chromosome 02, SDR8.1 (19.51 Mb) on chromosome 08 and SDR4.1 (15.99 Mb) on chromosome 04 (Fig. 5; Table 2). The number of SNPs per Mb varied considerably across the SDRs on each chromosome and ranged from 12,044 SNPs per Mb for chromosome 10 to 24,953 SNPs per Mb for chromosome 09 (Table 2). For InDels, the number of InDels per Mb ranged from 71 InDels per Mb for chromosome 08 to 162 per Mb for chromosome 04 (Table 2). The number of SNPs per Mb for SDRs on chromosomes 02, 04, 06, 07 and 09 was higher than that of the remaining regions or the whole chromosome. In contrast, the number of SNPs per Mb for SDRs on chromosomes 01, 03, 05, 08, 10 and 11was lower than that of the remaining regions or the whole chromosome (Table 2). The same results were also observed for InDels (Table 2). A mixture of dense and sparse SNP SDRs was also included in some of the chromosomes (Table 2). For example, on chromosome 07, the number of SNPs per Mb was from 14,429 SNPs per Mb for SDR7.3 to 10,241 SNPs per Mb for SDR7.2. On chromosome 11, the number of SNPs per Mb was from 14,634 SNPs per Mb for SDR11.5 to 11,198 SNP/Mb for SDR11.3. On chromosome 09, the number of SNPs per Mb was from 15,395 SNPs per Mb for SDR9.2 to 12,714 SNPs per Mb for SDR9.4 (Table 2). The distribution patterns of InDels densities also vary for SDRs on each chromosome (Table 2).
Discussion
Genetic linkage maps are highly valuable tools for the identification of novel genes/QTLs, map-based cloning or in marker-assisted breeding programs. Dense genetic maps based on sequence-derived markers, such as SSRs, InDels and SNPs, accelerate the process of fine mapping and map-based cloning of genes/QTLs. Among them, SNP markers have become increasingly popular in plant genetic and genomics studies due to their abundance, uniform genome distribution and cost-effectiveness (Rafalski 2002; Ganal et al. 2009). SNP markers have been used extensively over the last few years for genetic mapping in many crops such as wheat (Triticum aestivum L. em. Thell.) (Akhunov et al. 2009), maize (Zea mays L.) (Ganal et al. 2011) and rice (Oryza sativa L.) (Chen et al. 2014) as well as in other cucurbit species including melon (Cucumis melo L.) (Deleu et al. 2009; Harel-Beja et al. 2010; Garcia-Mas et al. 2012) and summer squash (Cucurbita pepo L.) (Zraidi et al. 2007; Esteras et al. 2012).
Cultivated watermelon has been reported to carry narrow genetic diversity (Levi et al. 2001a, b), resulting in low polymorphism of molecular markers between parents of mapping populations. In the present map, the mapping population was generated from a cross between cultivated genotype and a wild watermelon germplasm. The wide genetic diversity between the two parents provided higher frequency of polymorphisms in the map construction. Furthermore, the present map was based on a cross involving wild-type watermelon germplasm PI 189225 known to possess resistance to powdery mildew (Tetteh et al. 2010; 2013), anthracnose (Boyhan et al. 1994) and gummy stem blight (Gusmini et al. 2005). The parent K3 is an advanced breeding line with high fruit quality and has been used as a parental line for several commercial watermelon hybrids in Jiangsu Province of China; however, it is highly susceptible to fungal diseases. Therefore, the present map and the markers can facilitate the identification of new genes/QTLs for disease resistance within this wild watermelon germplasm.
