
Abstract
Using a metagenome library constructed from a bacterial associated with a marine sponge Hyrtios erecta, we identified a novel esterase that belongs to the SGNH hydrolase superfamily of esterases. The substrate specificity of EstHE1 was determined using p-nitrophenyl (pNP) ester (C2: acetate, C4: butylate, C6: caproate, C12: laurate, C16: palmitate). EstHE1 exhibited activity against C2 (5.6 U/mg), C4 (5.1 U/mg), and C6 (2.8 U/mg) substrates. The optimal temperature for EstHE1 esterase activity of the pNP acetate substrate was 40°C, and EstHE1 retained 60% of its enzymatic activity in the 30–50°C range. This esterase showed moderate thermostability, retaining 58% of its activity even after preincubation for 12 h at 40°C. EstHE1 also maintained activity in high concentrations of NaCl, indicating that this esterase is salt-tolerant. Thus, EstHE1 has the thermal stability and salt tolerance necessary for use as an industrial enzyme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Esterases and lipases are often used for the resolution of racemic mixtures, which are solutions containing equal amounts of left- and right-handed enantiomers of a chiral molecule, as well as for the synthesis of important ester compounds of pharmaceutical, food, biochemical, and biological interests. Thus, newly found esterases and lipases are potentially useful for industrial processes. Many esterases and lipases contain the pentapeptide motif GXSXG; however, not all of lipolytic enzymes have this motif (Akoh et al. 2004). GDSL motif is seen in a new subfamily of hydrolytic/lipolytic enzymes (Upton and Buckley 1995). GDSL hydrolases exhibit multifunctional properties and broad substrate specificity due to the flexibility of the active site that undergoes an induced-fit style conformational change upon substrate binding (Koshland 1958). GDSL enzymes all have a consensus sequence that is divided into five conserved sequence blocks (I–V), and the GDSL motif is in conserved block I. The GDSL family is further classified based on the strict conservation of the catalytic residues S, G, N, and H within conserved blocks I, II, III, and V, respectively (Dalrymple et al. 1997; Li et al. 2000; Molgaard et al. 2000). These four residues are the namesake of the SGNH superfamily (Ser, Gly, Asn, and His; SGNH) and utilize a different catalytic mechanism for hydrolase activity from that of common α/β-hydrolases (Lo et al. 2003). The catalytic triad of the common SGNH hydrolases is shaped by the Ser in the GDSL motif, the Asp, and the His in conserved block V (Dalrymple et al. 1997; Li et al. 2000; Lo et al. 2003). While SGNH hydrolases are well represented in eukaryotic organisms, the isolation and characterization of SGNH hydrolases from bacteria have been limited (Cho and Cronan 1993; Lo et al. 2003).
The screening of microorganisms for enzymes that may be useful in industrial processes is a common practice. However, standard culturing techniques support the growth of less than 1% of the bacterial found in the environment (Giovannoni et al. 1990). For this reason, metagenome analyses have gained popularity for the screening and identification of candidate industrial-use enzymes. Metagenomic approaches consist of the construction of metagenomic libraries by direct extraction and cloning of DNA from environmental samples and have opened the way to access gene sources of unknown or noncultivatable bacteria. To overcome the limitations of standard culture techniques, we have developed a metagenome preparation method from a limited number of environmental samples by multiple displacement amplification using ϕ29 phage polymerase (Yokouchi et al. 2006).
We are especially interested in the sponge metagenome. Marine sponges are filter feeders capable of processing thousands of liters of sea water per kilogram of sponge per day (Reiswig 1974). During this process, many microorganisms are taken up and digested by phagocytosis or harbored as a symbiont consortia. To date, many novel compounds from marine sponge have been discovered, and the symbiotic bacteria living within the sponge tissue may contribute in the production of bioactive compounds (Hentschel et al. 2001). Bacteria can sometimes reach 40% of the sponge tissue mass, suggesting that the contribution of the bacteria can be substantial (Vacelet and Donadey 1977). Phylogenetic analyses of these sponge symbiotic bacterial communities revealed that most of them are noncultivatable (Webster et al. 2001), so developing the production of potentially useful compounds on an industrial scale is challenging.
