
Abstract
Microorganisms associated with marine sponges are potential resources for marine enzymes. In this study, culture-independent metagenomic approach was used to isolate lipases from the complex microbiome of the sponge Ircinia sp. obtained from the South China Sea. A metagenomic library was constructed, containing 6568 clones, and functional screening on 1 % tributyrin agar resulted in the identification of a positive lipase clone (35F4). Following sequence analysis 35F4 clone was found to contain a putative lipase gene lipA. Sequence analysis of the predicted amino acid sequence of LipA revealed that it is a member of subfamily I.1 of lipases, with 63 % amino acid similarity to the lactonizing lipase from Aeromonas veronii (WP_021231793). Based on the predicted secondary structure, LipA was predicted to be an alkaline enzyme by sequence/structure analysis. Heterologous expression of lipA in E. coli BL21 (DE3) was performed and the characterization of the recombinant enzyme LipA showed that it is an alkaline enzyme with high tolerance to organic solvents. The isolated lipase LipA was active in the broad alkaline range, with the highest activity at pH 9.0, and had a high level of stability over a pH range of 7.0–12.0. The activity of LipA was increased in the presence of 5 mM Ca2+ and some organic solvents, e.g. methanol, acetone and isopropanol. The optimum temperature for the activity of LipA is 40 °C and the molecular weight of LipA was determined to be ~30 kDa by SDS-PAGE. LipA is an alkaline lipase and shows good tolerance to some organic solvents, which make it of potential utility in the detergent industry and enzyme mediated organic synthesis. The result of this study has broadened the diversity of known lipolytic genes and demonstrated that marine sponges are an important source for new enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipases, which are defined as carboxylesterases that catalyze both the hydrolysis and synthesis of long-chain acylglycerols (Gupta et al. 2004), are a group of important biocatalysts with uses in organic chemistry, pharmaceutical, food and leather industries, biodegradation of plastics and production of optically active compounds (Andree et al. 1980; Jaeger and Reetz 1998; Gombert et al. 1999; Beisson et al. 2000; Muralidhar et al. 2001; Gupta et al. 2003; Saxena et al. 2003; Kiran et al. 2008; Bajaj et al. 2010). Particularly, lipases have great value to produce biodiesel from biologically derived oil by transesterification (Watanabe et al. 2002; Bajaj et al. 2010) and esterification of free fatty acids.
The challenges of the lipase industry include the discovery of new lipases producers with good properties, for example, high stability of lipase in water-miscible organic solvents is desired in the pharmaceutical industry and for biodiesel production (Glogauer et al. 2011); halo-tolerant lipases are particularly useful for those industrial applications related to the production of marine products (Selvin et al. 2012); alkaline lipases in a detergent should have high activity and stability over a broad range of temperature and pH, and should also be compatible with different components in a detergent including metal ions, surfactants and oxidants (Wang et al. 1995).
The discovery of new esterases and lipases will increase the diversity of lipolytic enzymes and therefore aid the selection of suitable biocatalysts for challenging reactions (Ranjan et al. 2005). Microbial lipases have received a great deal of attention in the field of food technology, pharmaceutical sciences, chemical and detergent industries (Gupta et al. 2004; Jaeger et al. 1999). But it is known that the great majority of microorganisms in the natural environment can’t be cultured (Kennedy et al. 2008), which limits the searching for new enzymes from microorganisms. Thus, metagenomics has become an increasingly important option for finding novel enzymes from complex microbiomes (Hu et al. 2010; Jeon et al. 2009; Kim et al. 2009). For example, a novel cold-adapted alkaline lipase belonging to a new family of lipolytic enzyme was isolated from an intertidal flat metagenome (Kim et al. 2009); 15 different lipolytic genes were obtained from the microbial metagenomic library of the South China Sea marine sediment and two proteins probably representing a novel family of the bacterial lipolytic enzymes were discovered (Hu et al. 2010).
Using metagenomic approach, novel lipolytic enzymes were isolated from sponges Aplysina aerophoba and Hyrtios erecta (Okamura et al. 2010). A lipase with novel halo-tolerance was also isolated from the metagenome of the marine sponge Haliclona simulans (Selvin et al. 2012). Marine ecosystems represent a large and as yet largely unexplored reservoir of biodiversity with respect to industrially useful biocatalysts (Kiran et al. 2014), for example, marine sponge-associated microbes were found to be an important source for marine enzymes such as lipase, urease, protease, cellulase and chitinase (Kiran et al. 2008; Han et al. 2009; Zhang et al. 2009b; Feby and Nair 2010; Shanmughapriya et al. 2010).