In this study, we constructed a genetic map of watermelon using 1,161 bin markers representing 3,465 SNP markers by sequencing DArT tags using Illumina short read sequencing technology. The mapped SNP marker number was larger than recently reported watermelon genetic maps, containing 378 (Sandlin et al. 2012) and 386 SNP markers (Ren et al. 2014), respectively. The distribution of markers on the 11 chromosomes was generally uniform and, despite the presence of a 9.9 and 10.4 cM gaps in chromosome 01 and 11, respectively, greater number of SNP markers obtained in the present study will significantly expand marker pools in watermelon genetic and genomics studies. The total genetic length of the map was 1,099.2 cM, which is 37 % larger than the map (800 cM) constructed using 1,339 markers including 698 SSR, 21 InDel and 36 SV markers by Ren et al. (2012a) and the map (798 cM) of Ren et al. (2014) that was complemented by integrating four watermelon maps with 698 SSR, 219 InDel, 36 SV and 386 SNP markers. The length is considerably shorter (42.7 %) than that of the map reported by Sandlin et al. (2012) for contig sequence-derived 378 SNP mapped over 1,917.6 cM. The whole genome average marker density between bins was 0.9 cM, which is comparable to that of 0.8 cM obtained in Ren et al. (2012a) and 0.6 cM obtained in Ren et al. (2014).
The completion of the watermelon genomic sequence facilitated the comparison of genetic and physical distances. In the present study, the recombination rate in the population varied greatly among each chromosome, from 2.0 to 4.2 cM/Mb, corresponding to the actual physical length of the smallest and largest chromosomes 07 and 05, respectively. The whole genome recombination rate was 3.3 cM/Mb and this ratio was similar to that of cucumber (Cucumis sativus L.) (3.2 cM/Mb) (Huang et al. 2009; Ren et al. 2012a), but larger than the average ratio of 2.3 cM/Mb for watermelon reported earlier (Ren et al. 2012a). This suggests that there is an apparent inconsistency between the genetic positions and sequence-based physical positions for a segment on chromosome 11, and such inconsistency could be due to either structural variations (such as an inversion for this part of genome) or more likely incorrect assembly of the reference genome; however, further studies are needed to elucidate this phenomenon.
Segregation distortion in plants is a common phenomenon positively influenced by marker types, mapping population types and the relationships of the parents (Kianian and Quiros 1992; Lorieux et al. 1995). These factors may work simultaneously favoring alleles of the cultivated or wild parent in different genomic regions. In the present study, each bin marker was tested against the expected segregation ratio of 1:2:1 using a Chi squared goodness of fit and a distortion of 53.5 % of the SNP bin markers showing significant (P < 0.05) segregation distortion. Chromosomes 01, 02, 03, 04, 05, 07, 08, 09, 10 and 11 all contained distorted markers with different proportions in favor of cultivar alleles (K3) except chromosome 06. Ren et al. (2014) also reported distorted marker alleles in favor of the cultivar alleles (97103) in cultivar watermelon × wild watermelon population detected on chromosomes 01, 02, 03, 04, 05, 07, 08 and 09 except chromosome 06 and 11. In the present study, 513 (83.3 %) out of 616 distorted segregation markers were skewed toward the cultivated parent, while 103 markers were skewed toward the wild parent, PI 189225. This strong preference of the cultivated parent allele in an interspecific cross was also reported by Ren et al. (2014), who found that all marker alleles within the SDRs on chromosomes 01, 02, 03, 04, 05, 07, 08 and 09 were associated with the cultivated parent and only markers within the SDRs on chromosomes 10 were skewed toward the wild parent PI 296341-FR in their interspecific cross of 97103 (C. lanatus var. lanatus) and PI 296341-FR (C. lanatus var. citroides).