In this study, we found an esterase of the SGNH hydrolase superfamily in the metagenome library of a bacterial genome that is associated with the Hyrtios erecta marine sponge, collected from Okinawa. The catalytic activity of this newly identified esterase was stable against high-temperature and high-salt concentrations. Further, since the SGNH hydrolase superfamily contains few esterases, none of which exhibit high-thermostability or high-salt tolerance, this enzyme represents a novel member of the SGNH hydrolases (Lo et al. 2003). The unique biochemical properties of this enzyme suggest that it could be a valuable industrial tool. Further, the use of metagenomes is a useful resource to identify previously undiscovered enzymes.
Materials and Methods
Sponges, Bacterial Strains, and Vectors
Marine sponges H. erecta were collected from off the coast of Ishigaki Island, Okinawa, Japan. Escherichia coli strain DH10B (Invitrogen), strain EPI300 (Epicentre), and strain BL21(DE3) (Novagen) were used for library construction, cloning, and recombinant protein expression, respectively. The cloning vector pHSG398 (accession number M19087), which is 2,227 bp and carries the chloramphenicol resistance gene (Takara, Shiga, Japan), was used for library preparation. The expression vector pET22b (5,493 bp, ampicillin-resistant; Novagen) was utilized for purifying the recombinant enzyme.
Preparation of Sponge-Associated Bacterial Cells and DNA Extraction
H. erecta was homogenized with a mortar and pestle in TNE buffer (10 mM Tris, 3.5% NaCl, 50 mM EDTA, pH 7.5), and the sponge tissue was removed by filtration through a 100-µm mesh. Nuclei were removed from the filtrate by centrifugation at 700×g for 5 min. Bacterial cells were harvested from the resulting supernatant by centrifugation at 7,000×g for 10 min and washed three times with TNE buffer. DNA was then extracted from the bacteria according to the following method: collected cells (approximately 300 mg) were resuspended in 10 mL TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) containing 500 mg lysozyme and incubated for 2 h at 37°C. Then, 1.4 mg Proteinase K and 10 mg RNase were added and incubated for additional 1 h. To this mixture, 3.2 ml of 10% sodium dodecyl sulfate (SDS) was added and incubated at 37°C overnight. Then, 11 ml of 5 M NaCl and 8 ml of cetyl trimethylammonium bromide (CTAB)/NaCl solution (10% CTAB in 0.7 M NaCl) were added to the mixture and incubated at 65°C for 20 min. DNA was separated from the insoluble material by centrifugation at 10,000×g for 10 min, which resulted in the enrichment of DNA in the supernatant. DNA was purified by two standard phenol-chloroform extractions. The extracted DNA (metagenome DNA) was purified twice using cesium chloride density-gradient centrifugation at 55,000×g for 16 h. The resulting DNA was dialyzed for 24 h to remove the cesium chloride.
Library Construction
Blunted and 5′-phosphorylated metagenome DNA was size-separated by pulsed-field gel electrophoresis under the following conditions (1% LMP agarose/1×TBE gel, 1× TBE running buffer, 0.5 s pulse, 6 V/cm, 14°C, 120°, 10 h) and approximately 3–9.5 kb of DNA was excised from gel and extracted using GELase (Epicentre). DNA fragments purified were ligated into the HincII restriction site of the pHSG398 vector, and the resulting ligation reaction was used to transform DH10B. The transformations were plating on LB agar containing 12.5 µg/ml chloramphenicol, and colonies were picked by BioPick (GENOMIC SOLUTIONS). The library was stored at −80°C in 96-well microtiter plates.
Screening for Lipolytic Clones and Sequence Analysis
Screening the metagenome library for clones carrying lipolytic activity was performed by halo formation of fatty acid salts on 1.5% agar plates containing 1 mM isopropyl thio-β-galactoside (IPTG), 0.5% Tween-20, and 0.01% CaCl2 according to the method of Rondon et al. (2000).
Clones that exhibited lypolytic activity were sequenced by automatic DNA sequencer ABI PRISM 3100 (Applied Biosystems) and analyzed using the GENETYX 9.0 (GENETYX CORPORATION, Tokyo, Japan). Homology searches were performed using BLAST analysis (McGinnis and Madden 2004) against the GenBank DNA databases. Regions of sequence homology were identified by multiple alignments using the ClustalW program (Thompson et al. 1994).