In this study, a metagenomic library was constructed from the marine sponge Ircinia sp. from the South China Sea to obtain the lipolytic genes, and the gene was heterologously expressed and the recombinant protein was characterized.
Materials and methods
Sample collection and DNA extraction
Three specimens of the marine sponge Ircinia sp. were collected by scuba diving at a depth of ca. 10–20 m from Yongxing Island (latitude 112°20′E, longitude 16°50′N), South China Sea in the year of 2010. The sponges was transported in an ice-cooled box immediately to the laboratory. The microbes on the sponge surface and inner cavity were removed by washing three times with sterile artificial seawater (ASW) (1.1 g CaCl2, 10.2 g MgCl2·6H2O, 31.6 g NaCl, 0.75 g KCl, 1.0 g Na2SO4, 2.4 g Tris-HCl, 0.02 g NaHCO3, 1 L distilled water, pH 7.6). Then the sponge samples were stored at −20 °C until DNA extraction.
Three sponge cubes (~1 cm2) were dissociated in sterile ASW without Ca2+ and Mg2+ at 110 rpm and 20 °C for 3 h, and then ground into slurry using a mortar and pestle. Sponge slurry was centrifuged at 100×g for 15 min to separate the skeletal components. The supernatant was freeze-dried and ground into powder, which was washed twice with 1 mL TE buffer at 12,000×g for 1 min. DNA was extracted with AllPrep DNA/RNA Mini Kit (Qiagen, Germany). The quantity and purity of DNA was determined respectively by testing the OD260 and the OD260/OD280 with NanoDrop Spectrophotometer ND-1000 (Thermo Fisher Scientific, Wilmington, USA).
Construction of metagenomic library
The obtained DNA was partially digested with Sau 3AI and then separated by 0.5 % agarose gel electrophoresis to get the digested DNA fragments of 2–10 kb. The ligation of the DNA fragments with Bam HI-digested pUC18 (2686 bp, ampicillin-resistant, Takara), which had previously been dephosphorylated with calf intestinal alkaline phosphatase, was performed using T4 DNA ligase (NewEnglandBiolabs) for 10–12 h at 16 °C. The products were transformed into E. coli DH5α. The transformants were plated on LB agar containing 100 μg/mL ampicillin, X-Gal and isopropyl-β-D-thiogalactoside (IPTG). Then the white colonies were selected and transferred into 96-well plates and cultured in LB media containing 100 μg/mL ampicillin (Wei et al. 2009) at 37 °C for 24 h. The library was stored at −80 °C with 20 % glycerin in 96-well plates.
Screening for clones with lipolytic activity and sequence analysis
Clones in the library were replicated onto LB agar plates containing 100 μg/mL ampicillin and 1 % tributyrin, and cultured at 37 °C. The clones with a clear halo around were identified as positive ones. The insert DNA in the positive clones were sequenced with DNA sequencer ABI3730xl (Applied Biosystems). Sequence assembly and editing were performed with the CodonCode Aligner software (CodonCode Corporation, Dedham, MA, USA). The amino acid sequences were compared with the non-redundant sequence database deposited at the NCBI using BLAST (Altschul et al. 1997), and the open reading frames (ORFs) were identified with the ORF Finder tool (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi) (Wheeler et al. 2008). The unrooted phylogenetic tree was constructed with MEGA 5.0. Multiple alignments between protein sequences were performed with the Clustal W program (Thompson et al. 1994). Signal peptide was determined using SignalP 4.0 Server (http://www.cbs.dtu.dk/services/SignalP/) analysis. Secondary structure prediction of the enzyme sequence was carried out using the Predator program (Frishman and Argos 1997). A random forest model was used to predict acidic or alkaline enzyme (Zhang et al. 2009a).