Wild species of C. lanatus var. citroides and C. colocynthis and some germplasms of C. lanatus var. lanatus have been known to be important sources of novel resistance genes that can be used for introgression into elite cultivars of watermelon. Knowledge of the chromosomal locations of segregation distorted loci as well as the underlying mechanism will aid breeders in designing appropriate crossing schemes and predicting the frequency at which a given allele will be transmitted to the progeny. The characterized segregation distorted loci can be used to maximize transmission of desired alleles or preferentially exclude deleterious alleles. In a previous recent study of Ren et al. (2014) on 182 F2 plants derived from ZWRM50 (elite) × PI 244019 (citron), five SDRs were detected on linkage groups 03, 07, 09, 10 and 12. Clustering of distorted markers have also been reported in other watermelon linkage map studies (Hashizume et al. 1996; Hawkins et al. 2001; Zhang et al. 2004; Levi et al. 2006b; Ren et al. 2012a). In the present study, 616 distorted segregation markers were clustered in 44 SDRs on 11 chromosomes; thus, the severe segregation distortion may be caused by genomic differences between C. lanatus var. lanatus and C. lanatus var. citroides.
According to the results of genomic variance analysis of SDRs, the number of SNPs and InDels per Mb for some SDRs was higher than that of the remaining regions of the same chromosome, indicating that these SDRs are general hot spots for genetic variation between C. lanatus var. lanatus and C. lanatus var. citroides. A mixture of dense and sparse SNP and InDel SDRs coexisted on some chromosome, suggesting that SDRs were expected to be randomly distributed throughout the genome.
Male sterility or gametophytic and zygotic factors have been the most commonly reported genetic factors associated with distorted segregation ratio (Harushima et al. 2001; Li et al. 2011) and many segregation distortion regions exist at these genetic loci (Lyttle 1991; Taylor and Ingvarsson 2003). In the present study, two male sterility genes (Cla001678 and Cla001679) were identified from the sequenced genome of watermelon of 97103 (Guo et al. 2013) on chromosome 01 located in the middle of SDR1.1, and it is possible that the two genes were responsible for the segregation distortion. SDR4.1 and SDR5.1 were identified as the two SDRs in which most of the markers were skewed in favor of K3, and few markers (2 out of 43 in SDR4.1 and 2 out of 29 in SDR5.1) were skewed in favor of PI 189225. In contrast, most of the markers in SDR7.1 were skewed in favor of PI 189225 and few markers (7 out of 23) were skewed in favor of K3 (Fig. 5). It is therefore likely that genetic selection factors for gametophyte competition existed on these SDRs. Further studies investigating the segregation distortion on specific regions detected in this interspecific population can elucidate possible underlying mechanisms of the distorted loci.
In the present study, we constructed a genetic map in watermelon (Citrullus lanatus) from an F2 population developed from a cross between feral and cultivated-type watermelon. The utilized map construction method produced large number of SNPs leading to the development of high-density and accurate genetic linkage map. Further characterization identified a number of significantly distorted segregation regions on chromosomes and an inconsistency between their genetic and physical positions on the map for a segment on chromosome 11. The developed high-density and -quality genetic map will facilitate further basic and applied research on the genome structure in this plant species and assist breeding efforts for the selection of agronomically important traits in watermelon.