Protein Expression and Purification
The esterase gene, without the signal peptide sequence, was amplified by polymerase chain reaction (PCR) using forward primers that contain an NcoI site (5′-CCATGGATAGGGATGCGCCCGTATTG-3′) and reverse primers that contain a HindIII site (5′-AAGCTTATGGTCAAGTGGAGCACCCTGTTC-3′), and the resulting PCR product was ligated into the NcoI/HindIII site of downstream of lac promoter in pET22b (5,493 bp, ampicillin-resistant; Novagen). Correct insertion of the gene was confirmed by sequencing, and the vector was then introduced into BL21(DE3) E. coli. The transformed cells were cultured at 37°C in the presence of 1 mM IPTG to induce esterase expression. The cells were harvested 5 h post induction, resuspended in 100 mM phosphate buffer (pH 7.4), and disrupted by sonication. Recombinant protein was purified using Ni-NTA Agarose (Qiagen) according to manufacturer’s instructions. Imidazole in the eluted fraction was removed by dialysis against a 100-mM phosphate buffer (pH 7.4). Enzyme purity was analyzed using 12.5% SDS-polyacrylamide gel electrophoresis, visualized by Coomasie brilliant blue R-250 staining, and confirmed by immunoblotting using HisDetector Western Blot Kit and HRP Colorimetric (Kirkegaard & Perry Laboratories, Inc., Washington, D.C).
Esterase Activity Assay
Purified enzyme was added to 200 µl of reaction mixture consisting of 2 mM p-nitrophenyl ester (pNP ester) as a substrate and 0.5% DMSO in 100 mM phosphate buffer, pH 7.4 and incubated at 37°C. The amount of free p-nitrophenol was measured by absorbance at 410 nm using a spectrophotometer. One unit of enzyme activity was defined as 1 μmol of free p-nitrophenol generation in 1 min. The substrate specificity of the esterase was determined using pNP esters of acetate (C2), butyrate (C4), caproate (C6), laurate (C12), and palmitate (C16) according to the same method. The optimal temperature for esterase activity was determined by examining the enzyme activity at temperatures ranging from 20–60°C using pNP acetate as a substrate. The thermostability of the esterase was determined by preincubating the enzyme at 40°C for various intervals, up to 12 h, and then assessing the esterase activity using pNP acetate as a substrate. Effect of NaCl concentration on esterase activity was measured using pNP acetate as a substrate in reaction mixtures containing NaCl concentrations ranging from 0 to 3.8 M. All measurements were performed in triplicate.
Esterase Gene Accession Number
The nucleotide sequence of estHE1 gene has been assigned GenBank accession number AB432912.
Results
Screening the Metagenome Library for Clones with Esterase/Lipase Activity
The metagenome library constructed contained 26,496 clones, the majority of which contained inserts of 3 to 5 kb. The whole library contained 79.5–180 Mb of total sequence. Esterase/lipase activity of one clone was identified by growth of the bacteria on an agar plate containing Tween-20 and CaCl2 to form fatty acid salts. When this clone was grown on media containing tributyrin, no activity was observed, indicating that the encoded enzyme had esterase, but not lipase activity.
Genetic Characterization
The plasmid DNA was extracted from the esterase-positive clone, and the insert was sequenced by primer walking. The insert was approximately 3.4 kb and contained four putative ORFs. The third putative ORF had 54% sequence identity with arylesterase from Marinobacter aquaeolei VT8 (Fig. 1). This gene was designated estHE1. Alignment of the EstHE1 sequence with several other esterases revealed that EstHE1 had the characteristic GDSLS consensus sequence within its N terminus and five distinct blocks of sequence homology to the GDSL subfamily of esterases (Fig. 2). Moreover, the highly conserved catalytic residues S, G, N, and H were found in conserved blocks I, II, III, and V, respectively. This means that EstHE1 belongs to SGNH hydrolases superfamily. Additionally, a signal peptide (Met1-Ala29) was identified at the N terminus of EstHE1 by the SignalP 3.0 server (http://www.cbs.dtu.dk/services/SignalP/), suggesting that EstHE1 would be secreted and function outside of the cell.