Cloning and expression of lipA gene
The lipA gene without signal peptide sequence was amplified by PCR using the primers: 5′-GGGAATTCATATGGGCTATACCCAAACT-3′ (the Nde I site was underlined) and 5′-GGAAGGGCTCGAGTTATAAGCCTAATTT-3′ (the XhoI site was underlined). The PCR product was digested with NdeI and XhoI, cloned into the NdeI/XhoI digested pET28a (+) (5369 bp, kanamycin-resistant, Novagen), and then transformed into E. coli BL21 (DE3) (Novagen). The inserted gene was examined by DNA sequence analysis. The transformed cells carrying the pET28a (+) vector containing the lipA gene were cultured until the cell density reached OD600 of 0.6 and induced by the addition of IPTG to a final concentration of 1 mM. The induced culture was incubated for a further overnight at 16 °C. E. coli BL21(DE3) cells were then harvested by centrifugation at 4000×g for 20 min, washed twice with 50 mM Tris-HCl buffer (pH 8.0), and disrupted by ultrasonication (80 cycles of 3 s pulses, 200 W, with 5 s intervals). The supernatant was removed by centrifugation (12,000×g for 5 min at 4 °C). The obtained inclusion body was washed with 50 mM Tris-HCl buffer (pH 8.0) and suspended in the same buffer containing 8 M urea and shaken at 100 rpm for 2 h at 4 °C to make it dissolve. After centrifugation under the same conditions (12,000×g, 5 min, 4 °C), the supernatant containing the His-tagged protein was loaded onto a Ni–NTA affinity His-Bind resin column (GE Healthcare Life Sciences) (Zheng et al. 2011) previously equilibrated with washing buffer (50 mM Tris-HCl, pH 8.0, 8 M urea). The protein was eluted using an imidazole step gradient (10, 20, 50, 100, 200 and 500 mM) in washing buffer. Finally, the imidazoles and urea in the purified protein were removed by dialysis in 50 mM Tris-HCl, pH 8.0 containing different concentration of urea (8, 6, 5, 4, 2, 0 M) successively and the protein solution was concentrated by polyethylene glycerol. The purified enzyme was evaluated by 12 % sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) (Laemmli 1970). The protein concentration was determined by the Bradford assay (Bradford 1976).
Analysis of enzymatic properties of LipA
The enzymatic activity of LipA was determined with p-nitrophenyl palmitate (p-NPP) as substrate (Selvin et al. 2012). The substrate solution was prepared by mixing 12 μL stock solution of 4 mM p-NPP in acetonitrile/isopropanol (1/4 v/v) with an assay buffer of 50 mM Tris-HCl (pH 8.0), 0.3 % (v/v) Triton X and 1 mM CaCl2 to a final volume of 230 μL. Then 230 μL of the substrate solution was pipette into a 96-well microtiter plate and the reaction was initiated by the addition of 20 μL of the enzyme solution (50 mM Tris-HCl pH 9.0, 1 mM·CaCl2). The amount of p-nitrophenol released from p-NPP was continuously monitored spectrophotometrically at 405 nm over a period of 10 min at 40 °C. All experiments were performed in triplicate. The extinction coefficients of p-nitrophenol was determined to be 1 × 104 M−1× cm−1 at 405 nm. One unit of enzyme activity was defined as the activity required to release 1 μmol of p-nitrophenol per minute from p-nitrophenyl ester.
The optimum temperature of LipA was determined from 20 to 60 °C. The thermostability of the enzyme was determined by preincubating the enzyme at 4–60 °C for 120 min and then the residual activity was measured at 40 °C.
The activity of the recombinant enzyme was determined between pH 5.0–12.0. The pH stability of the enzyme was tested by preincubating the enzyme for 30 min in three different buffer systems: 20 mM disodiumhydrogen phosphate–citric acid buffer (pH 5.0–8.0), 50 mM Tris-HCl buffer (pH 9.0–10.0), 20 mM glycine-NaOH (pH 11.0–12.0).
To test the effect of metal ions on the enzymatic activity, 5 mM of CaCl2, NaCl, KCl, CuCl2, MgCl2, CoCl2 and EDTA were added (Lee et al. 2006). The effects of organic solvents on the LipA activity were assessed with 15 or 30 % of methanol, ethanol, isopropanol, acetone and dimethyl sulfoxide (DMSO).
Kinetic experiments were carried out with different concentration of various p-nitrophenyl esters: p-nitrophenyl caprylate (C8), p-nitrophenyl decanoate (C10), p- nitrophenyl dodecanoate (C12), p-nitrophenyl myristate (C14), p-NPP (C16), ranging from 0.2 to 3 mM. The Km values were determined by a linear least-squares fitting of a Lineweaver-Burke plot of the Michaelis–Menten equation.
Nucleotide sequence accession number
The lipA gene sequence of 921 bp was deposited in the GenBank database under the accession numbers KC152848.
Results
Screening of gene encoding lipase from the metagenomic library of sponge microbiome
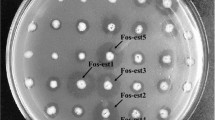
Approximately 6 μg DNA of sponge Ircinia sp. was used for the construction of metagenomic library. Consequently, a metagenomic library including 6568 clones (average insert size 2–10 kb) was constructed. The clones of the whole library were screened for lipolytic activity on 1 % tributyrin plates, and consequently a clone (35F4) with a clear halo around was found (Fig. 1).