References
Akhunov E, Nicolet C, Dvorak J (2009) Single nucleotide polymorphism genotyping in polyploid wheat with the Illumina GoldenGate assay. Theor Appl Genet 119:507–517
Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES (2000) An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407:513–516
Arumuganathan K, Earle E (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Report 9:208–218
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376
Boyhan G, Norton J, Abrahams B, Wen H (1994) A new source of resistance to anthracnose (Race 2) in watermelon. HortScience 29:111–112
Brookes AJ (1999) The essence of SNPs. Gene 234:177–186
Chen H, Xie W, He H, Yu H, Chen W, Li J, Yu R, Yao Y, Zhang W, He Y, Tang X, Zhou F, Deng XW, Zhang Q (2014) A high-density SNP genotyping array for rice biology and molecular breeding. Mol Plant 7:541–553
Courtois B, Audebert A, Dardou A, Roques S, Ghneim-Herrera T, Droc G, Frouin J, Rouan L, Gozé E, Kilian A (2013) Genome-wide association mapping of root traits in a japonica rice panel. PLoS One 8:e78037
Cruz VM, Kilian A, Dierig DA (2013) Development of DArT marker platforms and genetic diversity assessment of the U.S. collection of the new oilseed crop lesquerella and related species. PLoS One 8:e64062
Deleu W, Esteras C, Roig C, Gonzalez-To M, Fernandez-Silva I, Gonzalez-Ibeas D, Blanca J, Aranda MA, Arus P, Nuez F, Monforte AJ, Pico MB, Garcia-Mas J (2009) A set of EST-SNPs for map saturation and cultivar identification in melon. BMC Plant Biol 9:90
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Esteras C, Gomez P, Monforte AJ, Blanca J, Vicente-Dolera N, Roig C, Nuez F, Pico B (2012) High-throughput SNP genotyping in Cucurbita pepo for map construction and quantitative trait loci mapping. BMC Genom 13:80
Ganal MW, Altmann T, Röder MS (2009) SNP identification in crop plants. Curr Opin Plant Biol 12:211–217
Ganal MW, Durstewitz G, Polley A, Berard A, Buckler ES, Charcosset A, Clarke JD, Graner EM, Hansen M, Joets J, Le Paslier MC, McMullen MD, Montalent P, Rose M, Schon CC, Sun Q, Walter H, Martin OC, Falque M (2011) A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 6:e28334
Ganal MW, Polley A, Graner E-M, Plieske J, Wieseke R, Luerssen H, Durstewitz G (2012) Large SNP arrays for genotyping in crop plants. J Biosci 37:821–828
Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, Gonzalez VM, Henaff E, Camara F, Cozzuto L, Lowy E, Alioto T, Capella-Gutierrez S, Blanca J, Canizares J, Ziarsolo P, Gonzalez-Ibeas D, Rodriguez-Moreno L, Droege M, Du L, Alvarez-Tejado M, Lorente-Galdos B, Mele M, Yang L, Weng Y, Navarro A, Marques-Bonet T, Aranda MA, Nuez F, Pico B, Gabaldon T, Roma G, Guigo R, Casacuberta JM, Arus P, Puigdomenech P (2012) The genome of melon (Cucumis melo L.). Proc Natl Acad Sci U S A 109:11872–11877
Guo S, Liu J, Zheng Y, Huang M, Zhang H, Gong G, He H, Ren Y, Zhong S, Fei Z, Xu Y (2011) Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. BMC Genom 12:454
Guo S, Zhang J, Sun H, Salse J, Lucas WJ, Zhang H, Zheng Y, Mao L, Ren Y, Wang Z (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45:51–58
Gusmini G, Song R, Wehner TC (2005) New sources of resistance to gummy stem blight in watermelon. Crop Sci 45:582–588
Harel-Beja R, Tzuri G, Portnoy V, Lotan-Pompan M, Lev S, Cohen S, Dai N, Yeselson L, Meir A, Libhaber SE, Avisar E, Melame T, van Koert P, Verbakel H, Hofstede R, Volpin H, Oliver M, Fougedoire A, Stalh C, Fauve J, Copes B, Fei Z, Giovannoni J, Ori N, Lewinsohn E, Sherman A, Burger J, Tadmor Y, Schaffer AA, Katzir N (2010) A genetic map of melon highly enriched with fruit quality QTLs and EST markers, including sugar and carotenoid metabolism genes. Theor Appl Genet 121:511–533