Arrangement of the open reading frames in the DNA insert from the esterase positive clone (a) and homology analysis of each ORF (b). ORFs were identified using the Genetyx software, and homology search was performed using BLAST

Sequence alignment of EstHE1 with the GDSL family of esterases. Apart from the EstHE1 sequence (this work), the latter sequences are designated according to bacterial GDSL family esterases in NCBI (M. aquaeolei VT8 (YP_958323), Pseudomonas mendocina ymp (YP_001188046), H. halophila SL1 (YP_001003634), Delftia acidovorans SPH-1 (YP_001565051)). Identical residues are shown in the black boxes. Closed inverted triangles indicate amino acid residues conserved in the catalytic triad. The four consensus blocks I, II, III, and V of the SGNH subfamily are boxed
Expression and Purification of His-Tagged EstHE1
For biochemical characterization of EstHE1 esterase activity, a His-tagged EstHE1 was constructed and expressed in E. coli. The functional domain of EstHE1, without the signal sequence, was fused to downstream of the pelB signal sequence to direct periplasmic localization in the E. coli. The recombinant protein was partially purified by Ni-NTA column chromatography (Fig. 3a), and the identity of the eluted protein was confirmed by immunoblot analysis (Fig. 3b). The contaminant proteins in the EstHE1 preparation were nonspecifically co-purified, because they were also observed in the preparations from E. coli harboring the control vector (Fig. 3a). These contaminant proteins exhibited no esterase activities. The purified His-tagged EstHE1 was used for the characterization of EstHE1 esterase activity.

Expression and purification of recombinant His-tagged EstEH1. a SDS-PAGE analysis and b immunoblot analysis. Lane M marker; lane 1 purified and denatured EstEH1, lane 2 purified and denatured proteins of E. coli ( pET22b vector ), lane 3 immunoblot of purified and denatured EstEH1
Activity of Ester Hydrolysis of EstHE1 on Fatty Acid Variant Substrate
The substrate specificity of EstHE1 was determined using pNP-fatty acid substrates. Results are shown in Table 1. The activity is represented as the specific activity (EU/mg) using the total protein amount (including impurities). EstHE1 activity preferred short chain-fatty acid substrates (C6>). However, triglycerides and tributyrin (C4) were not hydrolyzed (data not shown). Based on substrate specificity, EstHE1 is an esterase but not a lipase.
Optimum Temperature and Thermostability of EstHE1
To determine the optimal temperature for esterase activity and the thermostability of EstHE1, pNP-acetate was used for a substrate. The temperature profile of enzymatic activity is presented in Fig. 4. The optimal temperature for EstHE1 activity was near 40°C; however, the enzyme was active over a wide range of temperature, retaining over 50% of its relative activity between 25 and 55°C. To test the thermostability of EstHE1, it was incubated at 40°C, and the retention of esterase activity was assayed. EstHE1 retained more than 80% of its relative activity after 3 h at 40°C, and 58% of its relative activity even after 12 h at 40°C (Fig. 5).

Effect of temperature on the activity of EstHE1. The enzymatic assay was performed at the indicated temperature for 5 min in 100 mM phosphate buffer (pH 7.4), using 2 mM p-nitrophenyl acetate as a substrate

Thermal stability of the purified EstHE1. After preincubation at 40°C for the indicated time, the enzymatic assay was performed at 40°C for 10 min in 100 mM phosphate buffer (pH 7.4), using 2 mM p-nitrophenyl acetate as a substrate. All measurements were performed in triplicate
Effect of Salt on EstHE1 Esterase Activity
Since EstHE1 is derived from bacteria that live in a marine environment, it is reasonable to think that this enzyme may be relatively stable in high-salt conditions. The stability of EstHE1 esterase activity was tested in various NaCl concentrations. Esterase activity of EstHE1 was inhibited by increasing the salt concentration up to 1.9 M NaCl. In the presence of 1.9 M NaCl, EstHE1 esterase activity was reduced to 55%, as compared to the no-NaCl condition. Interestingly, as the salt concentration increases from 1.9 to 3.8 M, the esterase activity recovered slightly (Fig. 6). At 3.8 M NaCl, the activity was approximately 62% of the no-NaCl condition. These results indicate that EstHE1 exhibits not only halotolerance but also reactivation by higher concentration of salt similar to the actinidain esterase found in kiwifruit (Morimoto et al. 2006).

The effect of the NaCl concentrations on EstHE1 activity. The enzymatic assay was performed at 37°C in 100 mM phosphate buffer (pH 7.4) supplemented with the indicated NaCl concentration using 2 mM p-nitrophenyl acetate as a substrate. All measurements were performed in triplicate
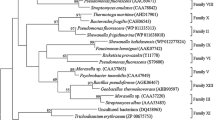
Phylogeny of EstHE1
Currently, there are only a few examples of the GDSL subfamily of serine esterases/lipases. To evaluate novelty of this esterase, the amino acid sequence of EstHE1 was analyzed by sequence alignment. Phylogenetic analysis showed that EstHE1 clustered into the same clade as an unknown marine γ-proteobacterium and Halorhodospira halophila, which is classified in a subdivision of γ-proteobacteria (Fig. 7). The high-salt tolerance of EstHE1 might be explained by its homology with the esterase of H. halophila. Of the esterases/lipases shown in Fig. 7, the enzymatic activities of all of these enzymes have not been evaluated experimentally. Thus, EstHE1 would never have been identified without the use of PCR-based metagenome analysis and screening for esterase activity.