Identification of a lipolytic clone, 35F4, that forms a hydrolysis halo in the tributyrin plate assay
The insert DNA (4602 bp) of 35F4 was sequenced. The sequence analysis revealed the presence of 921 bp lipA gene encoding lipase. The translated amino acid sequence of LipA showed 63 % similarity to lactonizing lipase from Aeromonas veronii (WP_021231793) and lactonizing lipase from Aeromonas veronii (WP_005356782), 62 % similarity to triacylglycerol lipase from Glaciecola polaris (WP_007104527), 62 % similarity to lipase from Aeromonas hydrophila ML09-11 (YP_008041659). The phylogenetic analysis showed that LipA is a member of subfamily I.1 and might be novel as it was not clustered with the reference sequences (Fig. 2).

Phylogenetic analysis of 35F4 LipA and closely related proteins. The protein sequences are retrieved from GenBank (NCBI). The phylogenetic tree is generated using MEGA 5. The scale represents the number of amino acid substitutions per site. Open circle represents the closely related proteins with 35F4 LipA; X with dots represents the proteins obtained through metagenomic approach (Glogauer et al. 2011; Lee et al. 2006; Kim et al. 2009; Jeon et al. 2009)
In addition, the sequence alignment showed that it belongs to α/β hydrolase superfamily, with conserved signature sequences (Gly-X-Ser-X-Gly) of lipolytic enzymes in the ORF (101–105, Fig. 3). The 35F4 LipA catalytic triad was predicted to be formed by Ser103, Asp250 and His272 and the nucleophilic Ser residue appears in the conserved pentapeptide Gly-X-Ser-X-Gly. Two Asp residues of 35F4 LipA (Asp232and Asp 274) form a calcium binding pocket that is conserved in lipases of subfamilies I.1 (Arpigny and Jaeger 1999). 35F4 LipA had two Cys residue (Cys205 and Cys256) and they were predicted to form a disulfide bridge (Nardini et al. 2000).

Characteristic residues in multiple sequence alignment between 35F4 LipA and lipases from subfamilies I.1. Conserved residues of the active site (as), cysteine residues forming the disulfide bridge (db) and aspartic residues involved in the calcium binding domains (cb) are annotated. Shaded regions indicate similar amino acids in all the aligned sequences. The accession number and the organism name of the aligned sequences are (from top to the bottom): Lipase from Proteus mirabilis ATCC 29906 (Genebank:ZP_03839679); Lipase from Aeromonas hydrophila ML09-11 (Genebank: YP_00804165); Lipase from Yersinia enterocolitica subsp. Palearctica (Genebank:CBY26912); Lipase from uncultured bacterium (Genebank: AEK97793); Lipase from Pseudomonas fragi [Genebank:CAC07191]; 35F4 LipA; Triacylglycerol lipase from Glaciecola polaris (GeneBank: WP_007104527); Lactonizing lipase from Aeromonas veronii (WP_021231793); Lactonizing lipase from Aeromonas veronii (GeneBank: WP_005356782)
Also, the secondary structure of 35F4 was predicted and the content of amino acid residues in helix, sheet and random coil regions were computed (Zhang et al. 2009a). 35F4 LipA was predicted to be an alkaline enzyme using the random forest model (Zhang et al. 2009a).
The enzymatic characteristics of LipA
After removing signal peptide sequence, LipA was expressed in E. coli BL21 (DE3). However, an insoluble inclusion body was formed. 8 M urea was added to make the inclusion body dissolve, and the recombinant LipA with a C-terminal histidine tag was purified by His-Bind resin affinity chromatography. The protein size was determined to be about 30 kDa by SDS-PAGE (12 %) (Fig. 4).

SDS-PAGE of purified LipA. M Molecular weight markers; 1: Induced cell lysate; 2: Flow through proteins; 3: Purified LipA. The size of the Molecular weight markers is 97.2, 66.4, 44.3, 29.0, 20.1, 14.3 kDa (from top to bottom)
The temperature profile of enzyme activity was showed in Fig. 5a. The enzyme was active over a wide range of temperature, retaining over 50 % of its relative activity between 30 and 55 °C (Fig. 5a) with the optimum temperature at 40 °C. After incubation for 120 min, the LipA retained 90, 42.39, 30.54 and 10 % of relative activity at 4, 40, 50 and 60 °C, respectively (Fig. 5b).