Harushima Y, Nakagahra M, Yano M, Sasaki T, Kurata N (2001) A genome-wide survey of reproductive barriers in an intraspecific hybrid. Genetics 159:883–892
Hashizume T, Shimamoto I, Harushima Y, Yui M, Sato T, Imai T, Hirai M (1996) Construction of a linkage map for watermelon (Citrullus lanatus (Thunb.) Matsum & Nakai) using random amplified polymorphic DNA (RAPD). Euphytica 90:265–273
Hashizume T, Shimamoto I, Hirai M (2003) Construction of a linkage map and QTL analysis of horticultural traits for watermelon [Citrullus lanatus (THUNB.) Matsum & Nakai] using RAPD, RFLP and ISSR markers. Theor Appl Genet 106:779–785
Hawkins LK, Dane F, Kubisiak TL, Rhodes BB, Jarret RL (2001) Linkage mapping in a watermelon population segregating for fusarium wilt resistance. J Am Soc Hortic Sci 126:344–350
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J, Lin K, Jin W, Fei Z, Li G, Staub J, Kilian A, van der Vossen EA, Wu Y, Guo J, He J, Jia Z, Ren Y, Tian G, Lu Y, Ruan J, Qian W, Wang M, Huang Q, Li B, Xuan Z, Cao J, Asan WuZ, Zhang J, Cai Q, Bai Y, Zhao B, Han Y, Li Y, Li X, Wang S, Shi Q, Liu S, Cho WK, Kim JY, Xu Y, Heller-Uszynska K, Miao H, Cheng Z, Zhang S, Wu J, Yang Y, Kang H, Li M, Liang H, Ren X, Shi Z, Wen M, Jian M, Yang H, Zhang G, Yang Z, Chen R, Liu S, Li J, Ma L, Liu H, Zhou Y, Zhao J, Fang X, Li G, Fang L, Li Y, Liu D, Zheng H, Zhang Y, Qin N, Li Z, Yang G, Yang S, Bolund L, Kristiansen K, Zheng H, Li S, Zhang X, Yang H, Wang J, Sun R, Zhang B, Jiang S, Wang J, Du Y, Li S (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Jansen J, De Jong A, Van Ooijen J (2001) Constructing dense genetic linkage maps. Theor Appl Genet 102:1113–1122
Jones N, Ougham H, Thomas H, Pasakinskiene I (2009) Markers and mapping revisited: finding your gene. New Phytol 183:935–966
Joobeur T, Gusmini G, Zhang X, Levi A, Xu Y, Wehner TC, Oliver M, Dean RA (2006) Construction of a watermelon BAC library and identification of SSRs anchored to melon or Arabidopsis genomes. Theor Appl Genet 112:1553–1562
Kianian S, Quiros C (1992) Generation of a Brassica oleracea composite RFLP map: linkage arrangements among various populations and evolutionary implications. Theor Appl Genet 84:544–554
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. In: Data production and analysis in population genomics. Springer, pp 67–89
Kosambi D (1943) The estimation of map distances from recombination values. Annal Eugen 12:172–175
Levi A, Thomas CE, Keinath AP, Wehner TC (2001a) Genetic diversity among watermelon (Citrullus lanatus and Citrullus colocynthis) accessions. Genet Resour Crop Evol 48:559–566
Levi A, Thomas CE, Wehner TC, Zhang X (2001b) Low genetic diversity indicates the need to broaden the genetic base of cultivated watermelon. HortScience 36:1096–1101
Levi A, Thomas C, Joobeur T, Zhang X, Davis A (2002) A genetic linkage map for watermelon derived from a testcross population:(Citrullus lanatus var. citroides × C. lanatus var. lanatus) × Citrullus colocynthis. Theor Appl Genet 105:555–563
Levi A, Davis A, Hernandez A, Wechter P, Thimmapuram J, Trebitsh T, Tadmor Y, Katzir N, Portnoy V, King S (2006a) Genes expressed during the development and ripening of watermelon fruit. Plant Cell Rep 25:1233–1245
Levi A, Thomas C, Trebitsh T, Salman A, King J, Karalius J, Newman M, Reddy O, Xu Y, Zhang X (2006b) An extended linkage map for watermelon based on SRAP, AFLP, SSR, ISSR, and RAPD markers. J Am Soc Hortic Sci 131:393–402
Li X, Wang X, Wei Y, Brummer EC (2011) Prevalence of segregation distortion in diploid alfalfa and its implications for genetics and breeding applications. Theor Appl Genet 123:667–679