Phylogenetic relationships between EstHE1 and other members of the GDSL family of serine esterases/lipases. Box indicates the isolated esterase from metagenome library
Discussion
Industrial processes require the development of enzymes with strict reaction characteristic. Therefore, recombinant enzymes were rigorously developed by protein engineering or evolutionary molecular engineering such as gene shuffling (Cherry and Fidantsef 2003). These enzymes have been modified to alter substrate recognition (Fukuda et al. 2007), cofactor requirement (Watanabe et al. 2007), pH dependence (Gibbs et al. 2001), or calcium dependence (Hashida and Bisgaard-Frantzen 2000) to fit specific reaction conditions. However, multiple modifications of an enzyme do not always lead to synergistic effects on the enzymatic activity. The engineering of enzymes often results in the loss of the native enzymatic activity. Rather than selective engineering of a recombinant enzyme to obtain a specific type of activity, classical and comprehensive screening techniques to identify and to clone new enzymes is a viable alternative. Further, if the correct screening techniques are used, then no further engineering would be necessary because the desired enzyme would have already undergone evolutionary selection for optimal activity. Thus, metagenomic screening for useful enzymes has gained popularity in the past 10 years.
EstHE1 exhibited esterase, but not lipase activity, and the enzymatic activity was stable in a wide range of temperatures and NaCl concentrations. Interestingly, salt inhibition of EstHE1 was greatest at a moderate NaCl concentration (1.9 M) but recovered slightly at higher salt concentrations. This behavior is similar to the enzymatic characteristic of actinidain. In the case of actinidain, the tryptophan fluorescence intensity was decreased by 0.5–3 M salt, while the esterase activity increased in same range of salt concentrations. Thus, structural changes in the environment of the tryptophan residues in the higher salt concentrations are thought to be associated with the reactivation of enzyme activity (Morimoto et al. 2006). Actinidain contains six tryptophan residues, one of which (W26) is located near the active residue C25 (Baker 1980). In contrast, EstHE1 only contains two tryptophan residues, which are well conserved in GDSL family enzyme. This NaCl reactivation behavior has not been reported for other GDSL family members, so the mechanism of this interesting characteristic is still unknown. However, unlike actinidain, the other SGNH-hydrolase superfamily members are thought to be stabilized by a unique hydrogen bond network located around the catalytic center (Lo et al. 2003). Further, GDSL family members are relatively flexible, which allows the active site to undergo conformation changes after substrate binding (Huang et al. 2001). Thus, the hydrogen bond network might allow for conformational changes by salt bridge formation and lead a recovery of activity at higher salt concentrations.
Stable enzymes that retain activity for a long period of time and can survive long-term storage are desired for the industrial processes. High thermostability is also desirable, because the enzymatic activity is amenable to a wide range of temperatures and retains activity for extended periods. While EstHE1 does not show thermophilic, it did exhibit stability to moderately high temperatures (up to 40°C) for an extended period of time (12 h). This suggests that EstHE1 is derived from a mesophilic, whose habitat tends to be at a higher temperature.
This study reveals that EstHE1 is both a member of a minor group of esterase/lipase and exhibits high-salt tolerance and moderate thermostability. These are favorable enzymatic characteristics, and it is improbable that PCR or selective evolution techniques would be able to engineer these characteristics from a previously known esterase enzyme. This study exemplifies the high potential that metagenome screening has for the identification and characterization of potentially novel and useful enzymes.