Enzymatic characteristics of LipA. a The effect of temperature on LipA. Enzymatic activity is assayed toward p-NPP (C16) in 50 mM Tris-HCl (pH 9.0) for 10 min; b Thermostability. The thermostability of purified LipA is determined by preincubating the LipA in 50 mM Tris-HCl buffer (pH 9.0) at 4 °C (filled diamond), 40 °C (filled circle), 50 °C (filled triangle), 60 °C (filled square) for 120 min; c The effect of pH on LipA. Enzymatic activity is measured at 40 °C in buffers ranging from pH 5.0–12.0 for 10 min; d The pH stability. The activity is determined after preincubating the LipA at 40 °C for 30 min in three different buffer systems: 20 mM disodiumhydrogen phosphate-citric acid buffer (pH 5.0–8.0), 50 mM Tris-HCl buffer (pH 9.0–10.0), 20 mM glycine-NaOH (pH 11.0–12.0). In each of the panels, the activity under optimum condition is set as 100 %
Over a pH range of 5–12, LipA displayed more than 60 % relative activity at pH values from 7.0 to 10.0, with the maximum activity at pH 9.0 (Fig. 5c). The lipase retained above 60 % of its relative activity after 30 min incubation at pH 7.0–12.0 in the pH stability study (Fig. 5d). These results showed that LipA is an alkaline lipase.
The effect tests of metal ions revealed that the relative activity of LipA was greatly inhibited by the addition of 5 mM Cu2+ and Co2+, respectively (Table 1), and in contrast, the relative activity of LipA was increased to 120.86 and 112.24 % with the addition of 5 mM Ca2+, Mg2+ respectively (Table 1), indicating the LipA activity was stimulated particularly by Ca2+. The activation observed with calcium is consistent with the inhibitory effect of EDTA, which preferentially binds calcium (Table 1). The presence of 5 mM Na+ enhanced LipA activity (Table 1) as well. The most likely explanation for this is it improves the solubility of pNPP within the emulsion. Evidence of this increase in solubility is given by the fact that at 1.5 M NaCl the pNPP emulsion remains transparent even without heating (Glogauer et al. 2011).
The addition of 15 or 30 % methanol or acetone greatly increased the activity of LipA. The increase of enzyme activity was also observed in 15 % isopropanol, while 30 % isopropanol inhibited the activity. In the other two solvents, ethanol and DMSO (15 and 30 %), the activity of LipA was inhibited (Table 2).
The activity of purified LipA was examined by using p-nitrophenyl esters with five different length acyl chains (C8, C10, C12, C14, C16) as substrate. Lipase activities and Km of the enzyme toward different p-nitrophenyl esters were showed in Table 3. The purified enzyme showed the highest activity towards p-nitrophenyl myristate (C14).
Discussion
Lipases which are active and stable in alkaline media have a great potential for applications in the detergent industry (Wang et al. 1995). For example, lipases that will work under alkaline conditions as fat stain removers are desirable (Cherif et al. 2011).
In this study, the obtained lipase (LipA) from the metagenomic library of marine sponge Ircinia sp. was active in the broad alkaline range, with the highest activity at pH 9.0, and had a high level of stability over a pH range of 7.0–12.0 (retaining above 60 % activity), indicating it is an alkaline enzyme. This phenomenon of marine derived lipases preferring alkaline pH as optimum was also reported previously through metagenomic approach and culturing of isolated microorganisms. A new lipase, LipEH 166, isolated from an intertidal flat metagenome had the highest activity at pH 8.0, and maintained more than 80 % activity in the pH range of 5–11 (Kim et al. 2009). Another lipase from the marine bacterium Oceanobacillus sp. PUMB02 was optimally active at pH 8.0 and was stable at alkaline pH (Kiran et al. 2014). Also a detergent compatible lipase produced by marine Bacillus smithii BTMS 11 was found to have pH 8.0 as optimum condition for maximal activity and was active over pH 7.0–10.0 (Lailaja and Chandrasekaran 2013). These alkaline lipases have desirable properties with regard to pH optimum and pH stability for a potential application in the detergent industry, supporting the faith that marine ecosystem is a large and as yet largely unexplored reservoir of biodiversity with respect to industrially useful biocatalysts.
Because enzyme instability at pH extreme is one of the main bottlenecks in extending the application of it, the stability mechanism of acidic and alkaline enzymes is quite important and interesting not only academically but also industrially. The stability of acidic and alkaline enzymes had been studied in the biophysical and biotechnological research areas, and it has been investigated whether acidophily and alkaliphily can be detected at the amino acid level (Dubnovitsky et al. 2005; Kelch et al. 2007; Geierstanger et al. 1998; Settembre et al. 2004; Shirai et al. 1997). It was proposed that the propensity of the individual residues to participate in different secondary structures might be a general stability mechanism for their adaptation to pH extremes. Base on it, a random forest model in conjunction with a feature named secondary structure amino acid composition was used to discriminate acidic and alkaline enzymes (Zhang et al. 2009a). Using this model, LipA was predicted to be an alkaline enzyme, which is consistent with the experimental data, supporting the proposed adaptation mechanism of enzymes to pH extremes. The new lipase could be further used to achieve a better understanding of enzyme mechanism and structure–function relationships.