Lorieux M, Perrier X, Goffinet B, Lanaud C, De León DG (1995) Maximum-likelihood models for mapping genetic markers showing segregation distortion. 2. F2 populations. Theor Appl Genet 90:81–89
Lyttle TW (1991) Segregation distorters. Annu Rev Genet 25:511–581
Rafalski JA (2002) Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci 162:329–333
Raman H, Raman R, Kilian A, Detering F, Carling J, Coombes N, Diffey S, Kadkol G, Edwards D, McCully M, Ruperao P, Parkin IA, Batley J, Luckett DJ, Wratten N (2014) Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS ONE 9:e101673
Ren R, Wang M, Chen X, Zhang Z (2012a) Characterization and molecular mapping of Yr52 for high-temperature adult-plant resistance to stripe rust in spring wheat germplasm PI 183527. Theor Appl Genet 125:847–857
Ren Y, Zhao H, Kou Q, Jiang J, Guo S, Zhang H, Hou W, Zou X, Sun H, Gong G, Levi A, Xu Y (2012b) A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS One 7:e29453
Ren Y, McGregor C, Zhang Y, Gong G, Zhang H, Guo S, Sun H, Cai W, Zhang J, Xu Y (2014) An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol 14:33
Sandlin K, Prothro J, Heesacker A, Khalilian N, Okashah R, Xiang W, Bachlava E, Caldwell DG, Taylor CA, Seymour DK, White V, Chan E, Tolla G, White C, Safran D, Graham E, Knapp S, McGregor C (2012) Comparative mapping in watermelon [Citrullus lanatus (Thunb.) Matsum. et Nakai]. Theor Appl Genet 125:1603–1618
Sansaloni C, Petroli C, Jaccoud D, Carling J, Detering F, Grattapaglia D, Kilian A (2011) Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. In: BMC Proceedings. BioMed Central Ltd, pp P54
Taylor DR, Ingvarsson PK (2003) Common features of segregation distortion in plants and animals. Genetica 117:27–35
Team RC (2012) R: A language and environment for statistical computing
Tetteh AY, Wehner TC, Davis AR (2010) Identifying resistance to powdery mildew race 2W in the USDA-ARS Watermelon Germplasm Collection. Crop Sci 50:933
Tetteh AY, Wehner TC, Davis AR (2013) Inheritance of resistance to powdery mildew race 2 in Citrullus lanatus var. lanatus. HortScience 48:1227–1230
Van Ooijen J (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res 93:343–349
Voorrips R (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wechter WP, Levi A, Harris KR, Davis AR, Fei Z, Katzir N, Giovannoni JJ, Salman-Minkov A, Hernandez A, Thimmapuram J, Tadmor Y, Portnoy V, Trebitsh T (2008) Gene expression in developing watermelon fruit. BMC Genom 9:275
Zhang R, Xu Y, Yi K, Zhang H, Liu L, Gong G, Levi A (2004) A genetic linkage map for watermelon derived from recombinant inbred lines. J Am Soc Hortic Sci 129:237–243
Zraidi A, Stift G, Pachner M, Shojaeiyan A, Gong L, Lelley T (2007) A consensus map for Cucurbita pepo. Mol Breed 20:375–388
Acknowledgments
The authors wish to thank Damian Jaccoud and Haitao Xiang for providing the SNP data and answering questions about data analysis. This research was supported by Grants from the China Postdoctoral Science Foundation (2013M541624), Jiangsu Province Postdoctoral Science Foundation (1301068B) and the Jiangsu Provincial Major Support Program for Agriculture (BE2012323).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2015_997_MOESM2_ESM.xlsx
Supplementary Table 2. Bin markers used to construct the genetic map of watermelon. Their chromosome, bin position, genetic distance, marker name, bin marker start, bin marker end and bin marker length are listed.(XLS) (XLSX 58 kb)
Rights and permissions
About this article

Cite this article
Ren, R., Ray, R., Li, P. et al. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Genet Genomics 290, 1457–1470 (2015). https://doi.org/10.1007/s00438-015-0997-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-0997-7