References
Akoh CC, Lee GC, Liaw YC, Huang TH, Shaw JF (2004) GDSL family of serine esterases/lipases. Prog Lipid Res 43:534–552
Baker EN (1980) Structure of actinidain, after refinement at 1.7 Å resolution. J Mol Biol 141:441–484
Cherry JR, Fidantsef AL (2003) Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol 14:438–443
Cho H, Cronan JE Jr (1993) Escherichia coli thioesterase I, molecular cloning and sequencing of the structural gene and identification as a periplasmic enzume. J Biol Chem 268:9238–9245
Dalrymple BP, Cybinski DH, Layton I, McSweeney CS, Xue GP, Swading YJ, Lowry JB (1997) Three Neocallimastix patriciarum esterases associated with the degradation of complex polysaccharides are members of a new family of hydrolases. Microbiology 143:2605–2614
Fukuda T, Kato-Murai M, Kadonosono T, Sahara H, Hata Y, Suye S, Ueda M (2007) Enhancement of substrate recognition ability by combinatorial mutation of β-glucosidase displayed on the yeast cell surface. Appl Microbiol Biotechnol 76:1027–1033
Gibbs MD, Nevalainen KM, Bergquist PL (2001) Degenerate oligonucleotide gene shuffling (DOGS): a method for enhancing the frequency of recombination with family shuffling. Gene 271:13–20
Giovannoni SJ, Britschgi TB, Moyer CL, Field KG (1990) Genetic diversity in Sargasso Sea bacterioplankton. Nature 345:60–63
Hashida M, Bisgaard-Frantzen H (2000) Protein engineering of new industrial amylases. Trends Glycosci Glycotechnol 12:389–401
Hentschel U, Schmid M, Wagner M, Fieseler L, Gernert C, Hacker J (2001) Isolation and phylogenetic analysis of bacteria with antimicrobial activities from the Mediterranean sponges Aplysina aerophoba and Aplysina cavernicola. FEMS Microbiol Ecol 35:305–312
Huang YT, Liaw YC, Gorbatyuk VK, Huang TH (2001) Backbone dynamics of Escherichia coli thioesterase/protease I: evidence of a flexible active-site environment for a serine protease. J Mol Biol 307:1075–1090
Koshland DE (1958) Application of a theory of enzyme specificity to protein synthesis (in Symposium on Amino Acid Activation). Proc Natl Acad Sci USA 44:98–104
Li J, Derewenda U, Dauter Z, Smith S, Derewenda ZS (2000) Crystal structure of the Escherichia coli esterase II, a homolog of the human Nef binding enzyme. Nat Struct Biol 7:555–559
Lo YC, Lin SC, Shaw JF, Liaw YC (2003) Crystal structure of the Escherichia coli thioesterase I/protease I/lysophospholipase L1: consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network. J Mol Biol 330:539–551
McGinnis S, Madden TL (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 1:32 (Web Server issue ): W20-5
Molgaard A, Kauppinen S, Larsen S (2000) Rhamnogalacturonan acetylesterase elucidated the structure and function of a new family of hydrolases. Struct Fold Des 8:373–383
Morimoto K, Furuta E, Hashimoto H, Inouye K (2006) Effects of high concentration of salts on the esterase activity and structure of a kiwifruit peptidase, actinidain. J Biochem 139:1065–1071
Reiswig HM (1974) Water transport, respiration and energetics of three tropical marine sponges. J Exp Mar Biol Ecol 14:231–249
Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacono KA, Lynch BA, MacNeil IA, Minor C, Tiong CL, Gilman M, Osburne MS, Clardy J, Handelsman J, Goodman RM (2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66:2541–2547
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Upton C, Buckley JT (1995) A new family of lipolytic enzymes? Trends Biochem Sci 20:178–179
Vacelet J, Donadey C (1977) Electron microscope study of the association between some sponges and bacteria. J Exp Mar Biol Ecol 30:301–314
Watanabe S, Saleh AA, Pack SP, Annaluru N, Kodaki T, Makino K (2007) Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein-engineered NADH-preferring xylose reductase from Pichia stipitis. Microbiol 153:3044–3054
Webster NS, Wilson KJ, Blackall LL, Hill RT (2001) Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl Environ Microbiol 67:434–444
Yokouchi H, Fukuoka Y, Mukoyama D, Calugay R, Takeyama H, Matsunaga T (2006) Whole-Metagenome Amplification of a Microbial Community Associated with Scleractinian Coral by Multiple Displacement Amplification Using ϕ29 Polymerase. Environ Microbiol 8:1155–1163
Acknowledgment
This work was funded in part by the New Energy and Industrial Technology Development Organization (NEDO) as part of a research and development project of the Industrial Science and Technology Frontier Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Okamura, Y., Kimura, T., Yokouchi, H. et al. Isolation and Characterization of a GDSL Esterase from the Metagenome of a Marine Sponge-associated Bacteria. Mar Biotechnol 12, 395–402 (2010). https://doi.org/10.1007/s10126-009-9226-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-009-9226-x