LipA activity was found to be enhanced in the presence of Ca2+, which was similar to the increases observed in the lipases from the metagenome of the marine sponge Haliclona simulans (Selvin et al. 2012) and the lipase isolated from the tidal flat sediment metagenomic library (Lee et al. 2006). Ca2+ ion is regarded as a ligand between amino acid residues in the enzyme active site (Salameh and Wiegel 2007) and the electrostatic interaction between calcium and fatty acids produced by the hydrolysis of the substrate leads to the “clearing” of the active site thereby allowing another substrate molecule to access the site (Voget et al. 2003). Similarly, LipC12 belonging to the subfamily I.1 of bacterial lipases was isolated through metagenomic approach and found to be calcium ion dependent (Glogauer et al. 2011).
In this study, the LipA activity was increased in the presence of organic solvents, such as methanol, isopropanol and acetone. Similarly, the stability of the lipase (Lpc53E1) from the marine sponge was increased upon the exposure to solvents, e.g. methanol, isopropanol and acetone (Selvin et al. 2012). Generally, enzymes, being proteins, lose their activity at organic co-solvent concentrations higher than 10–20 % (Gupta et al. 1997). Stability and activity of enzymes in organic solvents depend not only on the properties and concentration of the organic solvents, but also on the nature of the enzymes (Torres and Castro 2004). It is quite well known that the lipase activity can be increased in the presence of organic solvents, for example, Candida rugosa lipase is activated by organic solvents, which keep the lid of the enzyme in the open conformation, facilitating the access of the substrate to the active site (Grochulski et al. 1993; Colton et al. 1995). Thus the increased LipA activity suggests that the enzyme may possess a lid that is converted into an open conformation in the presence of organic solvents. These findings are particularly significant, due to the fact that organic solvents have been used in biodiesel production through biocatalysis (Glogauer et al. 2011). It was observed that B. smithii lipase with enhanced activity in the presence of 1–5 % organic solvents could effectively catalyze methyl-ester synthesis between fatty acids and methanol to obtain methyl esters of long-chain fatty acids which could be used as diesel fuels (Lailaja and Chandrasekaran 2013). In the present study, the activity of LipA was enhanced in 30 % methanol or 30 % acetone, suggesting that the enzyme holds potential for application in transesterification and ester synthesis.
LipA was found to have substrate specificity for p-nitrophenyl myristate (C14) followed by p-nitrophenyl dodecanoate (C12), p-nitrophenyl decanoate (C10) and p-NPP (C16). A low Km value represents a high affinity. For most industrially relevant enzymes, Km values range between 10−1 and 10−5 M, although the Km values of the enzyme generally vary widely (Fullbrook 1996). The marine derived B. smithii lipase had a value of 0.1 mM for the hydrolysis of p-nitrophenyl butyrate (Lailaja and Chandrasekaran 2013). In this study, the reaction kinetics of LipA with p-nitrophenyl esters with different length acyl chains indicated that the enzyme had the lowest Km value of 0.42 mM for the hydrolysis of p-nitrophenyl myristate (C14).
In conclusion, a metagenomic library of marine sponge Ircinia sp. from South China Sea was constructed. A lipolytic-active positive clone, 35F4, was discovered using an activity-based method from the metagenomic library of marine sponge Ircinia sp. The sequence analysis revealed that the predicted amino acid sequence displayed 63 % similarity to the lactonizing lipase from Aeromonas veronii (WP_021231793). Alignment of the protein sequence of LipA with known lipase families showed that LipA is a member of subfamily I.1 lipases. And LipA was predicted to be an alkaline enzyme by sequence/structure analysis, which was consistent with the characterization of the heterologously expressed enzyme that LipA was active and stable in the broad alkaline pH range. LipA showed high tolerance to some organic solvents, including methanol, acetone and isopropanol. So LipA has the potential applications in the detergent industry and enzyme mediated organic synthesis and could be used to understand the mechanism of acidophily and alkaliphily of enzymes. This study demonstrated that marine sponges are an important source for new enzymes.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Andree H, Mueller WR, Schmid RD (1980) Lipases as detergent components. J Appl Biochem 2:218–229
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Bajaj A, Lohan P, Jha PN, Mehrotra R (2010) Biodiesel production through lipase catalyzed transesterification: an overview. J Mol Catal B Enzym 62:9–14
Beisson F, Tiss A, Rivière C, Verger R (2000) Methods for lipase detection and assay: a critical review. Eur J Lipid Sci Technol 102:133–153
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cherif S, Mnif S, Hadrich F, Abdelkafi S, Sayadi S (2011) A newly high alkaline lipase: an ideal choice for application in detergent formulations. Lipids Health Dis 10:221
Colton I, Ahmed S, Kazlauskas R (1995) A 2-propanol treatment increases the enantioselectivity of Candida rugosa lipase toward esters of chiral carboxylic-acids. J Org Chem 60:212–217
Dubnovitsky AP, Kapetaniou EG, Papageorgiou AC (2005) Enzyme adaptation to alkaline pH: atomic resolution (1.08 Å) structure of phosphoserine amino-transferase from Bacillus alcalophilus. Protein Sci 14:97–110
Feby A, Nair S (2010) Sponge-associated bacteria of Lakshadweep coral reefs, India: resource for extracellular hydrolytic enzymes. Adv Bios Biotechnol 1:330–337
Frishman D, Argos P (1997) Seventy-five percent accuracy in protein secondary structure prediction. Proteins 27:329–335
Fullbrook PD (1996) Kinetics. In: Godfrey T, Reichelt J (eds) Industrial enzymology: the application of enzymes in industry, 2nd edn. Nature, New York
Geierstanger B, Jamin M, Volkman BF, Baldwin RL (1998) Protonation behavior of histidine 24 and histidine 119 in forming the pH 4 folding intermediate of apomyoglobin. Biochemistry 37:4254–4265
Glogauer A, Martini V, Faoro H, Couto G, Muller-Santos M, Monteiro R, Mitchell D, Souza E, Pedrosa F, Kireger N (2011) Identification and characterization of a new true lipase isolated through metagenomic approach. Microb Cell Fact 10:54
Gombert AK, Pinto AL, Castilho LR, Freirc DMG (1999) Lipase by Penicillium restrictum in solid state fermentation using Babassu oil cake as substrate. Process Biochem 35:85–90
Grochulski P, Li Y, Schrag J, Bouthillier F, Smith P (1993) Insights into interfacial activation from an open structure of Candida rugosa lipase. J Biol Chem 268:12843
Gupta M, Batra R, Tyagi R, Sharma A (1997) Polarity index: the guiding solvent parameter for enzyme stability in aqueous-organic cosolvent mixtures. Biotechnol Prog 13:284–288
Gupta R, Rathi P, Gupta N, Bradoo S (2003) Lipase assays for conventional and molecular screening: an overview. Biotechnol Appl Biochem 37:63–71
Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Appl Mocrobiol Biotechnol 64:763–781
Han Y, Yang B, Zhang F, Miao X, Li Z (2009) Characterization of antifungal chitinase from marine Streptomyces sp. DA11 associated with South China Sea sponge Craniella australiensis. Mar Biotechnol 11:132–140
Hu Y, Fu C, Huang Y, Yin Y, Cheng G, Lei F, Lu N, Li J, Ashforth E, Zhang L, Zhu B (2010) Novel lipolytic genes from the microbial metagenomic library of the South China Sea marine sediment. FEMS Microbiol Ecol 72:228–237
Jaeger KE, Reetz MT (1998) Microbial lipases form versatile tools for biotechnology. Trends Biotechnol 16:396–403
Jaeger KE, Dijkstra BW, Reetz MT (1999) Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol 53:315–351
Jeon JH, Kim JT, Kim YJ, Kim HK, Lee HS, Kang SG, Kim SJ, Lee JH (2009) Cloning and characterization of a new cold-active lipase from a deep-sea sediment metagenome. Appl Microbiol Biotechnol 81:865–874
Kelch BA, Eagen KP, Erciyas FP, Humphris EL, Thomason AR, Mitsuiki S (2007) Structural and mechanistic exploration of acid resistance: kinetic stability facilitates evolution of extremophilic behavior. J Mol Biol 368:870–883
Kennedy J, Marchesi JR, Dobson ADW (2008) Marine Metagenomics: strategies for the discovery of novel enzymes with biotechnological applications from marine ecosystems. Microb Cell Fact 7:27
Kim E, Oh K, Lee M, Kang C, Oh T, Yoon J (2009) Novel cold-adapted alkaline lipase from an intertidal flat metagenome and proposal for a new family of bacterial lipases. Appl Environ Microbiol 75(1):257–260
Kiran GS, Shanmughapriya S, Jayalakshmi J, Selvin J, Gandhimathi R, Sivaramakrishnan S, Arunkumar M, Thangavelu T, Natarajaseenivasan K (2008) Optimization of extracellular psychrophilic alkaline lipase produced by marine Pseudomonas sp. (MSI057). Bioprocess Biosyst Eng 31:483–492
Kiran GS, Lipton AN, Kennedy J, Dobson ADW, Selvin J (2014) A halotolerant thermostable lipase from the marine bacterium Oceanobacillus sp. PUMB02 with an ability to disrupt bacterial biofilms. Bioengineered 5(5):305–318
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lailaja VP, Chandrasekaran M (2013) Detergent compatible alkaline lipase produced by marine Bacillus smithii BTMS 11. World J Microbiol Biotechnol 29:1349–1360
Lee M-H, Lee CH, Oh T-K, Song JK, Yoon J-H (2006) Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: evidence for a new family of bacterial lipases. Appl Environ Microb 72:7406–7409
Muralidhar RV, Marchant R, Nigam P (2001) Lipases in racemic resolutions. J Chem Technol Biotechnol 76:3–8
Nardini M, Lang DA, Liebeton K, Jaeger KE, Dijkstra BW (2000) Crystal structure of Pseudomonas aeruginosa lipase in the open conformation. The prototype for family I.1 of bacterial lipases. J Biol Chem 275:31219–31225
Okamura Y, Kimura T, Yokouchi H, Meneses-Osori M, Katoh M, Matsunga T, Takeyama H (2010) Isolation and characterization of a GDSL esterase from the metagenome of a marine sponge-associated bacteria. Mar Biotechnol 12:395–402
Ranjan R, Grover A, Kapardar RK, Sharma R (2005) Isolation of novel lipolytic genes from uncultured bacteria of pond water. Biochem Bioph Res Co 335:57–65
Salameh MA, Wiegel J (2007) Purification and characterization of two highly thermophilic alkaline lipases from Thermosyntropha lipolytica. Appl Environ Microb 73:7725–7731
Saxena RK, Sheoran A, Giri B, Davidson S (2003) Purification strategies for microbial lipases. J Microbiol Meth 52:1–18
Selvin J, Kennedy J, Lejon D, Kiran G, Dobson A (2012) Isolation identification and biochemical characterization of a novel halo-tolerant lipase from the metagenome of the marine sponge Haliclona simulans. Microb Cell Fact 11:72
Settembre EC, Chittuluru JR, Mill CP, Kappock TJ, Ealick SE (2004) Acidophilic adaptations in the structure of Acetobacter aceti N5-carboxyaminoimidazole ribonucleotide mutase (PurE). Acta Crystallogr D Biol Crystallogr 60:1753–1760
Shanmughapriya S, Kiran GS, Selvin J, Thomas TA, Rani C (2010) Optimization, purification, and characterization of extracellular mesophilic alkaline cellulase from sponge-associated Marinobacter sp. MSI032. Appl Biochem Biotechnol 162:625–640
Shirai T, Suzuki A, Yamane T, Ashida T, Kobayashi T, Hitomi J (1997) High resolution crystal structure of M-protease: phylogeny aided analysis of the high-alkaline adaptation mechamism. Protein Eng 10:627–634
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Torres S, Castro G (2004) Non-aqueous biocatalysis in homogenous solvent systems. Food Technol Biotechnol 42(4):271–277
Voget S, Leggewie C, Uesbeck A, Raasch C, Jaeger KE, Streit WR (2003) Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol 69(10):6235–6242
Wang Y, Srivastava KC, Shen G-J, Wang HY (1995) Thermostable alkaline lipase from a newly isolated thermophilic Bacillus, strain A30-1 (ATCC 53841). J Ferment Bioeng 79:433–438
Watanabe Y, Shimada Y, Sugihara A, Tominaga Y (2002) Conversion of degummed soybean oil to biodiesel fuel with immobilized Candida antarctica lipase. J Mol Catal B Enzym 17:151–155
Wei P, Bai L, Song W, Hao G (2009) Characterization of two soil metagenome-derived lipases with high specificity for p-nitrophenyl palmitate. Arch Microbiol 191:233–240
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo LYG, Helberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko R, Tatusov L, Tatusova TA (2008) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36:D13–D21
Zhang GY, Li HC, Fang BS (2009a) Discriminating acidic and alkaline enzymes using a random forest model with secondary structure amino acid composition. Process Biochem 44(6):654–660
Zhang H, Zhang F, Li Z (2009b) Gene analysis, optimized production and property of marine lipase from Bacillus pumilus B106 associated with South China Sea sponge Halichondria rugosa. World J Microb Biotechol 25:1267–1274
Zheng X, Chu X, Zhang W, Wu N, Fan Y (2011) A novel cold-adapted lipase from Acinetobacter sp.XMZ-26: gene cloning and characterization. Appl Microbiol Biotechnol 90:971–978
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) (31000062).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Su, J., Zhang, F., Sun, W. et al. A new alkaline lipase obtained from the metagenome of marine sponge Ircinia sp.. World J Microbiol Biotechnol 31, 1093–1102 (2015). https://doi.org/10.1007/s11274-015-1859-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-015-1859-5